
Pancreatic Islet Cells Response to IFNγ Relies on Their Spatial Location within an Islet
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Procedure
2.2. Isolation, Culture and Treatment of Mouse Pancreatic Islets
2.3. Flow Cytometry
2.4. Reaggregation Experiments
2.5. RNA Extraction, Reverse Transcription and qPCR
2.6. Immunohistochemistry
2.7. Next Generation Sequencing and Bioinformatics
2.8. Statistical Analyses
3. Results
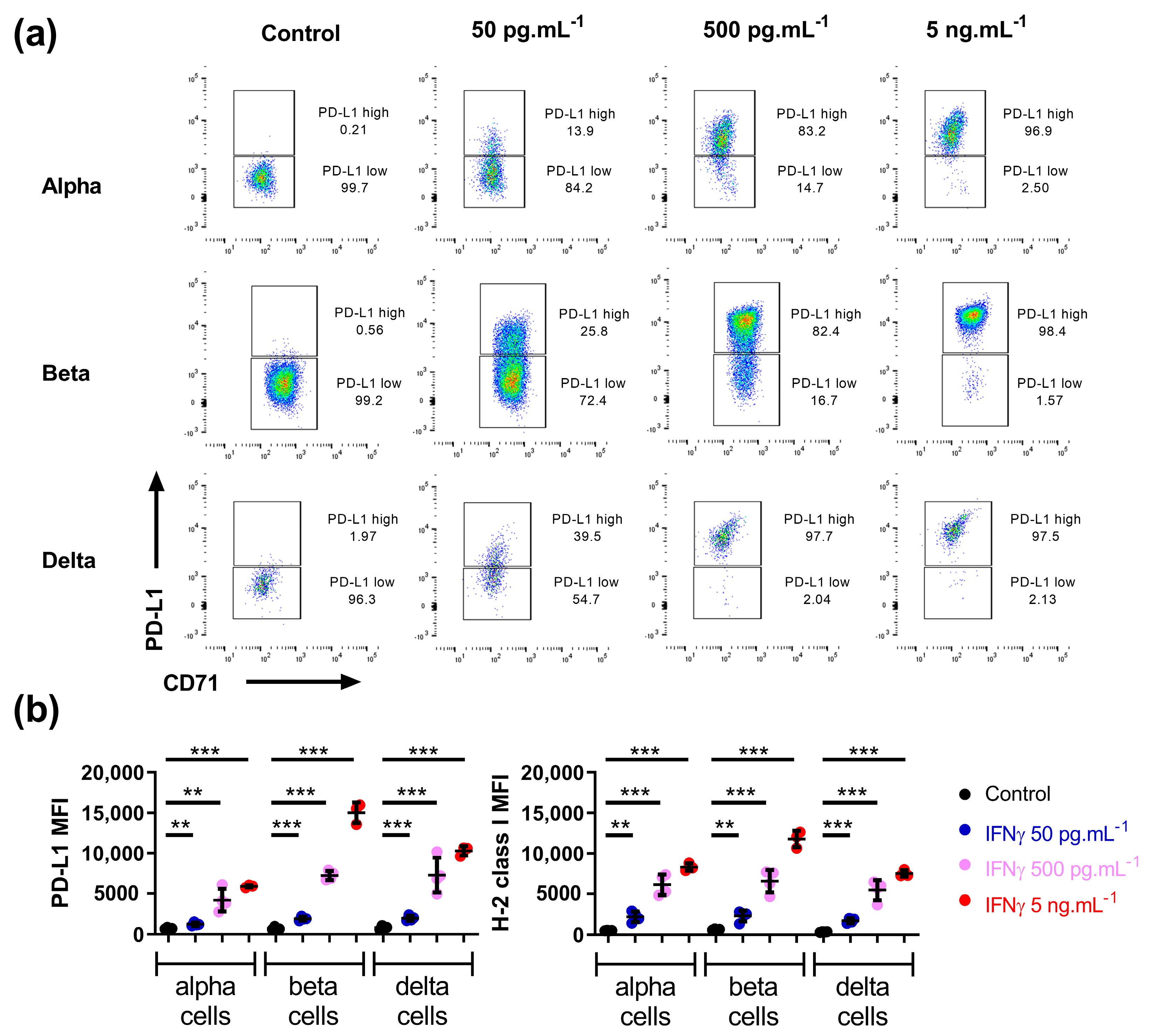
3.1. Mouse Alpha, Beta, and Delta Cells Respond to IFNγ in a Similar Manner
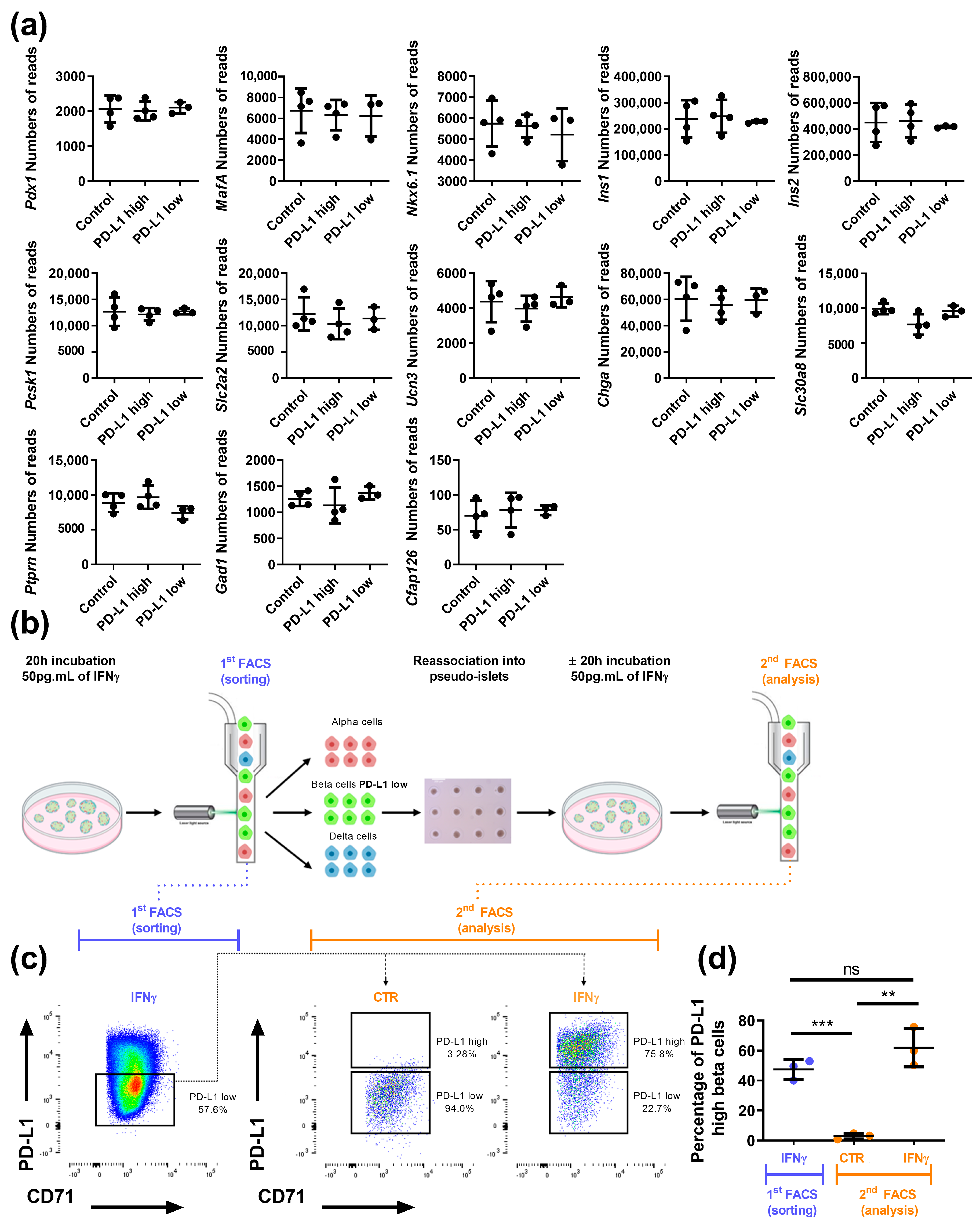
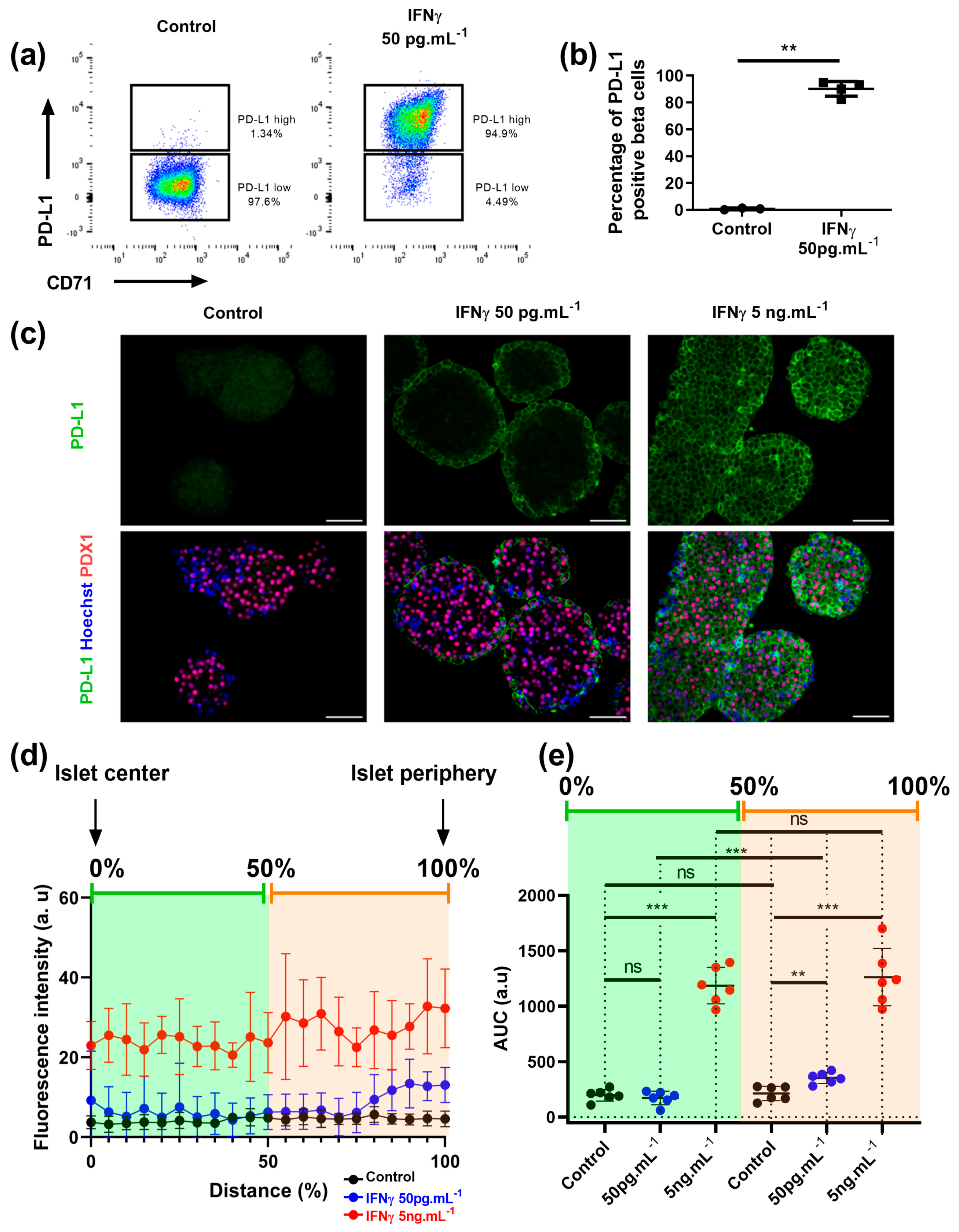
3.2. Beta Cell Heterogeneity in Response to IFNγ
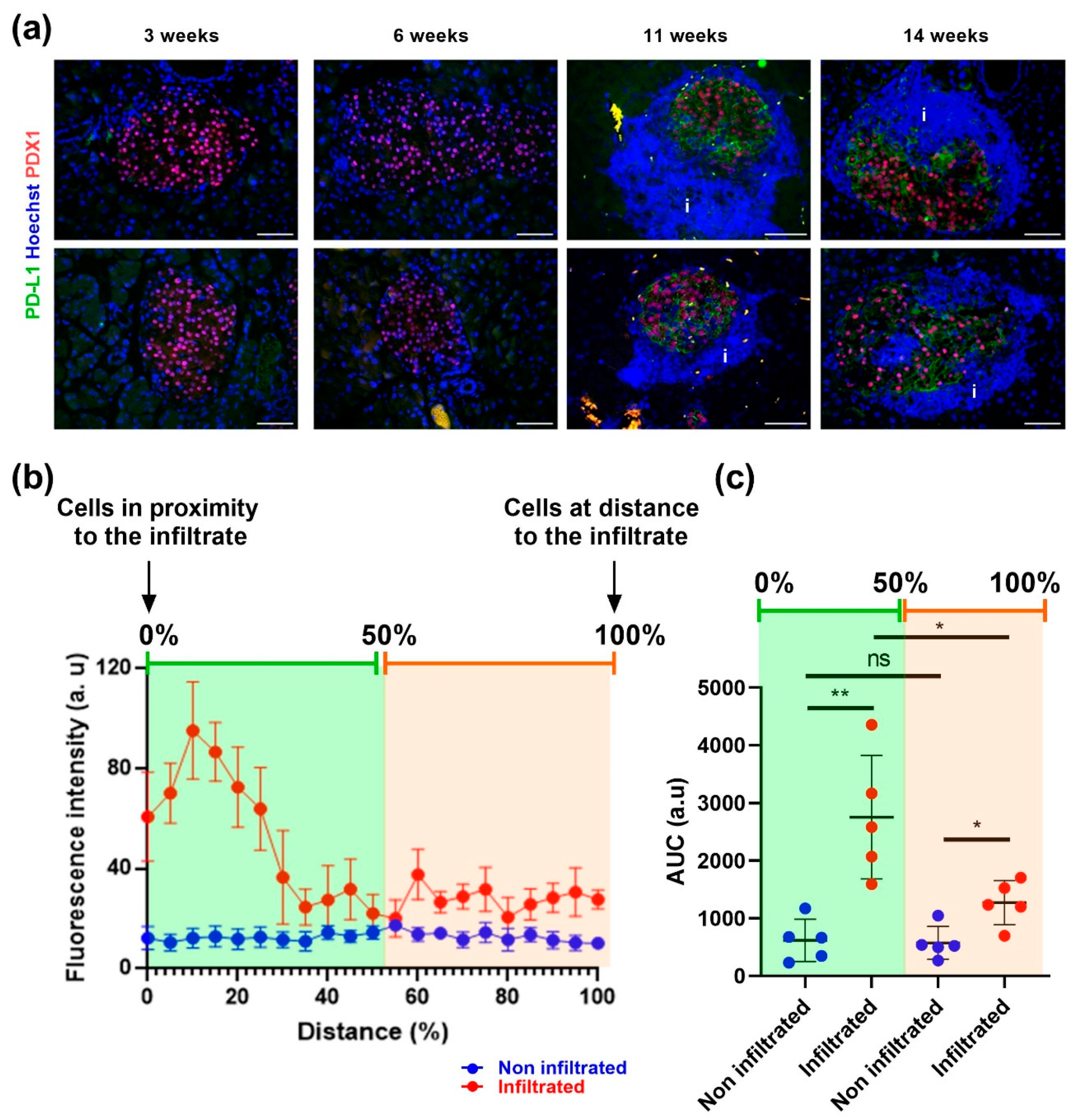
3.3. Beta Cells That Respond to IFNγ Are Located at the Islet Periphery
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kägi, D.; Odermatt, B.; Seiler, P.; Zinkernagel, R.M.; Mak, T.W.; Hengartner, H. Reduced Incidence and Delayed Onset of Diabetes in Perforin-Deficient Nonobese Diabetic Mice. J. Exp. Med. 1997, 186, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amrani, A.; Verdaguer, J.; Anderson, B.; Utsugi, T.; Bou, S.; Santamaria, P. Perforin-Independent β-Cell Destruction by Diabetogenic CD8+ T Lymphocytes in Transgenic Nonobese Diabetic Mice. J. Clin. Investig. 1999, 103, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Varanasi, V.; Avanesyan, L.; Schumann, D.M.; Chervonsky, A.V. Cytotoxic Mechanisms Employed by Mouse T Cells to Destroy Pancreatic B-Cells. Diabetes 2012, 61, 2862–2870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eizirik, D.L.; Mandrup-Poulsen, T. A Choice of Death—the Signal-Transduction of Immune-Mediated Beta-Cell Apoptosis. Diabetologia 2001, 44, 2115–2133. [Google Scholar] [CrossRef]
- Cnop, M.; Welsh, N.; Jonas, J.-C.; Jörns, A.; Lenzen, S.; Eizirik, D.L. Mechanisms of Pancreatic B-Cell Death in Type 1 and Type 2 Diabetes Many Differences, Few Similarities. Diabetes 2005, 54, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Pukel, C.; Baquerizo, H.; Rabinovitch, A. Destruction of Rat Islet Cell Monolayers by Cytokines.. Synergistic interactions of interferon-gamma, tumor necrosis factor, lymphotoxin, and interleukin 1. Diabetes 1988, 37, 133–136. [Google Scholar] [CrossRef]
- Campbell, I.L.; Iscaro, A.; Harrison, L.C. IFN-Gamma and Tumor Necrosis Factor-Alpha. Cytotoxicity to Murine Islets of Langerhans. J. Immunol. 1988, 141, 2325–2329. [Google Scholar]
- Delaney, C.A.; Pavlovic, D.; Hoorens, A.; Pipeleers, D.G.; Eizirik, D.L. Cytokines Induce Deoxyribonucleic Acid Strand Breaks and Apoptosis in Human Pancreatic Islet Cells. Endocrinology 1997, 138, 2610–2614. [Google Scholar] [CrossRef]
- Liu, D.; Pavlovic, D.; Chen, M.C.; Flodström, M.; Sandler, S.; Eizirik, D.L. Cytokines Induce Apoptosis in Beta-Cells Isolated from Mice Lacking the Inducible Isoform of Nitric Oxide Synthase (INOS-/-). Diabetes 2000, 49, 1116–1122. [Google Scholar] [CrossRef] [Green Version]
- Suk, K.; Kim, S.; Kim, Y.-H.; Kim, K.-A.; Chang, I.; Yagita, H.; Shong, M.; Lee, M.-S. IFN-γ/TNF-α Synergism as the Final Effector in Autoimmune Diabetes: A Key Role for STAT1/IFN Regulatory Factor-1 Pathway in Pancreatic β Cell Death. J. Immunol. 2001, 166, 4481–4489. [Google Scholar] [CrossRef] [Green Version]
- Rabinovitch, A.; Sumoski, W.; Rajotte, R.V.; Warnock, G.L. Cytotoxic Effects of Cytokines on Human Pancreatic Islet Cells in Monolayer Culture. J. Clin. Endocrinol. Metab. 1990, 71, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Gurgul-Convey, E.; Mehmeti, I.; Plötz, T.; Jörns, A.; Lenzen, S. Sensitivity Profile of the Human EndoC-ΒH1 Beta Cell Line to Proinflammatory Cytokines. Diabetologia 2016, 59, 2125–2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oleson, B.J.; McGraw, J.A.; Broniowska, K.A.; Annamalai, M.; Chen, J.; Bushkofsky, J.R.; Davis, D.B.; Corbett, J.A.; Mathews, C.E. Distinct Differences in the Responses of the Human Pancreatic β-Cell Line EndoC-ΒH1 and Human Islets to Proinflammatory Cytokines. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Foulis, A.K.; McGill, M.; Farquharson, M.A. Insulitis in Type 1 (Insulin-Dependent) Diabetes Mellitus in Man, Macrophages, Lymphocytes, and Interferon-γ Containing Cells. J. Pathol. 1991, 165, 97–103. [Google Scholar] [CrossRef]
- Rabinovitch, A.; Suarez-Pinzon, W.L.; Sorensen, O.; Bleackley, R.C.; Power, R.F. IFN-Gamma Gene Expression in Pancreatic Islet-Infiltrating Mononuclear Cells Correlates with Autoimmune Diabetes in Nonobese Diabetic Mice. J. Immunol. 1995, 154, 4874–4882. [Google Scholar] [PubMed]
- Rabinovitch, A.; Suarez-Pinzon, W.; El-Sheikh, A.; Sorensen, O.; Power, R.F. Cytokine Gene Expression in Pancreatic Islet-Infiltrating Leukocytes of BB Rats: Expression of Th1 Cytokines Correlates With β-Cell Destructive Insulitis and IDDM. Diabetes 1996, 45, 749–754. [Google Scholar] [CrossRef]
- Lundberg, M.; Krogvold, L.; Kuric, E.; Dahl-Jørgensen, K.; Skog, O. Expression of Interferon-Stimulated Genes in Insulitic Pancreatic Islets of Patients Recently Diagnosed With Type 1 Diabetes. Diabetes 2016, 65, 3104–3110. [Google Scholar] [CrossRef] [Green Version]
- Carrero, J.A.; Calderon, B.; Towfic, F.; Artyomov, M.N.; Unanue, E.R. Defining the Transcriptional and Cellular Landscape of Type 1 Diabetes in the NOD Mouse. PLoS ONE 2013, 8, e59701. [Google Scholar] [CrossRef]
- Anderson, M.S.; Bluestone, J.A. The NOD Mouse: A Model of Immune Dysregulation. Annu. Rev. Immunol. 2005, 23, 447–485. [Google Scholar] [CrossRef]
- Pearson, J.A.; Wong, F.S.; Wen, L. The Importance of the Non Obese Diabetic (NOD) Mouse Model in Autoimmune Diabetes. J. Autoimmun. 2016, 66, 76–88. [Google Scholar] [CrossRef] [Green Version]
- Campbell, I.L.; Kay, T.W.; Oxbrow, L.; Harrison, L.C. Essential Role for Interferon-Gamma and Interleukin-6 in Autoimmune Insulin-Dependent Diabetes in NOD/Wehi Mice. J. Clin. Investig. 1991, 87, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Von Herrath, M.G.; Oldstone, M.B.A. Interferon-g is Essential for Destruction of b Cells and Development of Insulin-Dependent Diabetes Mellitus. J. Exp. Med. 1997, 185, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Sarvetnick, N.; Liggitt, D.; Pitts, S.L.; Hansen, S.E.; Stewart, T.A. Insulin-Dependent Diabetes Mellitus Induced in Transgenic Mice by Ectopic Expression of Class II MHC and Interferon-Gamma. Cell 1988, 52, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.L.; Wong, G.H.W.; Schrader, J.W.; Harrisson, L.C. Interferon-Y Enhances the Expression of the Major Histocompatibility Class I Antigens on Mouse Pancreatic Beta Cells. Diabetes 1985, 34, 1205–1209. [Google Scholar] [CrossRef]
- Campbell, I.L.; Bizilj, K.; Colman, P.G.; Tuch, B.E.; Harrison, L.C. Interferon-γ Induces the Expression of HLA-A,B,C but Not HLA-DR on Human Pancreatic β-Cells. J. Clin. Endocrinol. Metab. 1986, 62, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Marroqui, L.; Dos Santos, R.S.; Op de beeck, A.; Coomans de Brachène, A.; Marselli, L.; Marchetti, P.; Eizirik, D.L. Interferon-α Mediates Human Beta Cell HLA Class I Overexpression, Endoplasmic Reticulum Stress and Apoptosis, Three Hallmarks of Early Human Type 1 Diabetes. Diabetologia 2017, 60, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Duque, S.; Azoury, M.E.; Colli, M.L.; Afonso, G.; Turatsinze, J.-V.; Nigi, L.; Lalanne, A.I.; Sebastiani, G.; Carré, A.; Pinto, S.; et al. Conventional and Neo-Antigenic Peptides Presented by β Cells Are Targeted by Circulating Naïve CD8+ T Cells in Type 1 Diabetic and Healthy Donors. Cell Metab. 2018, 28, 946–960. [Google Scholar] [CrossRef] [Green Version]
- Azoury, M.E.; Tarayrah, M.; Afonso, G.; Pais, A.; Colli, M.L.; Maillard, C.; Lavaud, C.; Alexandre-Heymann, L.; Gonzalez-Duque, S.; Verdier, Y.; et al. Peptides Derived From Insulin Granule Proteins Are Targeted by CD8+ T Cells Across MHC Class I Restrictions in Humans and NOD Mice. Diabetes 2020, 69, 2678–2690. [Google Scholar] [CrossRef]
- Osum, K.C.; Burrack, A.L.; Martinov, T.; Sahli, N.L.; Mitchell, J.S.; Tucker, C.G.; Pauken, K.E.; Papas, K.; Appakalai, B.; Spanier, J.A.; et al. Interferon-Gamma Drives Programmed Death-Ligand 1 Expression on Islet β Cells to Limit T Cell Function during Autoimmune Diabetes. Sci. Rep. 2018, 8, 8295. [Google Scholar] [CrossRef] [Green Version]
- Colli, M.L.; Hill, J.L.E.; Marroquí, L.; Chaffey, J.; Dos Santos, R.S.; Leete, P.; Coomans de Brachène, A.; Paula, F.M.M.; Op de Beeck, A.; Castela, A.; et al. PDL1 Is Expressed in the Islets of People with Type 1 Diabetes and Is Up-Regulated by Interferons-α and-γ via IRF1 Induction. EBioMedicine 2018, 36, 367–375. [Google Scholar] [CrossRef] [Green Version]
- Falcone, M.; Fousteri, G. Role of the PD-1/PD-L1 Dyad in the Maintenance of Pancreatic Immune Tolerance for Prevention of Type 1 Diabetes. Front. Endocrinol. 2020, 11, 569. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Liang, S.C.; Guleria, I.; Latchman, Y.E.; Qipo, A.; Albacker, L.A.; Koulmanda, M.; Freeman, G.J.; Sayegh, M.H.; Sharpe, A.H. Tissue Expression of PD-L1 Mediates Peripheral T Cell Tolerance. J. Exp. Med. 2006, 203, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Guleria, I.; Gubbels Bupp, M.; Dada, S.; Fife, B.; Tang, Q.; Ansari, M.J.; Trikudanathan, S.; Vadivel, N.; Fiorina, P.; Yagita, H.; et al. Mechanisms of PDL1-Mediated Regulation of Autoimmune Diabetes. Clin. Immunol. 2007, 125, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H.; Aguilar-Parada, E.; Müller, W.A.; Eisentraut, A.M. Studies of Pancreatic Alpha Cell Function in Normal and Diabetic Subjects. J. Clin. Investig. 1970, 49, 837–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerich, J.E.; Langlois, M.; Noacco, C.; Karam, J.H.; Forsham, P.H. Lack of Glucagon Response to Hypoglycemia in Diabetes: Evidence for an Intrinsic Pancreatic Alpha Cell Defect. Science 1973, 182, 171–173. [Google Scholar] [CrossRef]
- Brissova, M.; Haliyur, R.; Saunders, D.; Shrestha, S.; Dai, C.; Blodgett, D.M.; Bottino, R.; Campbell-Thompson, M.; Aramandla, R.; Poffenberger, G.; et al. α Cell Function and Gene Expression Are Compromised in Type 1 Diabetes. Cell Rep. 2018, 22, 2667–2676. [Google Scholar] [CrossRef] [Green Version]
- Doliba, N.M.; Rozo, A.V.; Roman, J.; Qin, W.; Traum, D.; Gao, L.; Liu, J.; Manduchi, E.; Liu, C.; Golson, M.L.; et al. α Cell Dysfunction in Islets from Nondiabetic, Glutamic Acid Decarboxylase Autoantibody–Positive Individuals. J. Clin. Investig. 2022, 132, e156243. [Google Scholar] [CrossRef]
- Orci, L.; Baetens, D.; Rufener, C.; Amherdt, M.; Ravazzola, M.; Studer, P.; Malaisse-Lagae, F.; Unger, R.H. Hypertrophy and Hyperplasia of Somatostatin-Containing D-Cells in Diabetes. Proc. Natl. Acad. Sci. USA 1976, 73, 1338–1342. [Google Scholar] [CrossRef] [Green Version]
- Kazumi, T.; Utsumi, M.; Yoshino, G.; Ishihara, K.; Hirose, Y.; Makimura, H.; Baba, S. Somatostatin Concentration Responds to Arginine in Portal Plasma: Effects of Fasting, Streptozotocin Diabetes, and Insulin Administration in Diabetic Rats. Diabetes 1980, 29, 71–73. [Google Scholar] [CrossRef]
- van der Meulen, T.; Donaldson, C.J.; Cáceres, E.; Hunter, A.E.; Cowing-Zitron, C.; Pound, L.D.; Adams, M.W.; Zembrzycki, A.; Grove, K.L.; Huising, M.O. Urocortin3 Mediates Somatostatin-Dependent Negative Feedback Control of Insulin Secretion. Nat. Med. 2015, 21, 769–776. [Google Scholar] [CrossRef] [Green Version]
- Xu, E.; Kumar, M.; Zhang, Y.; Ju, W.; Obata, T.; Zhang, N.; Liu, S.; Wendt, A.; Deng, S.; Ebina, Y.; et al. Intra-Islet Insulin Suppresses Glucagon Release via GABA-GABAA Receptor System. Cell Metab. 2006, 3, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Kawamori, D.; Kurpad, A.J.; Hu, J.; Liew, C.W.; Shih, J.L.; Ford, E.L.; Herrera, P.L.; Polonsky, K.S.; McGuinness, O.P.; Kulkarni, R.N. Insulin Signaling in α Cells Modulates Glucagon Secretion In Vivo. Cell Metab. 2009, 9, 350–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthault, C.; Staels, W.; Scharfmann, R. Purification of Pancreatic Endocrine Subsets Reveals Increased Iron Metabolism in Beta Cells. Mol. Metab. 2020, 42, 101060. [Google Scholar] [CrossRef]
- Duvillié, B.; Attali, M.; Aiello, V.; Quemeneur, E.; Scharfmann, R. Label-Retaining Cells in the Rat Pancreas. Diabetes 2003, 52, 2035–2042. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Thomas, H.E.; Parker, J.L.; Schreiber, R.D.; Kay, T.W. IFN-Gamma Action on Pancreatic Beta Cells Causes Class I MHC Upregulation but Not Diabetes. J. Clin. Investig. 1998, 102, 1249–1257. [Google Scholar] [CrossRef] [Green Version]
- Rui, J.; Deng, S.; Arazi, A.; Perdigoto, A.L.; Liu, Z.; Herold, K.C. β Cells That Resist Immunological Attack Develop during Progression of Autoimmune Diabetes in NOD Mice. Cell Metab. 2017, 25, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Bader, E.; Migliorini, A.; Gegg, M.; Moruzzi, N.; Gerdes, J.; Roscioni, S.S.; Bakhti, M.; Brandl, E.; Irmler, M.; Beckers, J.; et al. Identification of Proliferative and Mature β-Cells in the Islets of Langerhans. Nature 2016, 535, 430–434. [Google Scholar] [CrossRef]
- Stefan, Y.; Meda, P.; Neufeld, M.; Orci, L. Stimulation of Insulin Secretion Reveals Heterogeneity of Pancreatic B Cells in Vivo. J. Clin. Investig. 1987, 80, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Wojtusciszyn, A.; Armanet, M.; Morel, P.; Berney, T.; Bosco, D. Insulin Secretion from Human Beta Cells Is Heterogeneous and Dependent on Cell-to-Cell Contacts. Diabetologia 2008, 51, 1843–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorrell, C.; Schug, J.; Canaday, P.S.; Russ, H.A.; Tarlow, B.D.; Grompe, M.T.; Horton, T.; Hebrok, M.; Streeter, P.R.; Kaestner, K.H.; et al. Human Islets Contain Four Distinct Subtypes of β Cells. Nat. Commun. 2016, 7, 11756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salinno, C.; Büttner, M.; Cota, P.; Tritschler, S.; Tarquis-Medina, M.; Bastidas-Ponce, A.; Scheibner, K.; Burtscher, I.; Böttcher, A.; Theis, F.J.; et al. CD81 Marks Immature and Dedifferentiated Pancreatic β-Cells. Mol. Metab. 2021, 49, 101188. [Google Scholar] [CrossRef]
- Stancill, J.S.; Kasmani, M.Y.; Khatun, A.; Cui, W.; Corbett, J.A. Single-Cell RNA Sequencing of Mouse Islets Exposed to Proinflammatory Cytokines. Life Sci. Alliance 2021, 4, e202000949. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Gutierrez, G.; Xin, Y.; Gromada, J. Heterogeneity of Human Pancreatic B-Cells. Mol. Metab. 2019, 27, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Oyler-Yaniv, A.; Oyler-Yaniv, J.; Whitlock, B.M.; Liu, Z.; Germain, R.N.; Huse, M.; Altan-Bonnet, G.; Krichevsky, O. A Tunable Diffusion-Consumption Mechanism of Cytokine Propagation Enables Plasticity in Cell-to-Cell Communication in the Immune System. Immunity 2017, 46, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Gysemans, C.A.; Ladrière, L.; Callewaert, H.; Rasschaert, J.; Flamez, D.; Levy, D.E.; Matthys, P.; Eizirik, D.L.; Mathieu, C. Disruption of the Interferon-g Signaling Pathway at the Level of Signal Transducer and Activator of Transcription-1 Prevents Immune Destruction of b-Cells. Diabetes 2005, 54, 2396–2403. [Google Scholar] [CrossRef] [Green Version]
- Moore, F.; Naamane, N.; Colli, M.L.; Bouckenooghe, T.; Ortis, F.; Gurzov, E.N.; Igoillo-Esteve, M.; Mathieu, C.; Bontempi, G.; Thykjaer, T.; et al. STAT1 is a Master Regulator of Pancreatic β-Cell Apoptosis and Islet Inflammation. J. Biol. Chem. 2011, 286, 929–941. [Google Scholar] [CrossRef] [Green Version]
- Cardozo, A.K.; Kruhøffer, M.; Leeman, R.; Ørntoft, T.; Eizirik, D.L. Identification of Novel Cytokine-Induced Genes in Pancreatic B-Cells by High-Density Oligonucleotide Arrays. Diabetes 2001, 50, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Kutlu, B.; Cardozo, A.K.; Darville, M.I.; Kruhøffer, M.; Magnusson, N.; Ørntoft, T.; Eizirik, D.L. Discovery of Gene Networks Regulating Cytokine-Induced Dysfunction and Apoptosis in Insulin-Producing INS-1 Cells. Diabetes 2003, 52, 2701–2719. [Google Scholar] [CrossRef] [PubMed]
- Rasschaert, J.; Liu, D.; Kutlu, B.; Cardozo, A.K.; Kruhoffer, M.; Orntoft, T.F.; Eizirik, D.L. Global Profiling of Double Stranded RNA- and IFN-?-Induced Genes in Rat Pancreatic Beta Cells. Diabetologia 2003, 46, 1641–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colli, M.L.; Ramos-Rodríguez, M.; Nakayasu, E.S.; Alvelos, M.I.; Lopes, M.; Hill, J.L.E.; Turatsinze, J.-V.; Coomans de Brachène, A.; Russell, M.A.; Raurell-Vila, H.; et al. An Integrated Multi-Omics Approach Identifies the Landscape of Interferon-α-Mediated Responses of Human Pancreatic Beta Cells. Nat. Commun. 2020, 11, 2584. [Google Scholar] [CrossRef]
- Dusaulcy, R.; Handgraaf, S.; Visentin, F.; Howald, C.; Dermitzakis, E.T.; Philippe, J.; Gosmain, Y. High-Fat Diet Impacts More Changes in Beta-Cell Compared to Alpha-Cell Transcriptome. PLoS ONE 2019, 14, e0213299. [Google Scholar] [CrossRef] [PubMed]
- DiGruccio, M.R.; Mawla, A.M.; Donaldson, C.J.; Noguchi, G.M.; Vaughan, J.; Cowing-Zitron, C.; van der Meulen, T.; Huising, M.O. Comprehensive Alpha, Beta and Delta Cell Transcriptomes Reveal That Ghrelin Selectively Activates Delta Cells and Promotes Somatostatin Release from Pancreatic Islets. Mol. Metab. 2016, 5, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Mawla, A.M.; Huising, M.O. Navigating the Depths and Avoiding the Shallows of Pancreatic Islet Cell Transcriptomes. Diabetes 2019, 68, 1380–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.J.; Kaestner, K.H. Single-Cell RNA-Seq of the Pancreatic Islets––a Promise Not yet Fulfilled? Cell Metab. 2019, 29, 539–544. [Google Scholar] [CrossRef] [Green Version]
- Muraro, M.J.; Dharmadhikari, G.; Grün, D.; Groen, N.; Dielen, T.; Jansen, E.; van Gurp, L.; Engelse, M.A.; Carlotti, F.; de Koning, E.J.P.; et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 2016, 3, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Baron, M.; Veres, A.; Wolock, S.L.; Faust, A.L.; Gaujoux, R.; Vetere, A.; Ryu, J.H.; Wagner, B.K.; Shen-Orr, S.S.; Klein, A.M.; et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-Cell Population Structure. Cell Syst. 2016, 3, 346–360. [Google Scholar] [CrossRef] [Green Version]
- Segerstolpe, A.; Palasantza, A.; Eliasson, P.; Andersson, E.-M.; Andréasson, A.-C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Kryczek, I.; Wei, S.; Gong, W.; Shu, X.; Szeliga, W.; Vatan, L.; Chen, L.; Wang, G.; Zou, W. Cutting Edge: IFN-γ Enables APC to Promote Memory Th17 and Abate Th1 Cell Development. J. Immunol. 2008, 181, 5842–5846. [Google Scholar] [CrossRef]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 Pathway Contributes to Immune Escape in EGFR-Driven Lung Tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a Novel Direct Target of HIF-1α, and Its Blockade under Hypoxia Enhanced MDSC-Mediated T Cell Activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [Green Version]
- Perdigoto, A.L.; Deng, S.; Du, K.C.; Kuchroo, M.; Burkhardt, D.B.; Tong, A.; Israel, G.; Robert, M.E.; Weisberg, S.P.; Kirkiles-Smith, N.; et al. Immune Cells and Their Inflammatory Mediators Modify β Cells and Cause Checkpoint Inhibitor–Induced Diabetes. JCI Insight 2022, 7, e156330. [Google Scholar] [CrossRef]
- Rhode, A.; Pauza, M.E.; Barral, A.M.; Rodrigo, E.; Oldstone, M.B.A.; von Herrath, M.G.; Christen, U. Islet-Specific Expression of CXCL10 Causes Spontaneous Islet Infiltration and Accelerates Diabetes Development. J. Immunol. 2005, 175, 3516–3524. [Google Scholar] [CrossRef] [Green Version]
- Frigerio, S.; Junt, T.; Lu, B.; Gerard, C.; Zumsteg, U.; Hollander, G.A.; PIalli, L. β Cells Are Responsible for CXCR3-Mediated T-Cell Infiltration in Insulitis. Nat. Med. 2002, 8, 1414–1420. [Google Scholar] [CrossRef]
- Nigi, L.; Brusco, N.; Grieco, G.E.; Licata, G.; Krogvold, L.; Marselli, L.; Gysemans, C.; Overbergh, L.; Marchetti, P.; Mathieu, C.; et al. Pancreatic Alpha-Cells Contribute Together With Beta-Cells to CXCL10 Expression in Type 1 Diabetes. Front. Endocrinol. 2020, 11, 630. [Google Scholar] [CrossRef]
- Pineros, A.R.; Kulkarni, A.; Gao, H.; Glenn, L.; Huang, F.; liu, Y.; Gannon, M.; Syed, F.; Wu, W.; Anderson, C.M.; et al. Proinflammatory Signaling in Islet b Cells Propagates Invasion of Pathogenic Immune Cells in Autoimmune Diabetes. Cell Rep. 2022, 39, 111011. [Google Scholar] [CrossRef]
- Gurzov, E.N.; Ortis, F.; Cunha, D.A.; Gosset, G.; Li, M.; Cardozo, A.K.; Eizirik, D.L. Signaling by IL-1β+IFN-γ and ER Stress Converge on DP5/Hrk Activation: A Novel Mechanism for Pancreatic β-Cell Apoptosis. Cell Death Differ. 2009, 16, 1539–1550. [Google Scholar] [CrossRef] [Green Version]
- Iwahashi, H.; Hanafusa, T.; Eguchi, Y.; Nakajima, H.; Miyagawa, J.; Itoh, N.; Tomita, K.; Namba, M.; Kuwajima, M.; Noguchi, T.; et al. Cytokine-Induced Apoptotic Cell Death in a Mouse Pancreatic Beta-Cell Line: Inhibition by Bcl-2. Diabetologia 1996, 39, 530–536. [Google Scholar] [CrossRef]
- D’Angeli, F.; Scalia, M.; Cirnigliaro, M.; Satriano, C.; Barresi, V.; Musso, N.; Trovato-Salinaro, A.; Barbagallo, D.; Ragusa, M.; Di Pietro, C.; et al. PARP-14 Promotes Survival of Mammalian α but Not β Pancreatic Cells Following Cytokine Treatment. Front. Endocrinol. 2019, 10, 271. [Google Scholar] [CrossRef]
- Pipeleers, D.G. Heterogeneity in Pancreatic P-Cell Population. Diabetes 1992, 41, 777–781. [Google Scholar] [CrossRef]
- Schuit, F.C.; In’t Veld, P.A.; Pipeleers, D.G. Glucose Stimulates Proinsulin Biosynthesis by a Dose-Dependent Recruitment of Pancreatic Beta Cells. Proc. Natl. Acad. Sci. USA 1988, 85, 3865–3869. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.-W.; Johmura, Y.; Suzuki, N.; Omori, S.; Migita, T.; Yamaguchi, K.; Hatakeyama, S.; Yamazaki, S.; Shimizu, E.; Imoto, S.; et al. Blocking PD-L1–PD-1 Improves Senescence Surveillance and Ageing Phenotypes. Nature 2022, 611, 358–364. [Google Scholar] [CrossRef]
- Xin, Y.; Kim, J.; Okamoto, H.; Ni, M.; Wei, Y.; Adler, C.; Murphy, A.J.; Yancopoulos, G.D.; Lin, C.; Gromada, J. RNA Sequencing of Single Human Islet Cells Reveals Type 2 Diabetes Genes. Cell Metab. 2016, 24, 608–615. [Google Scholar] [CrossRef] [Green Version]
- Lawlor, N.; George, J.; Bolisetty, M.; Kursawe, R.; Sun, L.; Sivakamasundari, V.; Kycia, I.; Robson, P.; Stitzel, M.L. Single-Cell Transcriptomes Identify Human Islet Cell Signatures and Reveal Cell-Type–Specific Expression Changes in Type 2 Diabetes. Genome Res. 2017, 27, 208–222. [Google Scholar] [CrossRef] [Green Version]
- Szabat, M.; Pourghaderi, P.; Soukhatcheva, G.; Verchere, C.B.; Warnock, G.L.; Piret, J.M.; Johnson, J.D. Kinetics and Genomic Profiling of Adult Human and Mouse β-Cell Maturation. Islets 2011, 3, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.; Dominguez Gutierrez, G.; Okamoto, H.; Kim, J.; Lee, A.-H.; Adler, C.; Ni, M.; Yancopoulos, G.D.; Murphy, A.J.; Gromada, J. Pseudotime Ordering of Single Human β-Cells Reveals States of Insulin Production and Unfolded Protein Response. Diabetes 2018, 67, 1783–1794. [Google Scholar] [CrossRef] [Green Version]
- Oyler-Yaniv, J.; Oyler-Yaniv, A.; Shakiba, M.; Min, N.K.; Chen, Y.-H.; Cheng, S.; Krichevsky, O.; Altan-Bonnet, N.; Altan-Bonnet, G. Catch and Release of Cytokines Mediated by Tumor Phosphatidylserine Converts Transient Exposure into Long-Lived Inflammation. Mol. Cell 2017, 66, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Korpos, É.; Kadri, N.; Kappelhoff, R.; Wegner, J.; Overall, C.M.; Weber, E.; Holmberg, D.; Cardell, S.; Sorokin, L. The Peri-Islet Basement Membrane, a Barrier to Infiltrating Leukocytes in Type 1 Diabetes in Mouse and Human. Diabetes 2013, 62, 531–542. [Google Scholar] [CrossRef] [Green Version]
- Qin, T.; Hu, S.; Smink, A.M.; de Haan, B.J.; Silva-Lagos, L.A.; Lakey, J.R.T.; de Vos, P. Inclusion of Extracellular Matrix Molecules and Necrostatin-1 in the Intracapsular Environment of Alginate-Based Microcapsules Synergistically Protects Pancreatic β Cells against Cytokine-Induced Inflammatory Stress. Acta Biomater. 2022, 146, 434–449. [Google Scholar] [CrossRef] [PubMed]
- Llacua, L.A.; Haan, B.J.; Vos, P. Laminin and Collagen IV Inclusion in Immunoisolating Microcapsules Reduces Cytokine-mediated Cell Death in Human Pancreatic Islets. J. Tissue Eng. Regen. Med. 2018, 12, 460–467. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Burghgrave, M.; Lourenço, C.; Berthault, C.; Aiello, V.; Villalba, A.; Fouque, A.; Diedisheim, M.; You, S.; Oshima, M.; Scharfmann, R. Pancreatic Islet Cells Response to IFNγ Relies on Their Spatial Location within an Islet. Cells 2023, 12, 113. https://doi.org/10.3390/cells12010113
De Burghgrave M, Lourenço C, Berthault C, Aiello V, Villalba A, Fouque A, Diedisheim M, You S, Oshima M, Scharfmann R. Pancreatic Islet Cells Response to IFNγ Relies on Their Spatial Location within an Islet. Cells. 2023; 12(1):113. https://doi.org/10.3390/cells12010113
Chicago/Turabian StyleDe Burghgrave, Marine, Chloé Lourenço, Claire Berthault, Virginie Aiello, Adrian Villalba, Alexis Fouque, Marc Diedisheim, Sylvaine You, Masaya Oshima, and Raphaël Scharfmann. 2023. "Pancreatic Islet Cells Response to IFNγ Relies on Their Spatial Location within an Islet" Cells 12, no. 1: 113. https://doi.org/10.3390/cells12010113