
Emerging Roles of Extracellular Vesicles in Alzheimer’s Disease: Focus on Synaptic Dysfunction and Vesicle–Neuron Interaction


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Synaptic Dysfunction in AD
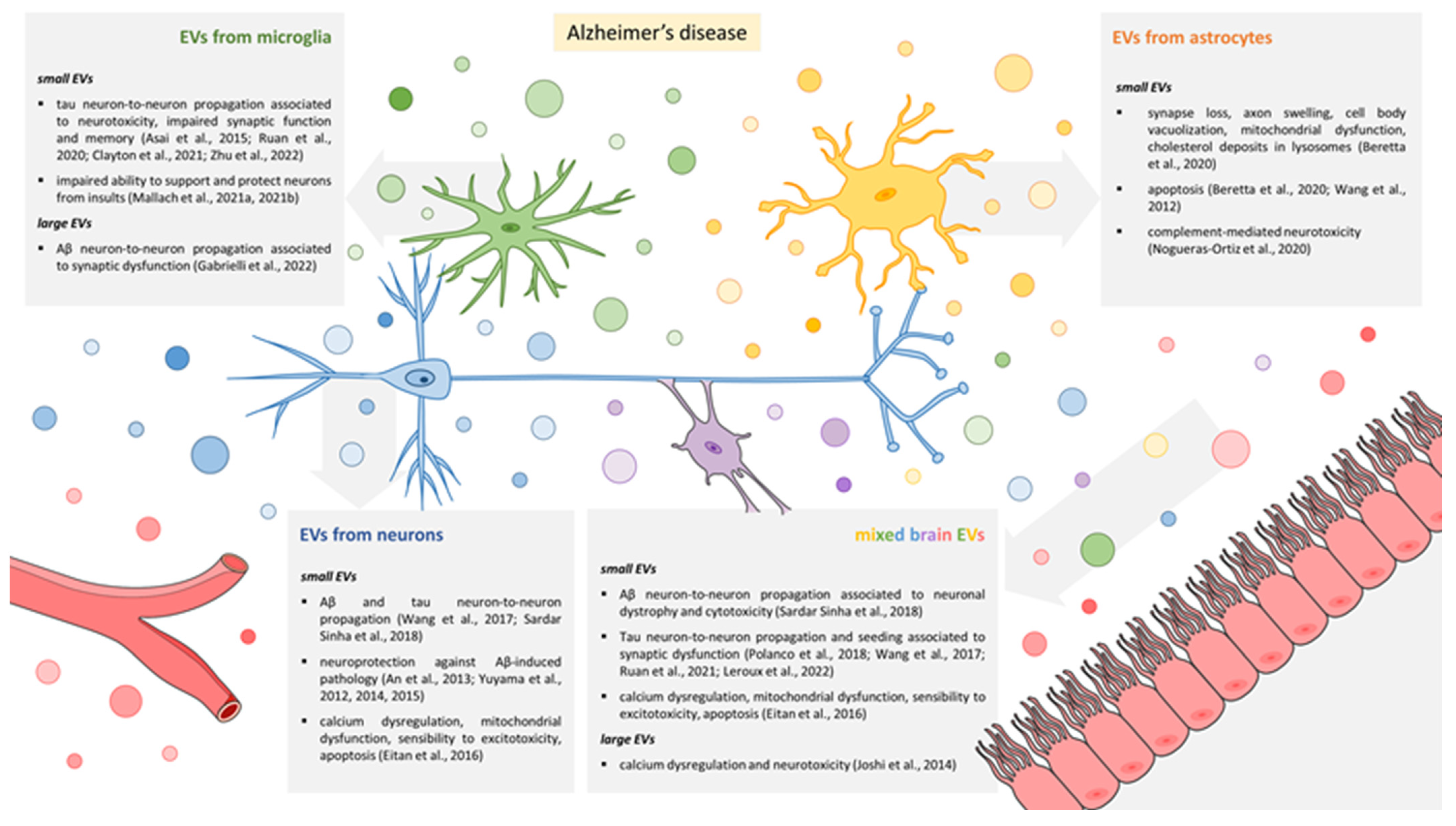
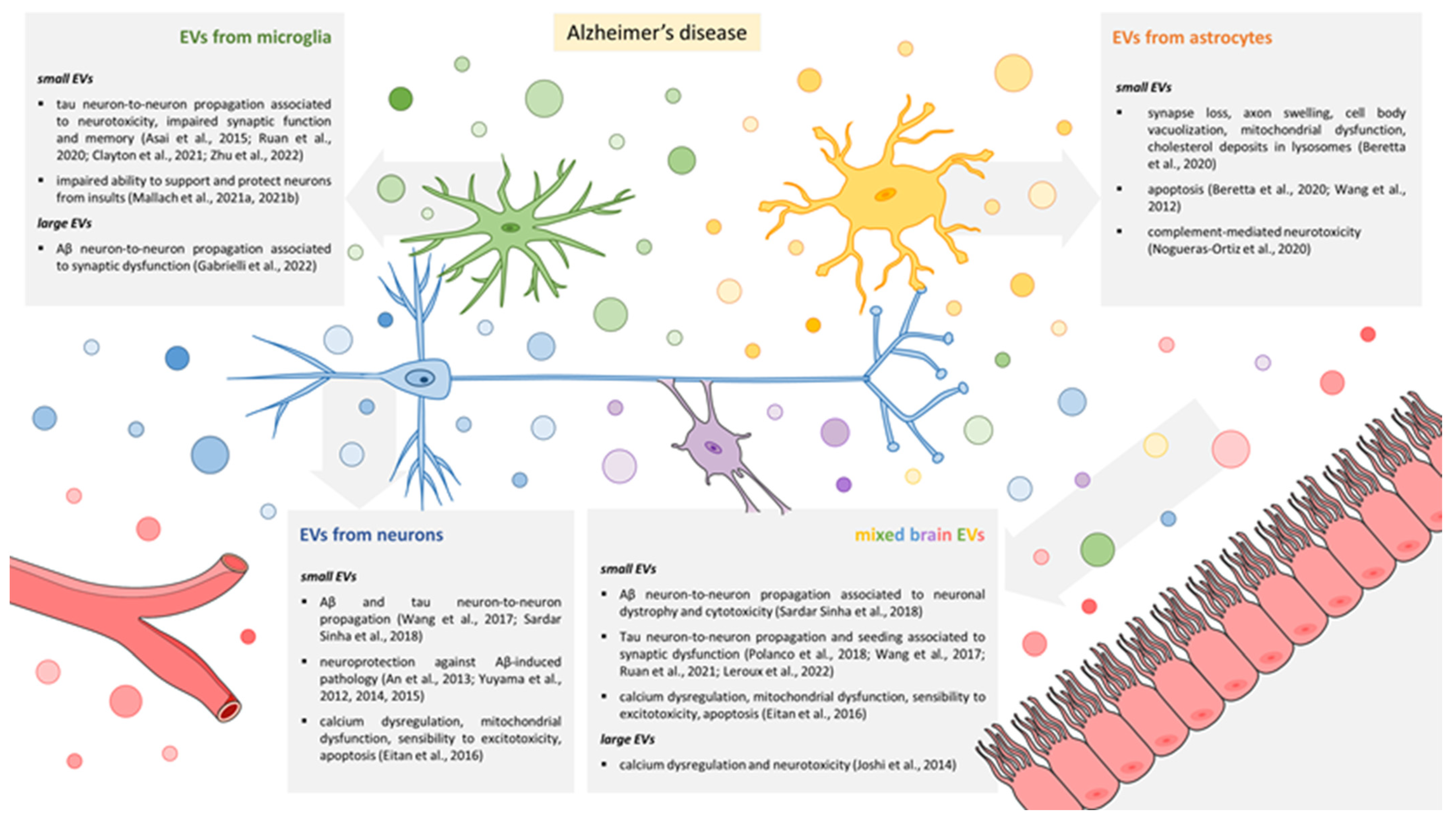
3. EVs and Synaptic Dysfunction in AD
3.1. EVs Released by Microglia

3.2. EVs Released by Astrocytes
3.3. EVs Released by Neurons
3.4. Mixed EV Populations Isolated from Body Fluids or Brain Tissue
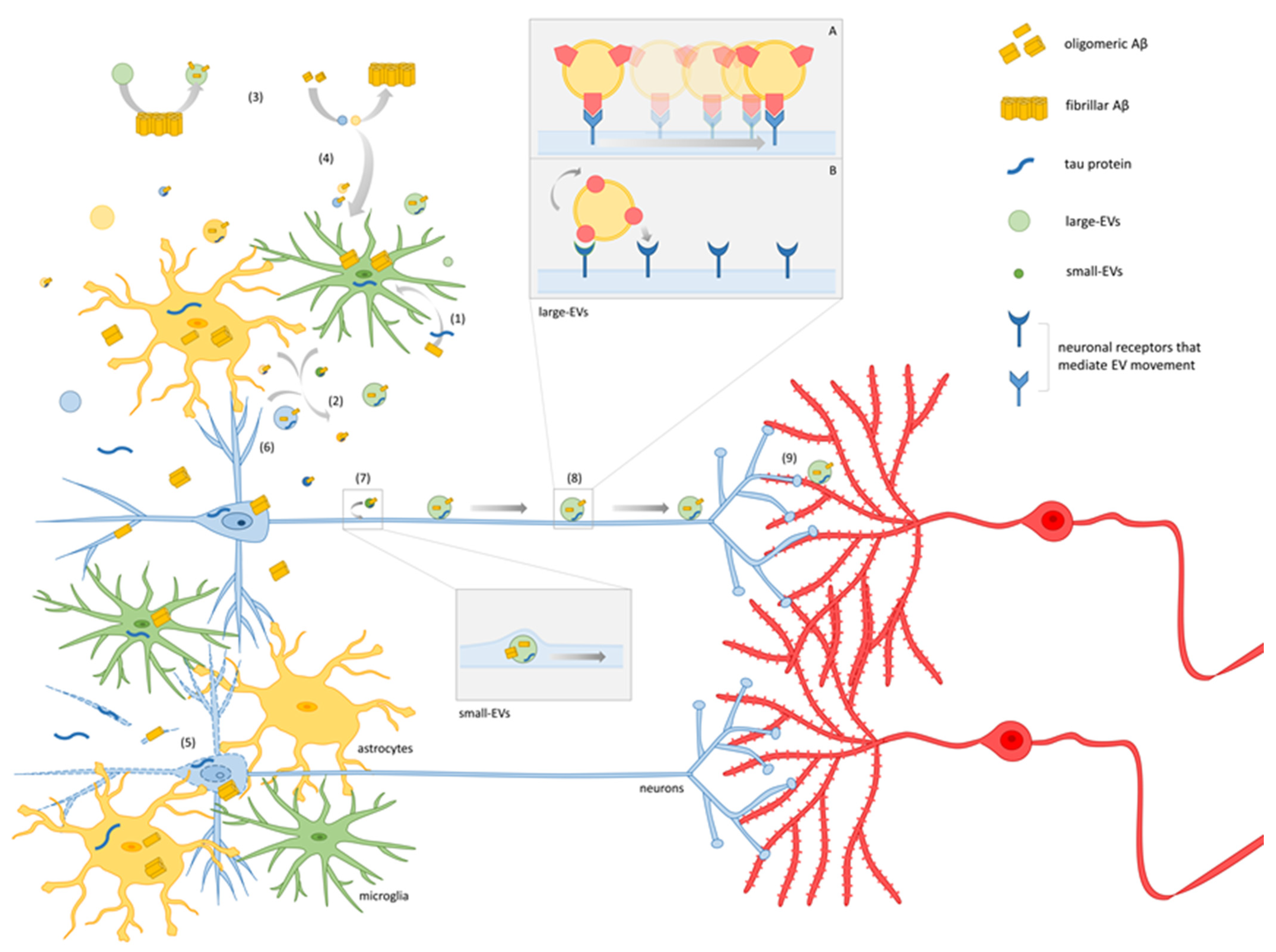
4. A Model for EV-Mediated Propagation of Synaptic Dysfunction
Molecular Mechanisms Underlying EV–Neuron Interaction
5. Discussion
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.W.; Stevens, B. Microglia Emerge as Central Players in Brain Disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Rub, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [Green Version]
- Sepulcre, J.; Masdeu, J.C. Advanced Neuroimaging Methods Towards Characterization of Early Stages of Alzheimer’s Disease. Methods Mol. Biol. 2016, 1303, 509–519. [Google Scholar] [CrossRef]
- Masliah, E. Mechanisms of synaptic dysfunction in Alzheimer’s disease. Histol. Histopathol. 1995, 10, 509–519. [Google Scholar] [PubMed]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godyn, J.; Jonczyk, J.; Panek, D.; Malawska, B. Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol. Rep. 2016, 68, 127–138. [Google Scholar] [CrossRef]
- Dujardin, S.; Lecolle, K.; Caillierez, R.; Begard, S.; Zommer, N.; Lachaud, C.; Carrier, S.; Dufour, N.; Auregan, G.; Winderickx, J.; et al. Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: Relevance to sporadic tauopathies. Acta Neuropathol. Commun. 2014, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.A.; Devidze, N.; Verret, L.; Ho, K.; Halabisky, B.; Thwin, M.T.; Kim, D.; Hamto, P.; Lo, I.; Yu, G.Q.; et al. Transsynaptic progression of amyloid-beta-induced neuronal dysfunction within the entorhinal-hippocampal network. Neuron 2010, 68, 428–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Drouet, V.; Wu, J.W.; Witter, M.P.; Small, S.A.; Clelland, C.; Duff, K. Trans-synaptic spread of tau pathology in vivo. PLoS ONE 2012, 7, e31302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, S.; Wegmann, S.; Cho, H.; DeVos, S.L.; Commins, C.; Roe, A.D.; Nicholls, S.B.; Carlson, G.A.; Pitstick, R.; Nobuhara, C.K.; et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat. Commun. 2015, 6, 8490. [Google Scholar] [CrossRef] [Green Version]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kugler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [Green Version]
- Delpech, J.C.; Pathak, D.; Varghese, M.; Kalavai, S.V.; Hays, E.C.; Hof, P.R.; Johnson, W.E.; Ikezu, S.; Medalla, M.; Luebke, J.I.; et al. Wolframin-1-expressing neurons in the entorhinal cortex propagate tau to CA1 neurons and impair hippocampal memory in mice. Sci. Transl. Med. 2021, 13, eabe8455. [Google Scholar] [CrossRef]
- Gabrielli, M.; Prada, I.; Joshi, P.; Falcicchia, C.; D’Arrigo, G.; Rutigliano, G.; Battocchio, E.; Zenatelli, R.; Tozzi, F.; Radeghieri, A.; et al. Microglial large extracellular vesicles propagate early synaptic dysfunction in Alzheimer’s disease. Brain 2022, 145, 2849–2868. [Google Scholar] [CrossRef]
- Ruan, Z.; Pathak, D.; Venkatesan Kalavai, S.; Yoshii-Kitahara, A.; Muraoka, S.; Bhatt, N.; Takamatsu-Yukawa, K.; Hu, J.; Wang, Y.; Hersh, S.; et al. Alzheimer’s disease brain-derived extracellular vesicles spread tau pathology in interneurons. Brain 2021, 144, 288–309. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Saman, S.; Kim, W.; Raya, M.; Visnick, Y.; Miro, S.; Saman, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.; et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 2012, 287, 3842–3849. [Google Scholar] [CrossRef]
- Joshi, P.; Turola, E.; Ruiz, A.; Bergami, A.; Libera, D.D.; Benussi, L.; Giussani, P.; Magnani, G.; Comi, G.; Legname, G.; et al. Microglia convert aggregated amyloid-beta into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2014, 21, 582–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouwens, L.K.; Ismail, M.S.; Rogers, V.A.; Zeller, N.T.; Garrad, E.C.; Amtashar, F.S.; Makoni, N.J.; Osborn, D.C.; Nichols, M.R. Abeta42 Protofibrils Interact with and Are Trafficked through Microglial-Derived Microvesicles. ACS Chem. Neurosci. 2018, 9, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Crotti, A.; Sait, H.R.; McAvoy, K.M.; Estrada, K.; Ergun, A.; Szak, S.; Marsh, G.; Jandreski, L.; Peterson, M.; Reynolds, T.L.; et al. BIN1 favors the spreading of Tau via extracellular vesicles. Sci. Rep. 2019, 9, 9477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiarini, A.; Armato, U.; Gardenal, E.; Gui, L.; Dal Pra, I. Amyloid beta-Exposed Human Astrocytes Overproduce Phospho-Tau and Overrelease It within Exosomes, Effects Suppressed by Calcilytic NPS 2143-Further Implications for Alzheimer’s Therapy. Front. Neurosci. 2017, 11, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eitan, E.; Hutchison, E.R.; Marosi, K.; Comotto, J.; Mustapic, M.; Nigam, S.M.; Suire, C.; Maharana, C.; Jicha, G.A.; Liu, D.; et al. Extracellular Vesicle-Associated Abeta Mediates Trans-Neuronal Bioenergetic and Ca2+-Handling Deficits in Alzheimer’s Disease Models. NPJ Aging Mech. Dis. 2016, 2, 16019. [Google Scholar] [CrossRef] [Green Version]
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjo, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Peraza, F.; Nogueras-Ortiz, C.J.; Volpert, O.; Liu, D.; Goetzl, E.J.; Mattson, M.P.; Greig, N.H.; Eitan, E.; Kapogiannis, D. Neuronal and Astrocytic Extracellular Vesicle Biomarkers in Blood Reflect Brain Pathology in Mouse Models of Alzheimer’s Disease. Cells 2021, 10, 993. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Mustapic, M.; Kapogiannis, D.; Eitan, E.; Lobach, I.V.; Goetzl, L.; Schwartz, J.B.; Miller, B.L. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer’s disease. FASEB J. 2016, 30, 3853–3859. [Google Scholar] [CrossRef] [Green Version]
- Muraoka, S.; DeLeo, A.M.; Sethi, M.K.; Yukawa-Takamatsu, K.; Yang, Z.; Ko, J.; Hogan, J.D.; Ruan, Z.; You, Y.; Wang, Y.; et al. Proteomic and biological profiling of extracellular vesicles from Alzheimer’s disease human brain tissues. Alzheimer’s Dement. 2020, 16, 896–907. [Google Scholar] [CrossRef]
- Muraoka, S.; Jedrychowski, M.P.; Iwahara, N.; Abdullah, M.; Onos, K.D.; Keezer, K.J.; Hu, J.; Ikezu, S.; Howell, G.R.; Gygi, S.P.; et al. Enrichment of Neurodegenerative Microglia Signature in Brain-Derived Extracellular Vesicles Isolated from Alzheimer’s Disease Mouse Models. J. Proteome Res. 2021, 20, 1733–1743. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimer’s Dement. 2015, 11, 600–607.e601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohn, W.; Melnik, M.; Huang, C.; Teter, B.; Chandra, S.; Zhu, C.; McIntire, L.B.; John, V.; Gylys, K.H.; Bilousova, T. Multi-Omics Analysis of Microglial Extracellular Vesicles From Human Alzheimer’s Disease Brain Tissue Reveals Disease-Associated Signatures. Front. Pharmacol. 2021, 12, 766082. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Wang, J.; Zhao, Y.; Feng, Y.; Han, S.; Dong, Q.; Cui, M.; Tieu, K. Microglial exosomes facilitate alpha-synuclein transmission in Parkinson’s disease. Brain 2020, 143, 1476–1497. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Pelucchi, S.; Gardoni, F.; Di Luca, M.; Marcello, E. Synaptic dysfunction in early phases of Alzheimer’s Disease. Handb. Clin. Neurol. 2022, 184, 417–438. [Google Scholar] [CrossRef]
- Masliah, E.; Mallory, M.; Alford, M.; DeTeresa, R.; Hansen, L.A.; McKeel, D.W., Jr.; Morris, J.C. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 2001, 56, 127–129. [Google Scholar] [CrossRef] [Green Version]
- Palop, J.J.; Mucke, L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat. Neurosci. 2010, 13, 812–818. [Google Scholar] [CrossRef] [Green Version]
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.Q.; Kreitzer, A.; et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 2007, 55, 697–711. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Zhang, M.; Yin, X.; Chen, K.; Hu, Z.; Zhou, Q.; Cao, X.; Chen, Z.; Liu, D. The role of pathological tau in synaptic dysfunction in Alzheimer’s diseases. Transl. Neurodegener. 2021, 10, 45. [Google Scholar] [CrossRef]
- Fa, M.; Puzzo, D.; Piacentini, R.; Staniszewski, A.; Zhang, H.; Baltrons, M.A.; Li Puma, D.D.; Chatterjee, I.; Li, J.; Saeed, F.; et al. Extracellular Tau Oligomers Produce An Immediate Impairment of LTP and Memory. Sci. Rep. 2016, 6, 19393. [Google Scholar] [CrossRef]
- Puzzo, D.; Piacentini, R.; Fa, M.; Gulisano, W.; Li Puma, D.D.; Staniszewski, A.; Zhang, H.; Tropea, M.R.; Cocco, S.; Palmeri, A.; et al. LTP and memory impairment caused by extracellular Abeta and Tau oligomers is APP-dependent. eLife 2017, 6, e26991. [Google Scholar] [CrossRef] [PubMed]
- Kaniyappan, S.; Chandupatla, R.R.; Mandelkow, E.M.; Mandelkow, E. Extracellular low-n oligomers of tau cause selective synaptotoxicity without affecting cell viability. Alzheimer’s Dement. 2017, 13, 1270–1291. [Google Scholar] [CrossRef] [PubMed]
- Regan, P.; Whitcomb, D.J.; Cho, K. Physiological and Pathophysiological Implications of Synaptic Tau. Neuroscientist 2017, 23, 137–151. [Google Scholar] [CrossRef] [Green Version]
- Ittner, L.M.; Gotz, J. Amyloid-beta and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Sperling, R.A.; Mormino, E.C.; Schultz, A.P.; Betensky, R.A.; Papp, K.V.; Amariglio, R.E.; Hanseeuw, B.J.; Buckley, R.; Chhatwal, J.; Hedden, T.; et al. The impact of amyloid-beta and tau on prospective cognitive decline in older individuals. Ann. Neurol. 2019, 85, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-beta and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Crimins, J.L.; Pooler, A.; Polydoro, M.; Luebke, J.I.; Spires-Jones, T.L. The intersection of amyloid beta and tau in glutamatergic synaptic dysfunction and collapse in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 757–763. [Google Scholar] [CrossRef] [Green Version]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Fonseca, M.I.; Zhou, J.; Botto, M.; Tenner, A.J. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2004, 24, 6457–6465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Chowdhury, S.; Ma, R.; Le, K.X.; Hong, S.; Caldarone, B.J.; Stevens, B.; Lemere, C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017, 9, eaaf6295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendran, L.; Paolicelli, R.C. Microglia-Mediated Synapse Loss in Alzheimer’s Disease. J. Neurosci. 2018, 38, 2911–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, Z.; Delpech, J.C.; Venkatesan Kalavai, S.; Van Enoo, A.A.; Hu, J.; Ikezu, S.; Ikezu, T. P2RX7 inhibitor suppresses exosome secretion and disease phenotype in P301S tau transgenic mice. Mol. Neurodegener. 2020, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Clayton, K.; Delpech, J.C.; Herron, S.; Iwahara, N.; Ericsson, M.; Saito, T.; Saido, T.C.; Ikezu, S.; Ikezu, T. Plaque associated microglia hyper-secrete extracellular vesicles and accelerate tau propagation in a humanized APP mouse model. Mol. Neurodegener. 2021, 16, 18. [Google Scholar] [CrossRef]
- Zhu, B.; Liu, Y.; Hwang, S.; Archuleta, K.; Huang, H.; Campos, A.; Murad, R.; Pina-Crespo, J.; Xu, H.; Huang, T.Y. Trem2 deletion enhances tau dispersion and pathology through microglia exosomes. Mol. Neurodegener. 2022, 17, 58. [Google Scholar] [CrossRef]
- Mallach, A.; Gobom, J.; Arber, C.; Piers, T.M.; Hardy, J.; Wray, S.; Zetterberg, H.; Pocock, J. Differential Stimulation of Pluripotent Stem Cell-Derived Human Microglia Leads to Exosomal Proteomic Changes Affecting Neurons. Cells 2021, 10, 2866. [Google Scholar] [CrossRef]
- Mallach, A.; Gobom, J.; Zetterberg, H.; Hardy, J.; Piers, T.M.; Wray, S.; Pocock, J.M. The influence of the R47H triggering receptor expressed on myeloid cells 2 variant on microglial exosome profiles. Brain Commun. 2021, 3, fcab009. [Google Scholar] [CrossRef]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Kruger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- An, K.; Klyubin, I.; Kim, Y.; Jung, J.H.; Mably, A.J.; O’Dowd, S.T.; Lynch, T.; Kanmert, D.; Lemere, C.A.; Finan, G.M.; et al. Exosomes neutralize synaptic-plasticity-disrupting activity of Abeta assemblies in vivo. Mol. Brain 2013, 6, 47. [Google Scholar] [CrossRef]
- Yuyama, K.; Sun, H.; Mitsutake, S.; Igarashi, Y. Sphingolipid-modulated exosome secretion promotes clearance of amyloid-beta by microglia. J. Biol. Chem. 2012, 287, 10977–10989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuyama, K.; Sun, H.; Sakai, S.; Mitsutake, S.; Okada, M.; Tahara, H.; Furukawa, J.; Fujitani, N.; Shinohara, Y.; Igarashi, Y. Decreased amyloid-beta pathologies by intracerebral loading of glycosphingolipid-enriched exosomes in Alzheimer model mice. J. Biol. Chem. 2014, 289, 24488–24498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuyama, K.; Sun, H.; Usuki, S.; Sakai, S.; Hanamatsu, H.; Mioka, T.; Kimura, N.; Okada, M.; Tahara, H.; Furukawa, J.; et al. A potential function for neuronal exosomes: Sequestering intracerebral amyloid-beta peptide. FEBS Lett. 2015, 589, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Polanco, J.C.; Li, C.; Durisic, N.; Sullivan, R.; Gotz, J. Exosomes taken up by neurons hijack the endosomal pathway to spread to interconnected neurons. Acta Neuropathol. Commun. 2018, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Leroux, E.; Perbet, R.; Caillierez, R.; Richetin, K.; Lieger, S.; Espourteille, J.; Bouillet, T.; Begard, S.; Danis, C.; Loyens, A.; et al. Extracellular vesicles: Major actors of heterogeneity in tau spreading among human tauopathies. Mol. Ther. 2022, 30, 782–797. [Google Scholar] [CrossRef]
- Beretta, C.; Nikitidou, E.; Streubel-Gallasch, L.; Ingelsson, M.; Sehlin, D.; Erlandsson, A. Extracellular vesicles from amyloid-beta exposed cell cultures induce severe dysfunction in cortical neurons. Sci. Rep. 2020, 10, 19656. [Google Scholar] [CrossRef]
- Wang, G.; Dinkins, M.; He, Q.; Zhu, G.; Poirier, C.; Campbell, A.; Mayer-Proschel, M.; Bieberich, E. Astrocytes secrete exosomes enriched with proapoptotic ceramide and prostate apoptosis response 4 (PAR-4): Potential mechanism of apoptosis induction in Alzheimer disease (AD). J. Biol. Chem. 2012, 287, 21384–21395. [Google Scholar] [CrossRef] [Green Version]
- Nogueras-Ortiz, C.J.; Mahairaki, V.; Delgado-Peraza, F.; Das, D.; Avgerinos, K.; Eren, E.; Hentschel, M.; Goetzl, E.J.; Mattson, M.P.; Kapogiannis, D. Astrocyte- and Neuron-Derived Extracellular Vesicles from Alzheimer’s Disease Patients Effect Complement-Mediated Neurotoxicity. Cells 2020, 9, 1618. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Cserep, C.; Posfai, B.; Lenart, N.; Fekete, R.; Laszlo, Z.I.; Lele, Z.; Orsolits, B.; Molnar, G.; Heindl, S.; Schwarcz, A.D.; et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 2020, 367, 528–537. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [Green Version]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jay, T.R.; Hirsch, A.M.; Broihier, M.L.; Miller, C.M.; Neilson, L.E.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2017, 37, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; Weiner, A.; Amit, I. The Physiology, Pathology, and Potential Therapeutic Applications of the TREM2 Signaling Pathway. Cell 2020, 181, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Criscuolo, C.; Fontebasso, V.; Middei, S.; Stazi, M.; Ammassari-Teule, M.; Yan, S.S.; Origlia, N. Entorhinal Cortex dysfunction can be rescued by inhibition of microglial RAGE in an Alzheimer’s disease mouse model. Sci. Rep. 2017, 7, 42370. [Google Scholar] [CrossRef] [Green Version]
- Origlia, N.; Bonadonna, C.; Rosellini, A.; Leznik, E.; Arancio, O.; Yan, S.S.; Domenici, L. Microglial receptor for advanced glycation end product-dependent signal pathway drives beta-amyloid-induced synaptic depression and long-term depression impairment in entorhinal cortex. J. Neurosci. 2010, 30, 11414–11425. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Gaiteri, C.; Bodea, L.G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, A.; Wake, H.; Ishikawa, A.W.; Eto, K.; Shibata, K.; Murakoshi, H.; Koizumi, S.; Moorhouse, A.J.; Yoshimura, Y.; Nabekura, J. Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 2016, 7, 12540. [Google Scholar] [CrossRef] [Green Version]
- Weinhard, L.; di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018, 9, 1228. [Google Scholar] [CrossRef] [Green Version]
- Antonucci, F.; Turola, E.; Riganti, L.; Caleo, M.; Gabrielli, M.; Perrotta, C.; Novellino, L.; Clementi, E.; Giussani, P.; Viani, P.; et al. Microvesicles released from microglia stimulate synaptic activity via enhanced sphingolipid metabolism. EMBO J. 2012, 31, 1231–1240. [Google Scholar] [CrossRef]
- Riganti, L.; Antonucci, F.; Gabrielli, M.; Prada, I.; Giussani, P.; Viani, P.; Valtorta, F.; Menna, E.; Matteoli, M.; Verderio, C. Sphingosine-1-Phosphate (S1P) Impacts Presynaptic Functions by Regulating Synapsin I Localization in the Presynaptic Compartment. J. Neurosci. 2016, 36, 4624–4634. [Google Scholar] [CrossRef] [Green Version]
- Marrone, M.C.; Morabito, A.; Giustizieri, M.; Chiurchiu, V.; Leuti, A.; Mattioli, M.; Marinelli, S.; Riganti, L.; Lombardi, M.; Murana, E.; et al. TRPV1 channels are critical brain inflammation detectors and neuropathic pain biomarkers in mice. Nat. Commun. 2017, 8, 15292. [Google Scholar] [CrossRef] [Green Version]
- Gabrielli, M.; Battista, N.; Riganti, L.; Prada, I.; Antonucci, F.; Cantone, L.; Matteoli, M.; Maccarrone, M.; Verderio, C. Active endocannabinoids are secreted on extracellular membrane vesicles. EMBO Rep. 2015, 16, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Prada, I.; Gabrielli, M.; Turola, E.; Iorio, A.; D’Arrigo, G.; Parolisi, R.; De Luca, M.; Pacifici, M.; Bastoni, M.; Lombardi, M.; et al. Glia-to-neuron transfer of miRNAs via extracellular vesicles: A new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol. 2018, 135, 529–550. [Google Scholar] [CrossRef]
- Vinuesa, A.; Bentivegna, M.; Calfa, G.; Filipello, F.; Pomilio, C.; Bonaventura, M.M.; Lux-Lantos, V.; Matzkin, M.E.; Gregosa, A.; Presa, J.; et al. Early Exposure to a High-Fat Diet Impacts on Hippocampal Plasticity: Implication of Microglia-Derived Exosome-like Extracellular Vesicles. Mol. Neurobiol. 2019, 56, 5075–5094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, P.; Benussi, L.; Furlan, R.; Ghidoni, R.; Verderio, C. Extracellular vesicles in Alzheimer’s disease: Friends or foes? Focus on abeta-vesicle interaction. Int. J. Mol. Sci. 2015, 16, 4800–4813. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Garcia-Garcia, E.; Royo, F.; Falcon-Perez, J.M.; Avila, J. Proteostasis of tau. Tau overexpression results in its secretion via membrane vesicles. FEBS Lett. 2012, 586, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Clayton, K.A.; Van Enoo, A.A.; Ikezu, T. Alzheimer’s Disease: The Role of Microglia in Brain Homeostasis and Proteopathy. Front. Neurosci. 2017, 11, 680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallart-Palau, X.; Guo, X.; Serra, A.; Sze, S.K. Alzheimer’s disease progression characterized by alterations in the molecular profiles and biogenesis of brain extracellular vesicles. Alzheimer’s Res. Ther. 2020, 12, 54. [Google Scholar] [CrossRef]
- D’Acunzo, P.; Hargash, T.; Pawlik, M.; Goulbourne, C.N.; Perez-Gonzalez, R.; Levy, E. Enhanced generation of intraluminal vesicles in neuronal late endosomes in the brain of a Down syndrome mouse model with endosomal dysfunction. Dev. Neurobiol. 2019, 79, 656–663. [Google Scholar] [CrossRef]
- Gauthier, S.A.; Perez-Gonzalez, R.; Sharma, A.; Huang, F.K.; Alldred, M.J.; Pawlik, M.; Kaur, G.; Ginsberg, S.D.; Neubert, T.A.; Levy, E. Enhanced exosome secretion in Down syndrome brain—A protective mechanism to alleviate neuronal endosomal abnormalities. Acta Neuropathol. Commun. 2017, 5, 65. [Google Scholar] [CrossRef] [Green Version]
- Agosta, F.; Dalla Libera, D.; Spinelli, E.G.; Finardi, A.; Canu, E.; Bergami, A.; Bocchio Chiavetto, L.; Baronio, M.; Comi, G.; Martino, G.; et al. Myeloid microvesicles in cerebrospinal fluid are associated with myelin damage and neuronal loss in mild cognitive impairment and Alzheimer disease. Ann. Neurol. 2014, 76, 813–825. [Google Scholar] [CrossRef]
- Gabrielli, M.; Raffaele, S.; Fumagalli, M.; Verderio, C. The multiple faces of extracellular vesicles released by microglia: Where are we 10 years after? Front. Cell Neurosci. 2022, 16, 984690. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. Tripartite synapses: Roles for astrocytic purines in the control of synaptic physiology and behavior. Neuropharmacology 2009, 57, 343–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Arrigo, G.; Gabrielli, M.; Scaroni, F.; Swuec, P.; Amin, L.; Pegoraro, A.; Adinolfi, E.; Di Virgilio, F.; Cojoc, D.; Legname, G.; et al. Astrocytes-derived extracellular vesicles in motion at the neuron surface: Involvement of the prion protein. J. Extracell. Vesicles 2021, 10, e12114. [Google Scholar] [CrossRef] [PubMed]
- Venturini, A.; Passalacqua, M.; Pelassa, S.; Pastorino, F.; Tedesco, M.; Cortese, K.; Gagliani, M.C.; Leo, G.; Maura, G.; Guidolin, D.; et al. Exosomes From Astrocyte Processes: Signaling to Neurons. Front. Pharmacol. 2019, 10, 1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta Chaudhuri, A.; Dasgheyb, R.M.; DeVine, L.R.; Bi, H.; Cole, R.N.; Haughey, N.J. Stimulus-dependent modifications in astrocyte-derived extracellular vesicle cargo regulate neuronal excitability. Glia 2020, 68, 128–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, M.R.; Weaver, A.M. Astrocyte-derived small extracellular vesicles promote synapse formation via fibulin-2-mediated TGF-beta signaling. Cell. Rep. 2021, 34, 108829. [Google Scholar] [CrossRef]
- You, Y.; Borgmann, K.; Edara, V.V.; Stacy, S.; Ghorpade, A.; Ikezu, T. Activated human astrocyte-derived extracellular vesicles modulate neuronal uptake, differentiation and firing. J. Extracell. Vesicles 2020, 9, 1706801. [Google Scholar] [CrossRef]
- Carter, S.F.; Scholl, M.; Almkvist, O.; Wall, A.; Engler, H.; Langstrom, B.; Nordberg, A. Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: A multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J. Nucl. Med. 2012, 53, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Zuo, Y.X.; Jiang, R.T. Astrocyte morphology: Diversity, plasticity, and role in neurological diseases. CNS Neurosci. Ther. 2019, 25, 665–673. [Google Scholar] [CrossRef]
- St-Pierre, M.K.; Vander Zwaag, J.; Loewen, S.; Tremblay, M.E. All roads lead to heterogeneity: The complex involvement of astrocytes and microglia in the pathogenesis of Alzheimer’s disease. Front. Cell. Neurosci. 2022, 16, 932572. [Google Scholar] [CrossRef]
- Sadick, J.S.; O’Dea, M.R.; Hasel, P.; Dykstra, T.; Faustin, A.; Liddelow, S.A. Astrocytes and oligodendrocytes undergo subtype-specific transcriptional changes in Alzheimer’s disease. Neuron 2022, 110, 1788–1805.e1710. [Google Scholar] [CrossRef]
- Sidoryk-Wegrzynowicz, M.; Gerber, Y.N.; Ries, M.; Sastre, M.; Tolkovsky, A.M.; Spillantini, M.G. Astrocytes in mouse models of tauopathies acquire early deficits and lose neurosupportive functions. Acta Neuropathol. Commun. 2017, 5, 89. [Google Scholar] [CrossRef] [PubMed]
- Richetin, K.; Steullet, P.; Pachoud, M.; Perbet, R.; Parietti, E.; Maheswaran, M.; Eddarkaoui, S.; Begard, S.; Pythoud, C.; Rey, M.; et al. Tau accumulation in astrocytes of the dentate gyrus induces neuronal dysfunction and memory deficits in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1567–1579. [Google Scholar] [CrossRef]
- Jones, R.S.; Minogue, A.M.; Connor, T.J.; Lynch, M.A. Amyloid-beta-induced astrocytic phagocytosis is mediated by CD36, CD47 and RAGE. J. Neuroimmune Pharmacol. 2013, 8, 301–311. [Google Scholar] [CrossRef]
- Chung, W.S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013, 504, 394–400. [Google Scholar] [CrossRef] [Green Version]
- Chang, G.H.; Barbaro, N.M.; Pieper, R.O. Phosphatidylserine-dependent phagocytosis of apoptotic glioma cells by normal human microglia, astrocytes, and glioma cells. Neuro Oncol. 2000, 2, 174–183. [Google Scholar] [CrossRef]
- Magnus, T.; Chan, A.; Linker, R.A.; Toyka, K.V.; Gold, R. Astrocytes are less efficient in the removal of apoptotic lymphocytes than microglia cells: Implications for the role of glial cells in the inflamed central nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 760–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolowski, J.D.; Nobles, S.L.; Heffron, D.S.; Park, D.; Ravichandran, K.S.; Mandell, J.W. Brain-specific angiogenesis inhibitor-1 expression in astrocytes and neurons: Implications for its dual function as an apoptotic engulfment receptor. Brain Behav. Immun. 2011, 25, 915–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loov, C.; Hillered, L.; Ebendal, T.; Erlandsson, A. Engulfing astrocytes protect neurons from contact-induced apoptosis following injury. PLoS ONE 2012, 7, e33090. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, H.M.; Mulder, S.D.; Belien, J.A.; Musters, R.J.; Eikelenboom, P.; Veerhuis, R. Astrocytic A beta 1-42 uptake is determined by A beta-aggregation state and the presence of amyloid-associated proteins. Glia 2010, 58, 1235–1246. [Google Scholar] [CrossRef]
- Nagele, R.G.; D’Andrea, M.R.; Lee, H.; Venkataraman, V.; Wang, H.Y. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003, 971, 197–209. [Google Scholar] [CrossRef]
- Loov, C.; Mitchell, C.H.; Simonsson, M.; Erlandsson, A. Slow degradation in phagocytic astrocytes can be enhanced by lysosomal acidification. Glia 2015, 63, 1997–2009. [Google Scholar] [CrossRef] [PubMed]
- Sollvander, S.; Nikitidou, E.; Brolin, R.; Soderberg, L.; Sehlin, D.; Lannfelt, L.; Erlandsson, A. Accumulation of amyloid-beta by astrocytes result in enlarged endosomes and microvesicle-induced apoptosis of neurons. Mol. Neurodegener. 2016, 11, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikitidou, E.; Khoonsari, P.E.; Shevchenko, G.; Ingelsson, M.; Kultima, K.; Erlandsson, A. Increased Release of Apolipoprotein E in Extracellular Vesicles Following Amyloid-beta Protofibril Exposure of Neuroglial Co-Cultures. J. Alzheimer’s Dis. 2017, 60, 305–321. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Molina, L.A.; Villar-Vesga, J.; Henao-Restrepo, J.; Villegas, A.; Lopera, F.; Cardona-Gomez, G.P.; Posada-Duque, R. Extracellular Vesicles from 3xTg-AD Mouse and Alzheimer’s Disease Patient Astrocytes Impair Neuroglial and Vascular Components. Front. Aging Neurosci. 2021, 13, 593927. [Google Scholar] [CrossRef]
- Dinkins, M.B.; Dasgupta, S.; Wang, G.; Zhu, G.; Bieberich, E. Exosome reduction in vivo is associated with lower amyloid plaque load in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1792–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetzl, E.J.; Schwartz, J.B.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D. High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann. Neurol. 2018, 83, 544–552. [Google Scholar] [CrossRef]
- Muraoka, S.; Jedrychowski, M.P.; Yanamandra, K.; Ikezu, S.; Gygi, S.P.; Ikezu, T. Proteomic Profiling of Extracellular Vesicles Derived from Cerebrospinal Fluid of Alzheimer’s Disease Patients: A Pilot Study. Cells 2020, 9, 1959. [Google Scholar] [CrossRef]
- You, Y.; Muraoka, S.; Jedrychowski, M.P.; Hu, J.; McQuade, A.K.; Young-Pearse, T.; Aslebagh, R.; Shaffer, S.A.; Gygi, S.P.; Blurton-Jones, M.; et al. Human neural cell type-specific extracellular vesicle proteome defines disease-related molecules associated with activated astrocytes in Alzheimer’s disease brain. J. Extracell. Vesicles 2022, 11, e12183. [Google Scholar] [CrossRef]
- Stronati, E.; Conti, R.; Cacci, E.; Cardarelli, S.; Biagioni, S.; Poiana, G. Extracellular Vesicle-Induced Differentiation of Neural Stem Progenitor Cells. Int. J. Mol. Sci. 2019, 20, 3691. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Li, C.; Huang, Y.; Wang, Y.; Xia, X.; Zheng, J.C. Exosomes released from neural progenitor cells and induced neural progenitor cells regulate neurogenesis through miR-21a. Cell. Commun. Signal. 2019, 17, 96. [Google Scholar] [CrossRef]
- Budnik, V.; Ruiz-Canada, C.; Wendler, F. Extracellular vesicles round off communication in the nervous system. Nat. Rev. Neurosci. 2016, 17, 160–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer-Albers, E.M.; Hill, A.F. Extracellular vesicles: Interneural shuttles of complex messages. Curr. Opin. Neurobiol. 2016, 39, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Bahrini, I.; Song, J.H.; Diez, D.; Hanayama, R. Neuronal exosomes facilitate synaptic pruning by up-regulating complement factors in microglia. Sci. Rep. 2015, 5, 7989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morel, L.; Regan, M.; Higashimori, H.; Ng, S.K.; Esau, C.; Vidensky, S.; Rothstein, J.; Yang, Y. Neuronal exosomal miRNA-dependent translational regulation of astroglial glutamate transporter GLT1. J. Biol. Chem. 2013, 288, 7105–7116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chivet, M.; Javalet, C.; Laulagnier, K.; Blot, B.; Hemming, F.J.; Sadoul, R. Exosomes secreted by cortical neurons upon glutamatergic synapse activation specifically interact with neurons. J. Extracell. Vesicles 2014, 3, 24722. [Google Scholar] [CrossRef] [Green Version]
- Lachenal, G.; Pernet-Gallay, K.; Chivet, M.; Hemming, F.J.; Belly, A.; Bodon, G.; Blot, B.; Haase, G.; Goldberg, Y.; Sadoul, R. Release of exosomes from differentiated neurons and its regulation by synaptic glutamatergic activity. Mol. Cell. Neurosci. 2011, 46, 409–418. [Google Scholar] [CrossRef] [Green Version]
- Chivet, M.; Javalet, C.; Hemming, F.; Pernet-Gallay, K.; Laulagnier, K.; Fraboulet, S.; Sadoul, R. Exosomes as a novel way of interneuronal communication. Biochem. Soc. Trans. 2013, 41, 241–244. [Google Scholar] [CrossRef] [Green Version]
- Chivet, M.; Hemming, F.; Pernet-Gallay, K.; Fraboulet, S.; Sadoul, R. Emerging role of neuronal exosomes in the central nervous system. Front. Physiol. 2012, 3, 145. [Google Scholar] [CrossRef] [Green Version]
- Faure, J.; Lachenal, G.; Court, M.; Hirrlinger, J.; Chatellard-Causse, C.; Blot, B.; Grange, J.; Schoehn, G.; Goldberg, Y.; Boyer, V.; et al. Exosomes are released by cultured cortical neurones. Mol. Cell. Neurosci. 2006, 31, 642–648. [Google Scholar] [CrossRef]
- Brenna, S.; Altmeppen, H.C.; Mohammadi, B.; Rissiek, B.; Schlink, F.; Ludewig, P.; Krisp, C.; Schluter, H.; Failla, A.V.; Schneider, C.; et al. Characterization of brain-derived extracellular vesicles reveals changes in cellular origin after stroke and enrichment of the prion protein with a potential role in cellular uptake. J. Extracell. Vesicles 2020, 9, 1809065. [Google Scholar] [CrossRef]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharples, R.A.; Vella, L.J.; Nisbet, R.M.; Naylor, R.; Perez, K.; Barnham, K.J.; Masters, C.L.; Hill, A.F. Inhibition of gamma-secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. FASEB J. 2008, 22, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Hamdane, M.; Loyens, A.; Gele, P.; Drobeck, H.; Begard, S.; Galas, M.C.; Delacourte, A.; Beauvillain, J.C.; Buee, L.; et al. Alkalizing drugs induce accumulation of amyloid precursor protein by-products in luminal vesicles of multivesicular bodies. J. Biol. Chem. 2007, 282, 18197–18205. [Google Scholar] [CrossRef] [Green Version]
- Ghidoni, R.; Paterlini, A.; Albertini, V.; Glionna, M.; Monti, E.; Schiaffonati, L.; Benussi, L.; Levy, E.; Binetti, G. Cystatin C is released in association with exosomes: A new tool of neuronal communication which is unbalanced in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 1435–1442. [Google Scholar] [CrossRef] [Green Version]
- Laulagnier, K.; Javalet, C.; Hemming, F.J.; Chivet, M.; Lachenal, G.; Blot, B.; Chatellard, C.; Sadoul, R. Amyloid precursor protein products concentrate in a subset of exosomes specifically endocytosed by neurons. Cell Mol. Life Sci. 2018, 75, 757–773. [Google Scholar] [CrossRef]
- Lauren, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [Green Version]
- Falker, C.; Hartmann, A.; Guett, I.; Dohler, F.; Altmeppen, H.; Betzel, C.; Schubert, R.; Thurm, D.; Wegwitz, F.; Joshi, P.; et al. Exosomal cellular prion protein drives fibrillization of amyloid beta and counteracts amyloid beta-mediated neurotoxicity. J. Neurochem. 2016, 137, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuyama, K.; Sun, H.; Igarashi, Y.; Monde, K.; Hirase, T.; Nakayama, M.; Makino, Y. Immuno-digital invasive cleavage assay for analyzing Alzheimer’s amyloid ss-bound extracellular vesicles. Alzheimer’s Res. Ther. 2022, 14, 140. [Google Scholar] [CrossRef] [PubMed]
- Yuyama, K.; Igarashi, Y. Linking glycosphingolipids to Alzheimer’s amyloid-ss: Extracellular vesicles and functional plant materials. Glycoconj. J. 2022, 39, 613–618. [Google Scholar] [CrossRef]
- Guix, F.X.; Corbett, G.T.; Cha, D.J.; Mustapic, M.; Liu, W.; Mengel, D.; Chen, Z.; Aikawa, E.; Young-Pearse, T.; Kapogiannis, D.; et al. Detection of Aggregation-Competent Tau in Neuron-Derived Extracellular Vesicles. Int. J. Mol. Sci. 2018, 19, 663. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Abner, E.L.; Jicha, G.A.; Kapogiannis, D.; Schwartz, J.B. Declining levels of functionally specialized synaptic proteins in plasma neuronal exosomes with progression of Alzheimer’s disease. FASEB J. 2018, 32, 888–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetzl, E.J.; Kapogiannis, D.; Schwartz, J.B.; Lobach, I.V.; Goetzl, L.; Abner, E.L.; Jicha, G.A.; Karydas, A.M.; Boxer, A.; Miller, B.L. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. 2016, 30, 4141–4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.R.; Yao, Y.X.; Jiang, X.Y.; Dong, Q.Y.; Yu, X.F.; Wang, T.; Cai, Y.N.; Han, Y. beta-Amyloid in blood neuronal-derived extracellular vesicles is elevated in cognitively normal adults at risk of Alzheimer’s disease and predicts cerebral amyloidosis. Alzheimer’s Res. Ther. 2022, 14, 66. [Google Scholar] [CrossRef] [PubMed]
- Kapogiannis, D.; Mustapic, M.; Shardell, M.D.; Berkowitz, S.T.; Diehl, T.C.; Spangler, R.D.; Tran, J.; Lazaropoulos, M.P.; Chawla, S.; Gulyani, S.; et al. Association of Extracellular Vesicle Biomarkers with Alzheimer Disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol. 2019, 76, 1340–1351. [Google Scholar] [CrossRef]
- Eren, E.; Hunt, J.F.V.; Shardell, M.; Chawla, S.; Tran, J.; Gu, J.; Vogt, N.M.; Johnson, S.C.; Bendlin, B.B.; Kapogiannis, D. Extracellular vesicle biomarkers of Alzheimer’s disease associated with sub-clinical cognitive decline in late middle age. Alzheimer’s Dement. 2020, 16, 1293–1304. [Google Scholar] [CrossRef]
- Tian, C.; Stewart, T.; Hong, Z.; Guo, Z.; Aro, P.; Soltys, D.; Pan, C.; Peskind, E.R.; Zabetian, C.P.; Shaw, L.M.; et al. Blood extracellular vesicles carrying synaptic function- and brain-related proteins as potential biomarkers for Alzheimer’s disease. Alzheimer’s Dement. 2022. [Google Scholar] [CrossRef]
- Perez-Gonzalez, R.; Kim, Y.; Miller, C.; Pacheco-Quinto, J.; Eckman, E.A.; Levy, E. Extracellular vesicles: Where the amyloid precursor protein carboxyl-terminal fragments accumulate and amyloid-beta oligomerizes. FASEB J. 2020, 34, 12922–12931. [Google Scholar] [CrossRef]
- Picciolini, S.; Gualerzi, A.; Carlomagno, C.; Cabinio, M.; Sorrentino, S.; Baglio, F.; Bedoni, M. An SPRi-based biosensor pilot study: Analysis of multiple circulating extracellular vesicles and hippocampal volume in Alzheimer’s disease. J. Pharm. Biomed. Anal. 2021, 192, 113649. [Google Scholar] [CrossRef]
- Zheng, T.; Pu, J.; Chen, Y.; Mao, Y.; Guo, Z.; Pan, H.; Zhang, L.; Zhang, H.; Sun, B.; Zhang, B. Plasma Exosomes Spread and Cluster Around beta-Amyloid Plaques in an Animal Model of Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 12. [Google Scholar] [CrossRef] [Green Version]
- Verweij, F.J.; Balaj, L.; Boulanger, C.M.; Carter, D.R.F.; Compeer, E.B.; D’Angelo, G.; El Andaloussi, S.; Goetz, J.G.; Gross, J.C.; Hyenne, V.; et al. The power of imaging to understand extracellular vesicle biology in vivo. Nat. Methods 2021, 18, 1013–1026. [Google Scholar] [CrossRef]
- Kur, I.M.; Prouvot, P.H.; Fu, T.; Fan, W.; Muller-Braun, F.; Das, A.; Das, S.; Deller, T.; Roeper, J.; Stroh, A.; et al. Neuronal activity triggers uptake of hematopoietic extracellular vesicles in vivo. PLoS Biol. 2020, 18, e3000643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neckles, V.N.; Morton, M.C.; Holmberg, J.C.; Sokolov, A.M.; Nottoli, T.; Liu, D.; Feliciano, D.M. A transgenic inducible GFP extracellular-vesicle reporter (TIGER) mouse illuminates neonatal cortical astrocytes as a source of immunomodulatory extracellular vesicles. Sci. Rep. 2019, 9, 3094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heusermann, W.; Hean, J.; Trojer, D.; Steib, E.; von Bueren, S.; Graff-Meyer, A.; Genoud, C.; Martin, K.; Pizzato, N.; Voshol, J.; et al. Exosomes surf on filopodia to enter cells at endocytic hot spots, traffic within endosomes, and are targeted to the ER. J. Cell. Biol. 2016, 213, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prada, I.; Amin, L.; Furlan, R.; Legname, G.; Verderio, C.; Cojoc, D. A new approach to follow a single extracellular vesicle-cell interaction using optical tweezers. Biotechniques 2016, 60, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Li, Z.; He, T.; Qu, M.; Jiang, L.; Li, W.; Shi, X.; Pan, J.; Zhang, L.; Wang, Y.; et al. M2 microglia-derived exosomes protect the mouse brain from ischemia-reperfusion injury via exosomal miR-124. Theranostics 2019, 9, 2910–2923. [Google Scholar] [CrossRef]
- Fevrier, B.; Vilette, D.; Archer, F.; Loew, D.; Faigle, W.; Vidal, M.; Laude, H.; Raposo, G. Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. USA 2004, 101, 9683–9688. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, M.; Gabrielli, M.; Adinolfi, E.; Verderio, C. Role of ATP in Extracellular Vesicle Biogenesis and Dynamics. Front. Pharmacol. 2021, 12, 654023. [Google Scholar] [CrossRef]
- Tonoli, E.; Verduci, I.; Gabrielli, M.; Prada, I.; Forcaia, G.; Coveney, C.; Savoca, M.P.; Boocock, D.J.; Sancini, G.; Mazzanti, M.; et al. Extracellular transglutaminase-2, nude or associated with astrocytic extracellular vesicles, modulates neuronal calcium homeostasis. Prog. Neurobiol. 2022, 216, 102313. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.M.; Tonoli, E.; Coveney, C.; Boocock, D.J.; Jongenelen, C.A.M.; Breve, J.J.P.; Verderio, E.A.M.; Drukarch, B. The Transglutaminase-2 Interactome in the APP23 Mouse Model of Alzheimer’s Disease. Cells 2022, 11, 389. [Google Scholar] [CrossRef]
- Williams, C.; Pazos, R.; Royo, F.; Gonzalez, E.; Roura-Ferrer, M.; Martinez, A.; Gamiz, J.; Reichardt, N.C.; Falcon-Perez, J.M. Assessing the role of surface glycans of extracellular vesicles on cellular uptake. Sci. Rep. 2019, 9, 11920. [Google Scholar] [CrossRef]
- Christianson, H.C.; Svensson, K.J.; van Kuppevelt, T.H.; Li, J.P.; Belting, M. Cancer cell exosomes depend on cell-surface heparan sulfate proteoglycans for their internalization and functional activity. Proc. Natl. Acad. Sci. USA 2013, 110, 17380–17385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barres, C.; Blanc, L.; Bette-Bobillo, P.; Andre, S.; Mamoun, R.; Gabius, H.J.; Vidal, M. Galectin-5 is bound onto the surface of rat reticulocyte exosomes and modulates vesicle uptake by macrophages. Blood 2010, 115, 696–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morelli, A.E.; Larregina, A.T.; Shufesky, W.J.; Sullivan, M.L.; Stolz, D.B.; Papworth, G.D.; Zahorchak, A.F.; Logar, A.J.; Wang, Z.; Watkins, S.C.; et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood 2004, 104, 3257–3266. [Google Scholar] [CrossRef] [Green Version]
- Sung, B.H.; Ketova, T.; Hoshino, D.; Zijlstra, A.; Weaver, A.M. Directional cell movement through tissues is controlled by exosome secretion. Nat. Commun. 2015, 6, 7164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purushothaman, A.; Bandari, S.K.; Liu, J.; Mobley, J.A.; Brown, E.E.; Sanderson, R.D. Fibronectin on the Surface of Myeloma Cell-derived Exosomes Mediates Exosome-Cell Interactions. J. Biol. Chem. 2016, 291, 1652–1663. [Google Scholar] [CrossRef] [Green Version]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell. Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Bianco, F.; Pravettoni, E.; Colombo, A.; Schenk, U.; Moller, T.; Matteoli, M.; Verderio, C. Astrocyte-derived ATP induces vesicle shedding and IL-1 beta release from microglia. J. Immunol. 2005, 174, 7268–7277. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, S.N.; Sun, L.; Cole, K.Y.; Ford, C.R., 3rd; Olcese, J.M.; Meckes, D.G., Jr. An optimized method for enrichment of whole brain-derived extracellular vesicles reveals insight into neurodegenerative processes in a mouse model of Alzheimer’s disease. J. Neurosci. Methods 2018, 307, 210–220. [Google Scholar] [CrossRef]
- Alim, M.A.; Ma, Q.L.; Takeda, K.; Aizawa, T.; Matsubara, M.; Nakamura, M.; Asada, A.; Saito, T.; Kaji, H.; Yoshii, M.; et al. Demonstration of a role for alpha-synuclein as a functional microtubule-associated protein. J. Alzheimer’s Dis. 2004, 6, 435–442; discussion 443-439. [Google Scholar] [CrossRef]
- Westergard, L.; Christensen, H.M.; Harris, D.A. The cellular prion protein (PrP(C)): Its physiological function and role in disease. Biochim. Biophys. Acta 2007, 1772, 629–644. [Google Scholar] [CrossRef]
- Deyts, C.; Thinakaran, G.; Parent, A.T. APP Receptor? To Be or Not to Be. Trends Pharmacol. Sci. 2016, 37, 390–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabo, S.L.; Ikin, A.F.; Buxbaum, J.D.; Greengard, P. The amyloid precursor protein and its regulatory protein, FE65, in growth cones and synapses in vitro and in vivo. J. Neurosci. 2003, 23, 5407–5415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brody, A.H.; Strittmatter, S.M. Synaptotoxic Signaling by Amyloid Beta Oligomers in Alzheimer’s Disease through Prion Protein and mGluR5. Adv. Pharmacol. 2018, 82, 293–323. [Google Scholar] [CrossRef] [PubMed]
- Nieznanski, K.; Surewicz, K.; Chen, S.; Nieznanska, H.; Surewicz, W.K. Interaction between prion protein and Abeta amyloid fibrils revisited. ACS Chem. Neurosci. 2014, 5, 340–345. [Google Scholar] [CrossRef]
- Corbett, G.T.; Wang, Z.; Hong, W.; Colom-Cadena, M.; Rose, J.; Liao, M.; Asfaw, A.; Hall, T.C.; Ding, L.; DeSousa, A.; et al. PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins. Acta Neuropathol. 2020, 139, 503–526. [Google Scholar] [CrossRef] [Green Version]
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fa, M.; Amato, A.; Palmeri, A.; D’Adamio, L.; Grassi, C.; Devanand, D.P.; Honig, L.S.; et al. Role of Amyloid-beta and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. J. Alzheimer’s Dis. 2018, 64, S611–S631. [Google Scholar] [CrossRef]
- Shimoda, M.; Khokha, R. Metalloproteinases in extracellular vesicles. Biochim. Biophys. Acta Mol. Cell. Res. 2017, 1864, 1989–2000. [Google Scholar] [CrossRef]
- Lewin, S.; Hunt, S.; Lambert, D.W. Extracellular vesicles and the extracellular matrix: A new paradigm or old news? Biochem. Soc. Trans. 2020, 48, 2335–2345. [Google Scholar] [CrossRef]
- Rilla, K.; Mustonen, A.M.; Arasu, U.T.; Harkonen, K.; Matilainen, J.; Nieminen, P. Extracellular vesicles are integral and functional components of the extracellular matrix. Matrix. Biol. 2019, 75–76, 201–219. [Google Scholar] [CrossRef]
- Rackov, G.; Garcia-Romero, N.; Esteban-Rubio, S.; Carrion-Navarro, J.; Belda-Iniesta, C.; Ayuso-Sacido, A. Vesicle-Mediated Control of Cell Function: The Role of Extracellular Matrix and Microenvironment. Front. Physiol. 2018, 9, 651. [Google Scholar] [CrossRef]
- Buzas, E.I.; Toth, E.A.; Sodar, B.W.; Szabo-Taylor, K.E. Molecular interactions at the surface of extracellular vesicles. Semin. Immunopathol. 2018, 40, 453–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, M.K.; Zaia, J. Extracellular matrix proteomics in schizophrenia and Alzheimer’s disease. Anal. Bioanal. Chem. 2017, 409, 379–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer-Albers, E.M. Extracellular vesicles in the oligodendrocyte microenvironment. Neurosci. Lett. 2020, 725, 134915. [Google Scholar] [CrossRef]
- Chen, X.D.; Zhao, J.; Yang, X.; Zhou, B.W.; Yan, Z.; Liu, W.F.; Li, C.; Liu, K.X. Gut-Derived Exosomes Mediate Memory Impairment After Intestinal Ischemia/Reperfusion via Activating Microglia. Mol. Neurobiol. 2021, 58, 4828–4841. [Google Scholar] [CrossRef] [PubMed]
- Cambria, C.; Ingegnoli, F.; Borzi, E.; Cantone, L.; Coletto, L.A.; Rizzuto, A.S.; De Lucia, O.; Briguglio, S.; Ruscica, M.; Caporali, R.; et al. Synovial Fluid-Derived Extracellular Vesicles of Patients with Arthritides Contribute to Hippocampal Synaptic Dysfunctions and Increase with Mood Disorders Severity in Humans. Cells 2022, 11, 2276. [Google Scholar] [CrossRef] [PubMed]
- Vandendriessche, C.; Kapogiannis, D.; Vandenbroucke, R.E. Biomarker and therapeutic potential of peripheral extracellular vesicles in Alzheimer’s disease. Adv. Drug Deliv. Rev. 2022, 190, 114486. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gabrielli, M.; Tozzi, F.; Verderio, C.; Origlia, N. Emerging Roles of Extracellular Vesicles in Alzheimer’s Disease: Focus on Synaptic Dysfunction and Vesicle–Neuron Interaction. Cells 2023, 12, 63. https://doi.org/10.3390/cells12010063
Gabrielli M, Tozzi F, Verderio C, Origlia N. Emerging Roles of Extracellular Vesicles in Alzheimer’s Disease: Focus on Synaptic Dysfunction and Vesicle–Neuron Interaction. Cells. 2023; 12(1):63. https://doi.org/10.3390/cells12010063
Chicago/Turabian StyleGabrielli, Martina, Francesca Tozzi, Claudia Verderio, and Nicola Origlia. 2023. "Emerging Roles of Extracellular Vesicles in Alzheimer’s Disease: Focus on Synaptic Dysfunction and Vesicle–Neuron Interaction" Cells 12, no. 1: 63. https://doi.org/10.3390/cells12010063
APA StyleGabrielli, M., Tozzi, F., Verderio, C., & Origlia, N. (2023). Emerging Roles of Extracellular Vesicles in Alzheimer’s Disease: Focus on Synaptic Dysfunction and Vesicle–Neuron Interaction. Cells, 12(1), 63. https://doi.org/10.3390/cells12010063