
Cysteine-Rich LIM-Only Protein 4 (CRP4) Promotes Atherogenesis in the ApoE−/− Mouse Model

Abstract
:1. Introduction
2. Material & Methods
2.1. Animal Care
2.2. In Vivo Atherosclerosis Mouse Model, Aorta Isolation and Plasma Lipid Profiles
2.3. En Face Oil Red O Staining
2.4. Cryosectioning of Aortic Vessels
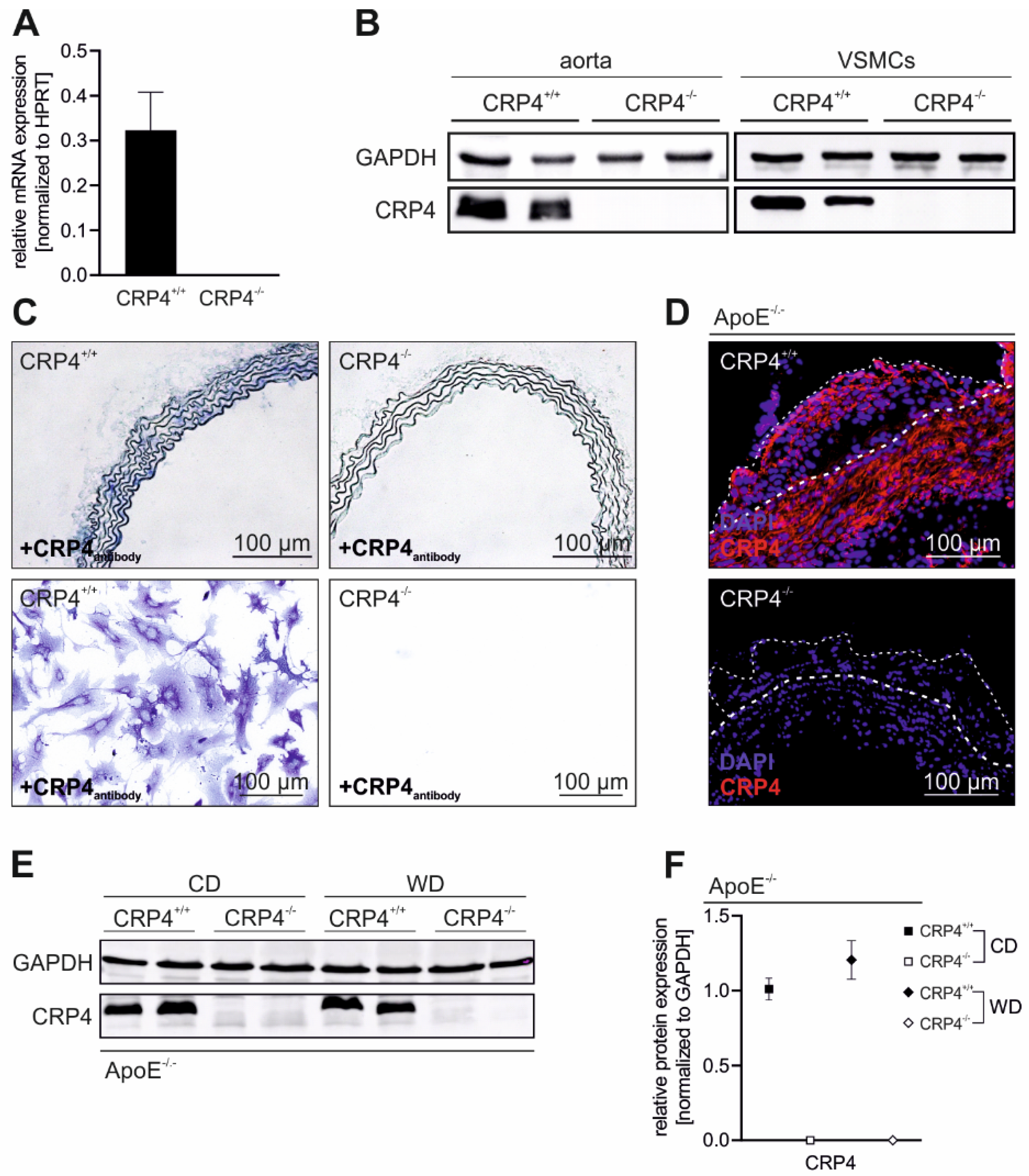
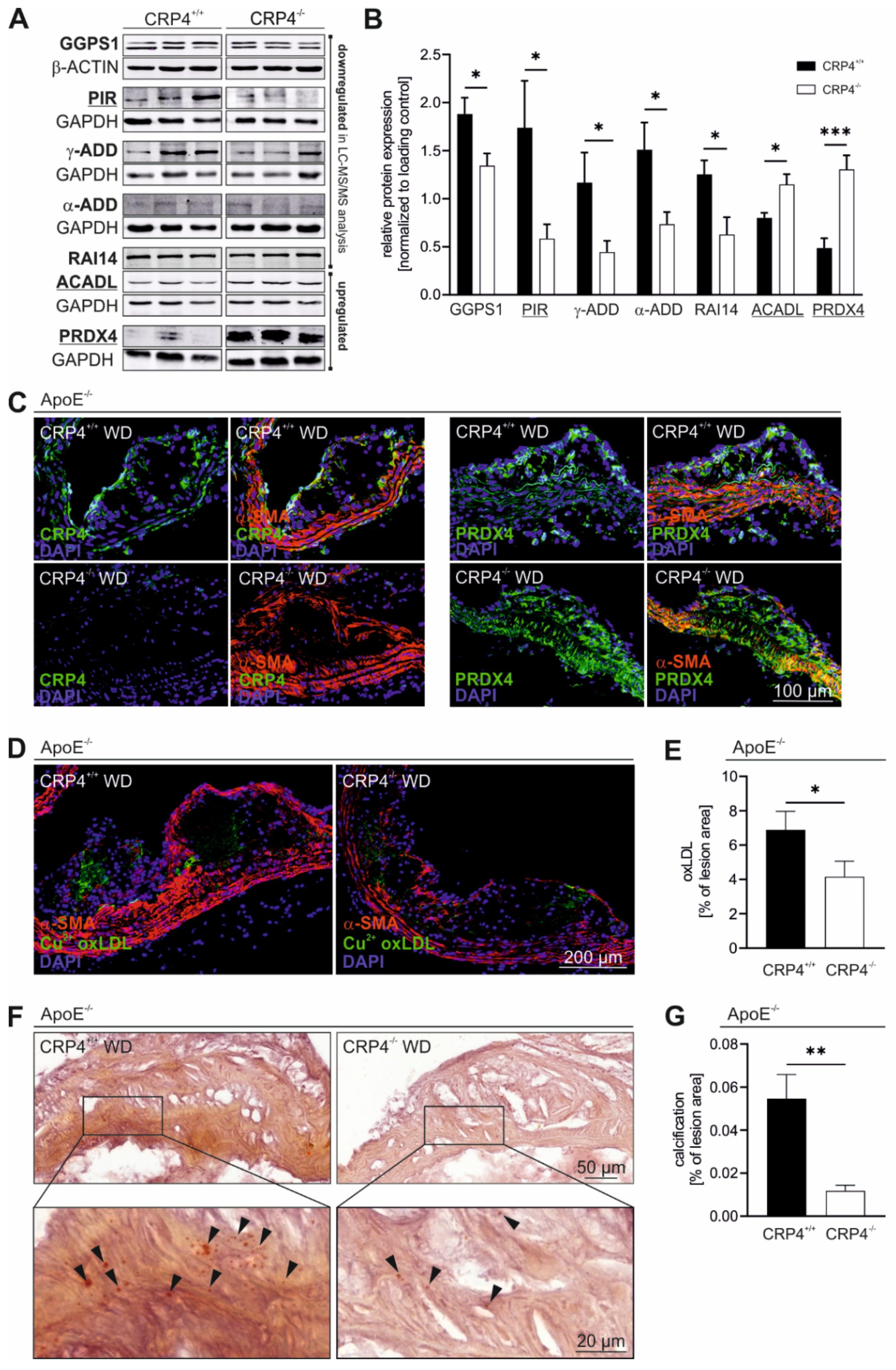
2.5. Immunohistochemistry and Immunofluorescence
2.6. Histological Staining of Aortic Lesions
2.7. Isolation of VSMCs from Mouse Aorta
2.8. mRNA Transcript Analysis
2.9. Immunoblot Analyses
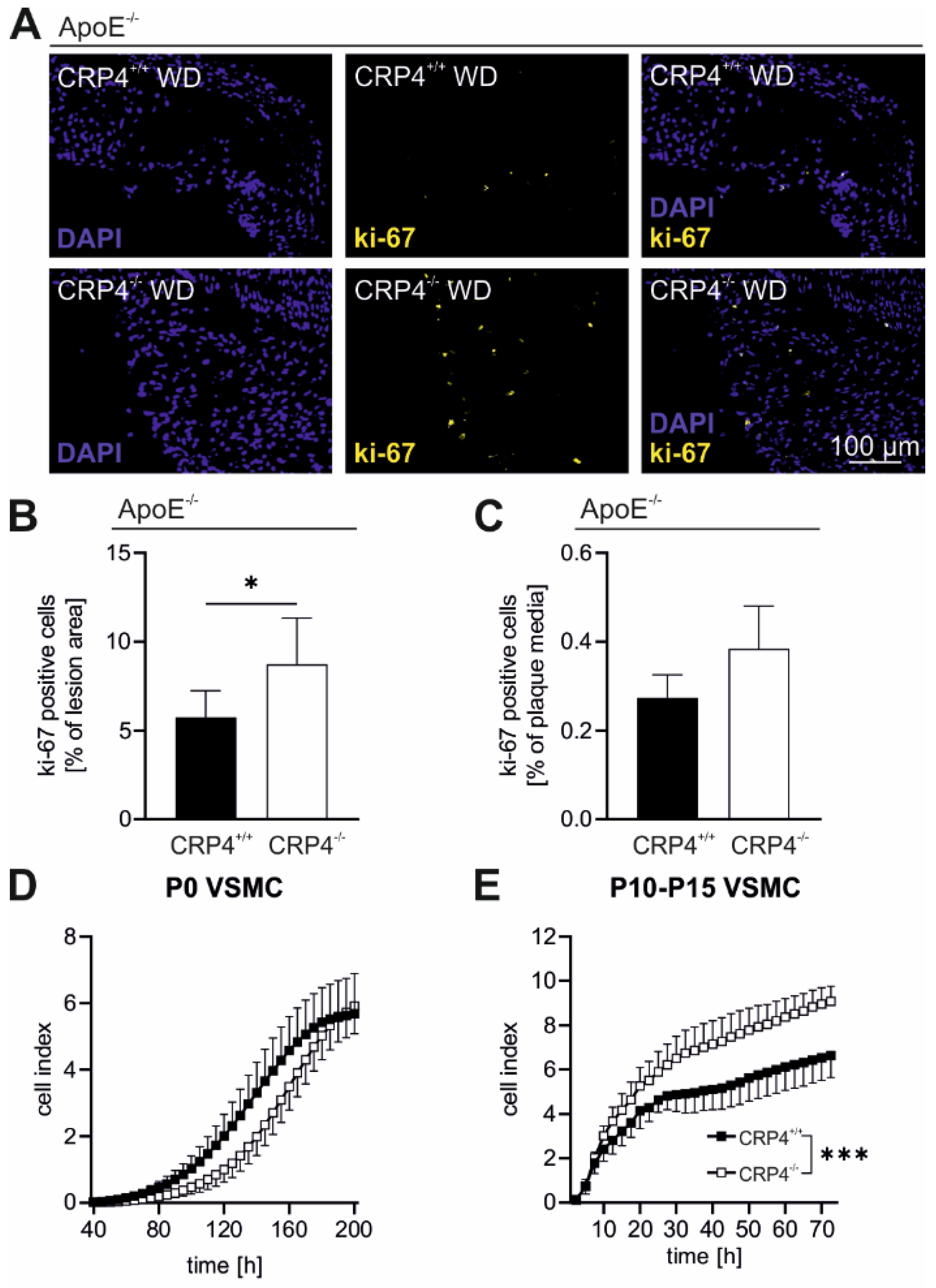
2.10. Migration Assay of Primary VSMCs
2.11. xCELLigence Proliferation Assay
2.12. Proteome Analysis
2.13. Statistical Analyses
3. Results
3.1. CRP4 Is Expressed in Medial VSMCs and in Atherosclerotic Lesions
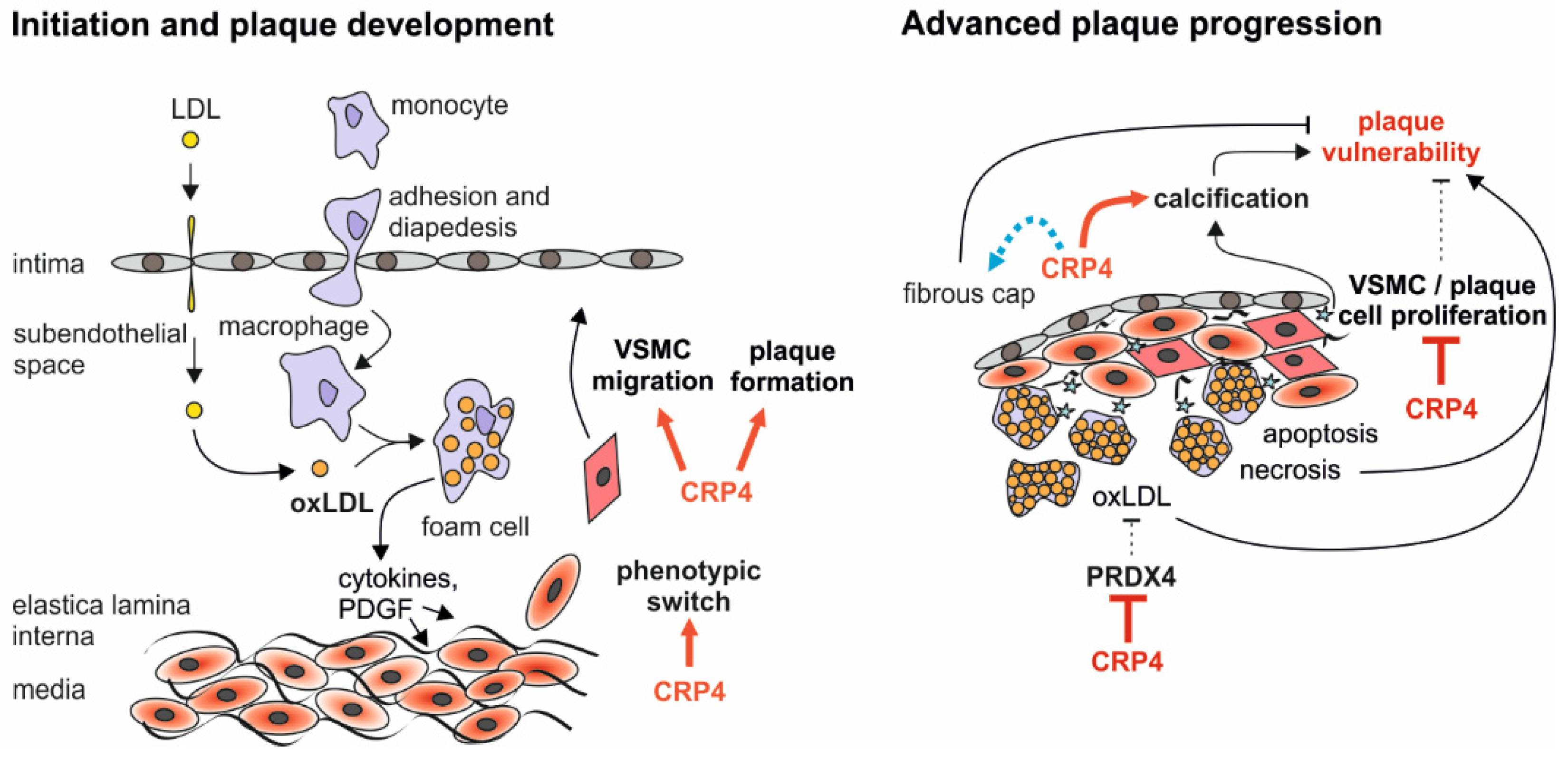
3.2. Lack of CRP4 Attenuates Atherosclerotic Plaque Progression in WD-Fed ApoE−/− Mice
3.3. Loss of CRP4 Results in a Reduced α-SMA Content in Atherosclerotic Vessels and in Lower Migratory Capacities of Primary VSMCs
3.4. Elevated Proliferation Rates in Synthetic VSMCs In Vitro and in Atherosclerotic Plaque Cells Lacking CRP4
3.5. Loss of CRP4 Causes Significant Alterations in the VSMC Proteome and has Adverse Effects on Oxidative Stress and Vascular Calcification
4. Discussion
5. Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tabas, I.; García-Cardeña, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Basatemur, G.L.; Jørgensen, H.F.; Clarke, M.C.; Bennett, M.R.; Mallat, Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M.R.; Owens, G.K. Epigenetic Control of Smooth Muscle Cell Differentiation and Phenotypic Switching in Vascular Development and Disease. Annu. Rev. Physiol. 2012, 74, 13–40. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Roostalu, U.; Wong, J.K.F. Arterial smooth muscle dynamics in development and repair. Dev. Biol. 2018, 435, 109–121. [Google Scholar] [CrossRef]
- Sakakura, K.; Nakano, M.; Otsuka, F.; Ladich, E.; Kolodgie, F.D.; Virmani, R. Pathophysiology of Atherosclerosis Plaque Progression. Heart Lung Circ. 2013, 22, 399–411. [Google Scholar] [CrossRef] [Green Version]
- Allahverdian, S.; Chaabane, C.; Boukais, K.; Francis, G.A.; Bochaton-Piallat, M.-L. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc. Res. 2018, 114, 540–550. [Google Scholar] [CrossRef] [Green Version]
- Van Rooy, M.-J.; Pretorius, E. Obesity, hypertension and hypercholesterolemia as risk factors for atherosclerosis leading to ischemic events. Curr. Med. Chem. 2014, 21, 2121–2129. [Google Scholar] [CrossRef]
- Glowinska, B.; Urban, M.; Koput, A.; Galar, M. New atherosclerosis risk factors in obese, hypertensive and diabetic children and adolescents. Atherosclerosis 2003, 167, 275–286. [Google Scholar] [CrossRef]
- Harman, J.L.; Jørgensen, H.F. The role of smooth muscle cells in plaque stability: Therapeutic targeting potential. Br. J. Pharmacol. 2019, 176, 3741–3753. [Google Scholar] [CrossRef] [PubMed]
- Sitia, S.; Tomasoni, L.; Atzeni, F.; Ambrosio, G.; Cordiano, C.; Catapano, A.; Tramontana, S.; Perticone, F.; Naccarato, P.; Camici, P.; et al. From endothelial dysfunction to atherosclerosis. Autoimmun. Rev. 2010, 9, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of Vascular Smooth Muscle Cells to Macrophage-Like Cells During Atherogenesis. Circ. Res. 2014, 115, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L. Metalloproteinases in atherosclerosis. Eur. J. Pharmacol. 2017, 816, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Moore, K.J.; Tabas, I. Macrophages in the Pathogenesis of Atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.C.H.; Figg, N.; Maguire, J.J.; Davenport, A.P.; Goddard, M.; Littlewood, T.D.; Bennett, M.R. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat. Med. 2006, 12, 1075–1080. [Google Scholar] [CrossRef]
- Gomez, D.; Shankman, L.S.; Nguyen, A.T.; Owens, G.K. Detection of histone modifications at specific gene loci in single cells in histological sections. Nat. Methods 2013, 10, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Dubland, J.A.; Allahverdian, S.; Asonye, E.; Sahin, B.; Jaw, J.E.; Sin, D.D.; Seidman, M.A.; Leeper, N.J.; Francis, G.A. Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 876–887. [Google Scholar] [CrossRef]
- Liu, R.; Leslie, K.L.; Martin, K.A. Epigenetic regulation of smooth muscle cell plasticity. Biochim. Et Biophys. Acta (BBA) Gene Regul. Mech. 2015, 1849, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhuang, S.; Casteel, D.E.; Looney, D.J.; Boss, G.R.; Pilz, R.B. A Cysteine-rich LIM-only Protein Mediates Regulation of Smooth Muscle-specific Gene Expression by cGMP-dependent Protein Kinase. J. Biol. Chem. 2007, 282, 33367–33380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-García, I.; Rabbits, T.H. The LIM domain: A new structural motif found in zinc-finger-like proteins. Trends Genet. 1994, 10, 315–320. [Google Scholar] [CrossRef]
- Huber, A.; Neuhuber, W.L.; Klugbauer, N.; Ruth, P.; Allescher, H.-D. Cysteine-rich Protein 2, a Novel Substrate for cGMP Kinase I in Enteric Neurons and Intestinal Smooth Muscle. J. Biol. Chem. 2000, 275, 5504–5511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiskirchen, R.; Pino, J.D.; Macalma, T.; Bister, K.; Beckerle, M.C. The Cysteine-rich Protein Family of Highly Related LIM Domain Proteins. J. Biol. Chem. 1995, 270, 28946–28954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiskirchen, R.; Günther, K. The CRP/MLP/TLP family of LIM domain proteins: Acting by connecting. BioEssays 2003, 25, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Okano, I.; Yamamoto, T.; Kaji, A.; Kimura, T.; Mizuno, K.; Nakamura, T. Cloning of CRP2, a novel member of the cysteine-rich protein family with two repeats of an unusual LIM/double zinc-finger motif. FEBS Lett. 1993, 333, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Straubinger, J.; Boldt, K.; Kuret, A.; Deng, L.; Krattenmacher, D.; Bork, N.; Desch, M.; Feil, R.; Feil, S.; Nemer, M.; et al. Amplified pathogenic actions of angiotensin II in cysteine-rich LIM-only protein 4–negative mouse hearts. FASEB J. 2017, 31, 1620–1638. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.-S.; Moctezuma-Anaya, M.; Kubo, A.; Keller, G.; Robertson, S. The heart LIM protein gene (Hlp), expressed in the developing and adult heart, defines a new tissue-specific LIM-only protein family. Mech. Dev. 2002, 116, 187–192. [Google Scholar] [CrossRef]
- Langst, N.; Adler, J.; Schweigert, O.; Kleusberg, F.; Cruz Santos, M.; Knauer, A.; Sausbier, M.; Zeller, T.; Ruth, P.; Lukowski, R. Cyclic GMP-Dependent Regulation of Vascular Tone and Blood Pressure Involves Cysteine-Rich LIM-Only Protein 4 (CRP4). Int. J. Mol. Sci. 2021, 22, 9925. [Google Scholar] [CrossRef]
- Liebhaber, S.A.; Emery, J.G.; Urbanek, M.; Wang, X.; Cooke, N.E. Characterization of a human cDNA encoding a widely expressed and highly conserved cysteine-rich protein with an unusual zinc-finger motif. Nucleic Acids Res. 1990, 18, 3871–3879. [Google Scholar] [CrossRef] [Green Version]
- Weiskirchen, R.; Bister, K. Suppression in transformed avian fibroblasts of a gene (crp) encoding a cysteine-rich protein containing LIM domains. Oncogene 1993, 8, 2317–2324. [Google Scholar] [PubMed]
- Arber, S.; Halder, G.; Caroni, P. Muscle LIM protein, a novel essential regulator of myogenesis, promotes myogenic differentiation. Cell 1994, 79, 221–231. [Google Scholar] [CrossRef]
- Lilly, B.; Clark, K.A.; Yoshigi, M.; Pronovost, S.; Wu, M.-L.; Periasamy, M.; Chi, M.; Paul, R.J.; Yet, S.-F.; Beckerle, M.C. Loss of the Serum Response Factor Cofactor, Cysteine-Rich Protein 1, Attenuates Neointima Formation in the Mouse. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Gorman, T.E.; Liu, X.; Ith, B.; Tseng, A.; Chen, Z.; Simon, D.I.; Layne, M.D.; Yet, S.-F. Increased Neointima Formation in Cysteine-Rich Protein 2–Deficient Mice in Response to Vascular Injury. Circ. Res. 2005, 97, 1323–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, D.F.; Belaguli, N.S.; Iyer, D.; Roberts, W.B.; Wu, S.-P.; Dong, X.-R.; Marx, J.G.; Moore, M.S.; Beckerle, M.C.; Majesky, M.W.; et al. Cysteine-Rich LIM-Only Proteins CRP1 and CRP2 Are Potent Smooth Muscle Differentiation Cofactors. Dev. Cell 2003, 4, 107–118. [Google Scholar] [CrossRef] [Green Version]
- Ignarro, L.J.; Kadowitz, P.J. The pharmacological and physiological role of cyclic GMP in vascular smooth muscle relaxation. Annu. Rev. Pharmacol. Toxicol. 1985, 25, 171–191. [Google Scholar] [CrossRef]
- Sausbier, M.; Schubert, R.; Voigt, V.; Hirneiss, C.; Pfeifer, A.; Korth, M.; Kleppisch, T.; Ruth, P.; Hofmann, F. Mechanisms of NO/cGMP-Dependent Vasorelaxation. Circ. Res. 2000, 87, 825–830. [Google Scholar] [CrossRef] [Green Version]
- Wolfsgruber, W.; Feil, S.; Brummer, S.; Kuppinger, O.; Hofmann, F.; Feil, R. A proatherogenic role for cGMP-dependent protein kinase in vascular smooth muscle cells. Proc. Natl. Acad. Sci. 2003, 100, 13519–13524. [Google Scholar] [CrossRef] [Green Version]
- Lukowski, R.; Weinmeister, P.; Bernhard, D.; Feil, S.; Gotthardt, M.; Herz, J.; Massberg, S.; Zernecke, A.; Weber, C.; Hofmann, F.; et al. Role of Smooth Muscle cGMP/cGKI Signaling in Murine Vascular Restenosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1244–1250. [Google Scholar] [CrossRef] [Green Version]
- Segura-Puimedon, M.; Mergia, E.; Al-Hasani, J.; Aherrahrou, R.; Stoelting, S.; Kremer, F.; Freyer, J.; Koesling, D.; Erdmann, J.; Schunkert, H.; et al. Proatherosclerotic Effect of the α1-Subunit of Soluble Guanylyl Cyclase by Promoting Smooth Muscle Phenotypic Switching. Am. J. Pathol. 2016, 186, 2220–2231. [Google Scholar] [CrossRef] [Green Version]
- Lehners, M.; Dobrowinski, H.; Feil, S.; Feil, R. cGMP Signaling and Vascular Smooth Muscle Cell Plasticity. J. Cardiovasc. Dev. Dis. 2018, 5, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidtko, A.; Gao, W.; Sausbier, M.; Rauhmeier, I.; Sausbier, U.; Niederberger, E.; Scholich, K.; Huber, A.; Neuhuber, W.; Allescher, H.-D.; et al. Cysteine-Rich Protein 2, a Novel Downstream Effector of cGMP/cGMP-Dependent Protein Kinase I-Mediated Persistent Inflammatory Pain. J. Neurosci. 2008, 28, 1320–1330. [Google Scholar] [CrossRef] [PubMed]
- Mohanta, S.; Yin, C.; Weber, C.; Hu, D.; Habenicht, A., Jr. Aorta Atherosclerosis Lesion Analysis in Hyperlipidemic Mice. Bio Protoc 2016, 6, e1833. [Google Scholar] [CrossRef] [PubMed]
- Weinmeister, P.; Lukowski, R.; Linder, S.; Traidl-Hoffmann, C.; Hengst, L.; Hofmann, F.; Feil, R. Cyclic guanosine monophosphate-dependent protein kinase I promotes adhesion of primary vascular smooth muscle cells. Mol. Biol. Cell 2008, 19, 4434–4441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroux-Berger, M.; Queguiner, I.; Maciel, T.T.; Ho, A.; Relaix, F.; Kempf, H. Pathologic calcification of adult vascular smooth muscle cells differs on their crest or mesodermal embryonic origin. J. Bone Miner. Res. 2011, 26, 1543–1553. [Google Scholar] [CrossRef] [PubMed]
- Boldt, K.; van Reeuwijk, J.; Lu, Q.; Koutroumpas, K.; Nguyen, T.M.; Texier, Y.; van Beersum, S.E.; Horn, N.; Willer, J.R.; Mans, D.A.; et al. An organelle-specific protein landscape identifies novel diseases and molecular mechanisms. Nat Commun 2016, 7, 11491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Owens, G.K. Regulation of differentiation of vascular smooth muscle cells. Physiol. Rev. 1995, 75, 487–517. [Google Scholar] [CrossRef]
- Chappell, J.; Harman, J.L.; Narasimhan, V.M.; Yu, H.; Foote, K.; Simons, B.D.; Bennett, M.R.; Jørgensen, H.F. Extensive Proliferation of a Subset of Differentiated, yet Plastic, Medial Vascular Smooth Muscle Cells Contributes to Neointimal Formation in Mouse Injury and Atherosclerosis Models. Circ. Res. 2016, 119, 1313–1323. [Google Scholar] [CrossRef]
- Jacobsen, K.; Lund, M.B.; Shim, J.; Gunnersen, S.; Füchtbauer, E.-M.; Kjolby, M.; Carramolino, L.; Bentzon, J.F. Diverse cellular architecture of atherosclerotic plaque derives from clonal expansion of a few medial SMCs. JCI Insight 2017, 2, e95890. [Google Scholar] [CrossRef]
- Wamhoff, B.R.; Hoofnagle, M.H.; Burns, A.; Sinha, S.; McDonald, O.G.; Owens, G.K. A G/C Element Mediates Repression of the SM22α Promoter within Phenotypically Modulated Smooth Muscle Cells in Experimental Atherosclerosis. Circ. Res. 2004, 95, 981–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonkman, J.E.; Cathcart, J.A.; Xu, F.; Bartolini, M.E.; Amon, J.E.; Stevens, K.M.; Colarusso, P. An introduction to the wound healing assay using live-cell microscopy. Cell Adh. Migr. 2014, 8, 440–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamley, J.H.; Campbell, G.R.; McConnell, J.D.; Gröschel-Stewart, U. Comparison of vascular smooth muscle cells from adult human, monkey and rabbit in primary culture and in subculture. Cell Tissue Res. 1977, 177, 503–522. [Google Scholar] [CrossRef] [PubMed]
- Kramer, N.; Walzl, A.; Unger, C.; Rosner, M.; Krupitza, G.; Hengstschläger, M.; Dolznig, H. In vitro cell migration and invasion assays. Mutat. Res. /Rev. Mutat. Res. 2013, 752, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Yang, X.; Lin, Q.; Liu, S.; Xie, Y.; Xia, Y.; Li, H.H. Inhibition of the Ubiquitin-Activating Enzyme UBA1 Suppresses Diet-Induced Atherosclerosis in Apolipoprotein E-Knockout Mice. J. Immunol Res. 2020, 2020, 7812709. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Yamada, S.; Tanimoto, A.; Ding, Y.; Wang, K.; Shimajiri, S.; Murata, Y.; Kimura, S.; Tasaki, T.; Nabeshima, A.; et al. Overexpression of Peroxiredoxin 4 Attenuates Atherosclerosis in Apolipoprotein E Knockout Mice. Antioxid. Redox Signal. 2012, 17, 1362–1375. [Google Scholar] [CrossRef] [Green Version]
- De Keyzer, D.; Karabina, S.A.; Wei, W.; Geeraert, B.; Stengel, D.; Marsillach, J.; Camps, J.; Holvoet, P.; Ninio, E. Increased PAFAH and oxidized lipids are associated with inflammation and atherosclerosis in hypercholesterolemic pigs. Arter. Thromb. Vasc. Biol. 2009, 29, 2041–2046. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, D.M.; Rinaldo, P.; Rhead, W.J.; Tian, L.; Millington, D.S.; Vockley, J.; Hamm, D.A.; Brix, A.E.; Lindsey, J.R.; Pinkert, C.A.; et al. Targeted disruption of mouse long-chain acyl-CoA dehydrogenase gene reveals crucial roles for fatty acid oxidation. Proc. Natl. Acad. Sci. USA 1998, 95, 15592–15597. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Huff, L.P.; Fujii, M.; Griendling, K.K. Redox regulation of the actin cytoskeleton and its role in the vascular system. Free Radic. Biol. Med. 2017, 109, 84–107. [Google Scholar] [CrossRef]
- Lundquist, M.R.; Storaska, A.J.; Liu, T.-C.; Larsen, S.D.; Evans, T.; Neubig, R.R.; Jaffrey, S.R. Redox Modification of Nuclear Actin by MICAL-2 Regulates SRF Signaling. Cell 2014, 156, 563–576. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, S.; Pfeuffer, S.; Arnold, P.; Treitz, C.; Aden, K.; Ebsen, H.; Falk-Paulsen, M.; Gisch, N.; Fazio, A.; Kuiper, J.; et al. Prdx4 limits caspase-1 activation and restricts inflammasome-mediated signaling by extracellular vesicles. EMBO J. 2019, 38, e101266. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Rehmani, I.; Esaki, S.; Fu, R.; Chen, L.; de Serrano, V.; Liu, A. Pirin is an iron-dependent redox regulator of NF-kappaB. Proc. Natl. Acad. Sci. USA 2013, 110, 9722–9727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, K.; Fukata, Y.; Matsuoka, Y.; Bennett, V.; Matsuura, Y.; Okawa, K.; Iwamatsu, A.; Kaibuchi, K. Regulation of the association of adducin with actin filaments by Rho-associated kinase (Rho-kinase) and myosin phosphatase. J. Biol. Chem. 1998, 273, 5542–5548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuoka, Y.; Li, X.; Bennett, V. Adducin: Structure, function and regulation. Cell Mol. Life Sci. 2000, 57, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.F.; Mandai, K.; Sakisaka, T.; Okabe, N.; Yamamoto, Y.; Yokoyama, S.; Mizoguchi, A.; Shiozaki, H.; Monden, M.; Takai, Y. Ankycorbin: A novel actin cytoskeleton-associated protein. Genes Cells 2000, 5, 1001–1008. [Google Scholar] [CrossRef]
- Rahuel, C.; Filipe, A.; Ritie, L.; El Nemer, W.; Patey-Mariaud, N.; Eladari, D.; Cartron, J.P.; Simon-Assmann, P.; Le Van Kim, C.; Colin, Y. Genetic inactivation of the laminin alpha5 chain receptor Lu/BCAM leads to kidney and intestinal abnormalities in the mouse. Am. J. Physiol. Renal. Physiol. 2008, 294, F393–F406. [Google Scholar] [CrossRef]
- Kainou, T.; Kawamura, K.; Tanaka, K.; Matsuda, H.; Kawamukai, M. Identification of the GGPS1 genes encoding geranylgeranyl diphosphate synthases from mouse and human. Biochim. Biophys. Acta 1999, 1437, 333–340. [Google Scholar] [CrossRef]
- Foley, A.R.; Zou, Y.; Dunford, J.E.; Rooney, J.; Chandra, G.; Xiong, H.; Straub, V.; Voit, T.; Romero, N.; Donkervoort, S.; et al. GGPS1 Mutations Cause Muscular Dystrophy/Hearing Loss/Ovarian Insufficiency Syndrome. Ann. Neurol. 2020, 88, 332–347. [Google Scholar] [CrossRef]
- Zani, A.; Yount, J.S. Antiviral Protection by IFITM3 In Vivo. Curr. Clin. Microbiol. Rep. 2018, 5, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Kenney, A.D.; McMichael, T.M.; Imas, A.; Chesarino, N.M.; Zhang, L.; Dorn, L.E.; Wu, Q.; Alfaour, O.; Amari, F.; Chen, M.; et al. IFITM3 protects the heart during influenza virus infection. Proc. Natl. Acad. Sci. USA 2019, 116, 18607–18612. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Zhang, T.; Yao, F.; Liao, Y.; Liu, F.; Ren, Z.; Han, L.; Diao, L.; Li, Y.; Zhou, B.; et al. THO Complex-Dependent Posttranscriptional Control Contributes to Vascular Smooth Muscle Cell Fate Decision. Circ. Res. 2018, 123, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Cui, B.; Jin, J.; Song, M.; Zhou, B.; Guo, H.; Qian, D.; He, Y.; Huang, L. The ubiquitin-activating enzyme E1 as a novel therapeutic target for the treatment of restenosis. Atherosclerosis 2016, 247, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Iuchi, Y.; Okada, F.; Tsunoda, S.; Kibe, N.; Shirasawa, N.; Ikawa, M.; Okabe, M.; Ikeda, Y.; Fujii, J. Peroxiredoxin 4 knockout results in elevated spermatogenic cell death via oxidative stress. Biochem. J. 2009, 419, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Guo, X. Peroxiredoxin 4 (PRDX4): Its critical in vivo roles in animal models of metabolic syndrome ranging from atherosclerosis to nonalcoholic fatty liver disease. Pathol. Int. 2018, 68, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Pinho Melo, E.; Lopes, C.; Mehmeti, I.; Lenzen, S.; Ron, D.; Avezov, E. ERO1-independent production of H2O2 within the endoplasmic reticulum fuels Prdx4-mediated oxidative protein folding. J. Cell Biol. 2015, 211, 253–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byon, C.H.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative Stress Induces Vascular Calcification through Modulation of the Osteogenic Transcription Factor Runx2 by AKT Signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mody, N.; Parhami, F.; Sarafian, T.A.; Demer, L.L. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic. Biol. Med. 2001, 31, 509–519. [Google Scholar] [CrossRef]
- Farrokhi, E.; Chaleshtori, M.H.; Samani, K.G. Oxidized Low-Density Lipoprotein Increases Bone Sialoprotein Expression in Vascular Smooth Muscle Cells Via Runt-Related Transcription Factor 2. Am. J. Med. Sci. 2015, 349, 240–243. [Google Scholar] [CrossRef]
- Kelly-Arnold, A.; Maldonado, N.; Laudier, D.; Aikawa, E.; Cardoso, L.; Weinbaum, S. Revised microcalcification hypothesis for fibrous cap rupture in human coronary arteries. Proc. Natl. Acad. Sci. USA 2013, 110, 10741–10746. [Google Scholar] [CrossRef] [Green Version]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of Intimal Smooth Muscle Cells to Cholesterol Accumulation and Macrophage-Like Cells in Human Atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef] [Green Version]
- Hui, D.Y.; Basford, J.E. Distinct signaling mechanisms for apoE inhibition of cell migration and proliferation. Neurobiol. Aging 2005, 26, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Oppi, S.; Lüscher, T.F.; Stein, S. Mouse Models for Atherosclerosis Research—Which Is My Line? Front. Cardiovasc. Med. 2019, 6, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Vascular smooth muscle cell in atherosclerosis. Acta Physiol. 2015, 214, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Parhami, F.; Morrow, A.D.; Balucan, J.; Leitinger, N.; Watson, A.D.; Tintut, Y.; Berliner, J.A.; Demer, L.L. Lipid Oxidation Products Have Opposite Effects on Calcifying Vascular Cell and Bone Cell Differentiation. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 680–687. [Google Scholar] [CrossRef]
- Lukowski, R.; Cruz Santos, M.; Kuret, A.; Ruth, P. cGMP and mitochondrial K+ channels-Compartmentalized but closely connected in cardioprotection. Br. J. Pharmacol. 2021. [CrossRef]
- Iuliano, L. The oxidant stress hypothesis of atherogenesis. Lipids 2001, 36, S41–S44. [Google Scholar] [CrossRef]
- D’Archivio, M.; Annuzzi, G.; Varì, R.; Filesi, C.; Giacco, R.; Scazzocchio, B.; Santangelo, C.; Giovannini, C.; Rivellese, A.A.; Masella, R. Predominant role of obesity/insulin resistance in oxidative stress development. Eur. J. Clin. Investig. 2012, 42, 70–78. [Google Scholar] [CrossRef]
- Vendrov, A.E.; Madamanchi, N.R.; Hakim, Z.S.; Rojas, M.; Runge, M.S. Thrombin and NAD(P)H Oxidase–Mediated Regulation of CD44 and BMP4-Id Pathway in VSMC, Restenosis, and Atherosclerosis. Circ. Res. 2006, 98, 1254–1263. [Google Scholar] [CrossRef] [Green Version]
- Barry-Lane, P.A.; Patterson, C.; van der Merwe, M.; Hu, Z.; Holland, S.M.; Yeh, E.T.H.; Runge, M.S. p47phox is required for atherosclerotic lesion progression in ApoE–/– mice. J. Clin. Investig. 2001, 108, 1513–1522. [Google Scholar] [CrossRef]
- Harrison, D.; Griendling, K.K.; Landmesser, U.; Hornig, B.; Drexler, H. Role of oxidative stress in atherosclerosis. Am. J. Cardiol. 2003, 91, 7–11. [Google Scholar] [CrossRef]
- Tavender, T.J.; Bulleid, N.J. Peroxiredoxin IV protects cells from oxidative stress by removing H2O2 produced during disulphide formation. J. Cell Sci. 2010, 123, 2672–2679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, D.-Y.; Chae, H.Z.; Rhee, S.G.; Jeang, K.-T. Regulatory Role for a Novel Human Thioredoxin Peroxidase in NF-κB Activation. J. Biol. Chem. 1997, 272, 30952–30961. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.-M.; Chun, A.C.; Kok, K.; Zhou, Y.; Fung, P.C.; Kung, H.-F.; Jeang, K.-T.; Jin, D.-Y. Characterization of human and mouse peroxiredoxin IV: Evidence for inhibition by Prx-IV of epidermal growth factor-and p53-induced reactive oxygen species. Antioxid. Redox Signal. 2000, 2, 507–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichart, D.; Gobom, J.; Klopfleisch, S.; Häsler, R.; Gustavsson, N.; Billmann, S.; Lehrach, H.; Seegert, D.; Schreiber, S.; Rosenstiel, P. Analysis of NOD2-mediated Proteome Response to Muramyl Dipeptide in HEK293 Cells. J. Biol. Chem. 2006, 281, 2380–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Mu, Y.; Ao, J.; Chen, X. Peroxiredoxin IV Regulates Pro-Inflammatory Responses in Large Yellow Croaker (Pseudosciaena crocea) and Protects against Bacterial Challenge. J. Proteome Res. 2010, 9, 1424–1436. [Google Scholar] [CrossRef] [PubMed]
- Pamukcu, B.; Lip, G.Y.H.; Shantsila, E. The nuclear factor—kappa B pathway in atherosclerosis: A potential therapeutic target for atherothrombotic vascular disease. Thromb. Res. 2011, 128, 117–123. [Google Scholar] [CrossRef]
- Kisucka, J.; Chauhan, A.K.; Patten, I.S.; Yesilaltay, A.; Neumann, C.; Etten, R.A.V.; Krieger, M.; Wagner, D.D. Peroxiredoxin1 Prevents Excessive Endothelial Activation and Early Atherosclerosis. Circ. Res. 2008, 103, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-G.; Yoo, J.-Y.; Jeong, S.-J.; Choi, J.-H.; Lee, M.-R.; Lee, M.-N.; Lee, J.H.; Kim, H.C.; Jo, H.; Yu, D.-Y.; et al. Peroxiredoxin 2 Deficiency Exacerbates Atherosclerosis in Apolipoprotein E–Deficient Mice. Circ. Res. 2011, 109, 739–749. [Google Scholar] [CrossRef]
- Wang, X.; Phelan, S.A.; Petros, C.; Taylor, E.F.; Ledinski, G.; Jürgens, G.; Forsman-Semb, K.; Paigen, B. Peroxiredoxin 6 deficiency and atherosclerosis susceptibility in mice: Significance of genetic background for assessing atherosclerosis. Atherosclerosis 2004, 177, 61–70. [Google Scholar] [CrossRef]
- Hung, R.-J.; Pak, C.W.; Terman, J.R. Direct Redox Regulation of F-Actin Assembly and Disassembly by Mical. Science 2011, 334, 1710–1713. [Google Scholar] [CrossRef] [Green Version]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Gene Name | t-Test p-Value | Significance B Value | Regulation in CRP4−/− | Selected Reference(s) |
|---|---|---|---|---|---|
| Oxidation Reduction Related Pathways GO: 0055114 | |||||
| Acyl-CoA dehydrogenase long chain (ACADL) | Acadl | 0.027 | 0.004 |  | [58] |
| Microtubule associated monooxygenase, Calponin and LIM domain containing 2 (MICAL2) | Mical2 | 0.025 | 0.002 | | [59,60] |
| Peroxiredoxin-4 (PRDX4) | Prdx4 | 0.016 | 0.001 | | [56,61] |
| Pirin (PIR) | Pir | 0.012 | 0.000 |  | [62] |
| Other pathways | |||||
| α-Adducin (α-ADD) | Add1 | 0.045 | 1.01 × 10−6 | | [63,64] |
| Ankycorbin (RAI14) | Rai14 | 0.029 | 3.5 × 10−4 | | [65] |
| Basal cell adhesion molecule (BCAM) | Bcam | 0.012 | 1.11 × 10−320 | | [66] |
| γ-Adducin (γ-ADD) | Add3 | 0.024 | 1.01 × 10−8 | | [64] |
| Geranylgeranyl diphosphate synthase 1 (GGPS1) | Ggps1 | 3.9 × 10−12 | 3.12 × 10−73 | | [67,68] |
| Interferon-induced transmembrane protein 3 (IFITM3) | Ifitm3 | 0.038 | 0.002 | | [69,70] |
| Platelet activating factor acetylhydrolase 1b catalytic subunit 3 (PAFAH1B3) | Pafah1b3 | 0.021 | 0.004 | | [57] |
| THO complex subunit 2 (THOC2) | Thoc2 | 0.008 | 1.58 × 10−61 | | [71] |
| Ubiquitin like modifier activating enzyme 1 (UBA1) | Uba1 | 0.010 | 4.57 × 10−5 | | [55,72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Längst, N.; Adler, J.; Kuret, A.; Peter, A.; Ruth, P.; Boldt, K.; Lukowski, R. Cysteine-Rich LIM-Only Protein 4 (CRP4) Promotes Atherogenesis in the ApoE−/− Mouse Model. Cells 2022, 11, 1364. https://doi.org/10.3390/cells11081364
Längst N, Adler J, Kuret A, Peter A, Ruth P, Boldt K, Lukowski R. Cysteine-Rich LIM-Only Protein 4 (CRP4) Promotes Atherogenesis in the ApoE−/− Mouse Model. Cells. 2022; 11(8):1364. https://doi.org/10.3390/cells11081364
Chicago/Turabian StyleLängst, Natalie, Julia Adler, Anna Kuret, Andreas Peter, Peter Ruth, Karsten Boldt, and Robert Lukowski. 2022. "Cysteine-Rich LIM-Only Protein 4 (CRP4) Promotes Atherogenesis in the ApoE−/− Mouse Model" Cells 11, no. 8: 1364. https://doi.org/10.3390/cells11081364