
Cardiomyocyte Ploidy, Metabolic Reprogramming and Heart Repair


Abstract
:1. Introduction
2. Cardiomyocyte Cell Cycle Dynamics during Development, Adulthood and Injury
3. Polyploidization and Nucleation
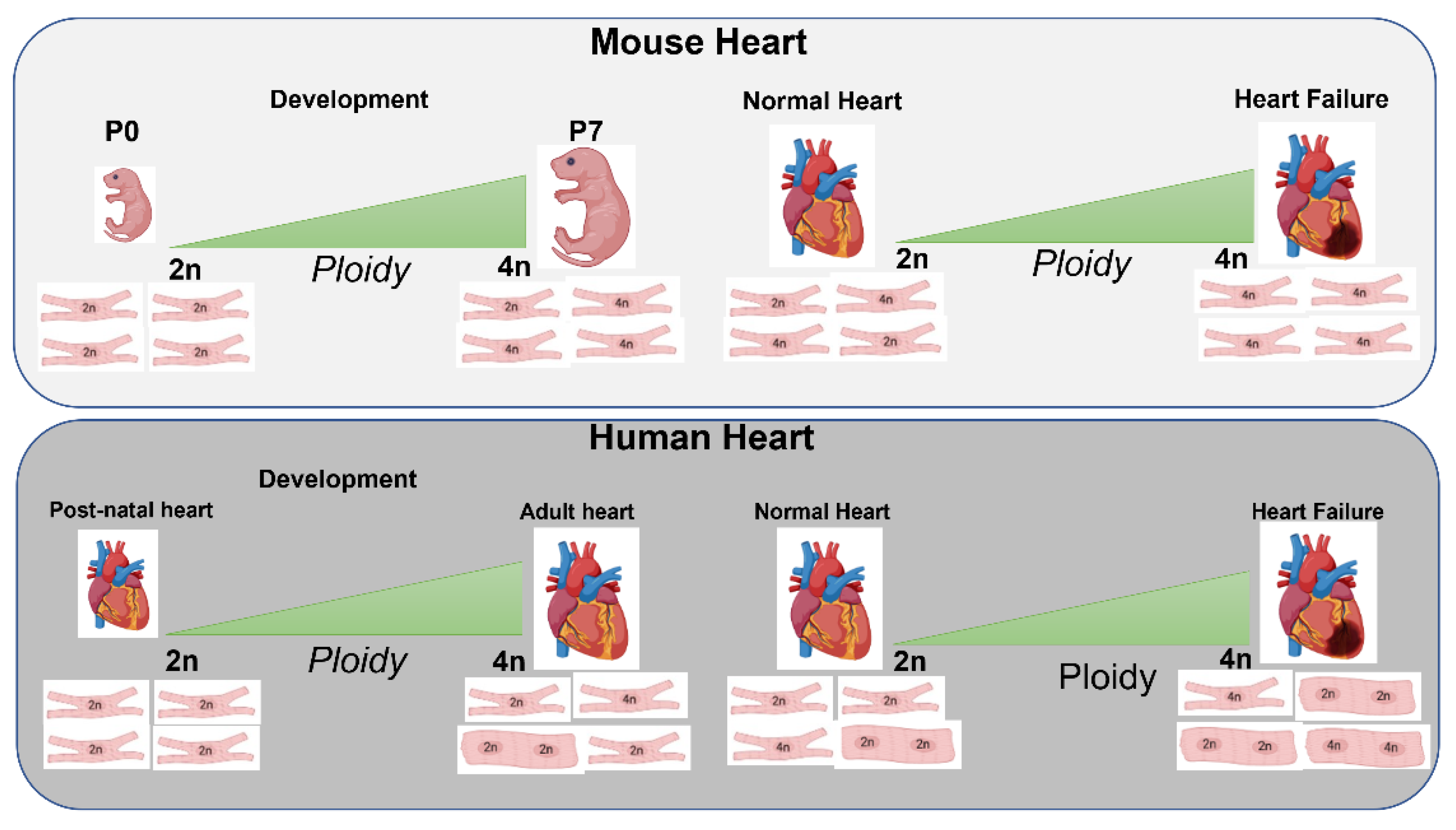
3.1. Role of Ploidy/Nucleation during Development, Postnatal and Adult Phases
3.2. Nucleation/Ploidy after Myocardial Injury
3.3. Strategies Targeting Ploidy/Nucleation and Their Effect on Myocardial Regeneration
4. Ploidy, Cell-Cycle Arrest, and Cell Death
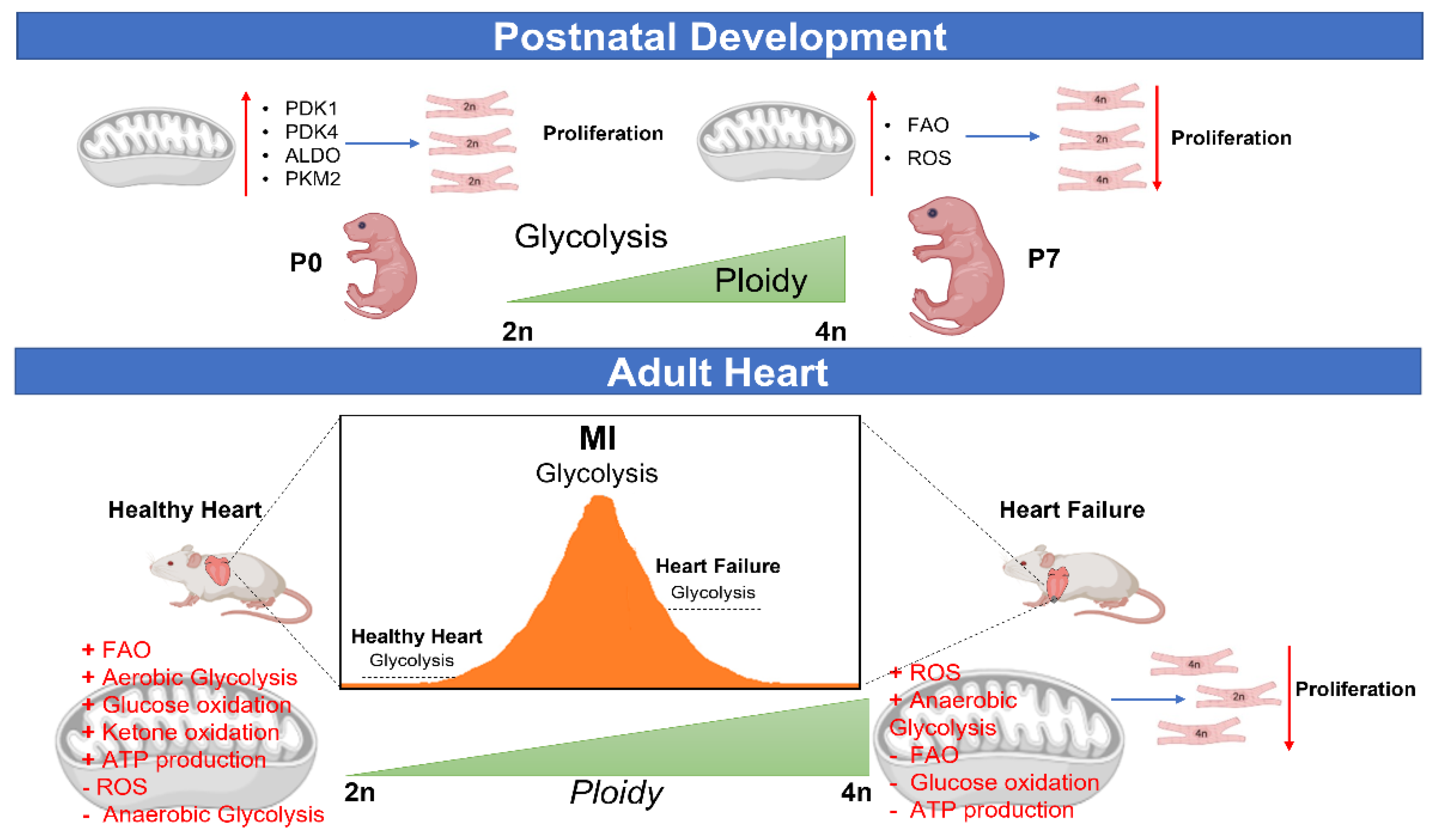
5. Metabolic Control of CM Ploidy
5.1. CM Ploidy and Metabolism during Development and Disease
5.2. Metabolic Reprogramming and CM Ploidy
6. Future Direction, Challenges and Limitations
Author Contributions
Funding
Conflicts of Interest
References
- Ahuja, P.; Sdek, P.; MacLellan, W.R. Cardiac Myocyte Cell Cycle Control in Development, Disease, and Regeneration. Physiol. Rev. 2007, 87, 521–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, R.; Jiang, Z.; Zagidullin, N.; Liu, T.; Cai, B. Regulation of cardiomyocyte fate plasticity: A key strategy for cardiac regeneration. Signal Transduct. Target. Ther. 2021, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Olson, E.N.; Bassel-Duby, R. Mending broken hearts: Cardiac development as a basis for adult heart regeneration and repair. Nat. Rev. Mol. Cell Biol. 2013, 14, 529–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, M.C.; Palmer, B.M.; Stauffer, B.L.; Leinwand, L.A.; Moore, R.L. Morphological and Functional Alterations in Ventricular Myocytes from Male Transgenic Mice with Hypertrophic Cardiomyopathy. Circ. Res. 2004, 94, 201–207. [Google Scholar] [CrossRef] [Green Version]
- Heallen, T.R.; Kadow, Z.; Kim, J.H.; Wang, J.; Martin, J.F. Stimulating Cardiogenesis as a Treatment for Heart Failure. Circ. Res. 2019, 124, 1647–1657. [Google Scholar] [CrossRef] [PubMed]
- Porrello, E.R.; Olson, E.N. A neonatal blueprint for cardiac regeneration. Stem Cell Res. 2014, 13, 556–570. [Google Scholar] [CrossRef] [Green Version]
- Uygur, A.; Lee, R.T. Mechanisms of Cardiac Regeneration. Dev. Cell 2016, 36, 362–374. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Bloomekatz, J.; Galvez-Santisteban, M.; Chi, N.C. Myocardial plasticity: Cardiac development, regeneration and disease. Curr. Opin. Genet. Dev. 2016, 40, 120–130. [Google Scholar] [CrossRef]
- Mauretti, A.; Spaans, S.; Bax, N.A.M.; Sahlgren, C.; Bouten, C.V.C. Cardiac Progenitor Cells and the Interplay with Their Microenvironment. Stem Cells Int. 2017, 2017, 7471582. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabé-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for Cardiomyocyte Renewal in Humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Maillet, M.; van Berlo, J.; Molkentin, J.D. Molecular basis of physiological heart growth: Fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasumarthi, K.B.; Field, L.J. Cardiomyocyte Cell Cycle Regulation. Circ. Res. 2002, 90, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef]
- Olson, E.N. Gene Regulatory Networks in the Evolution and Development of the Heart. Science 2006, 313, 1922–1927. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Nam, Y.-J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef] [Green Version]
- Zaglia, T.; Dedja, A.; Candiotto, C.; Cozzi, E.; Schiaffino, S.; Ausoni, S. Cardiac interstitial cells express GATA4 and control dedifferentiation and cell cycle re-entry of adult cardiomyocytes. J. Mol. Cell. Cardiol. 2009, 46, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, H.W.; Dashoush, N.H.; Tang, H.; Zhang, L.; Wang, X.; Wu, E.X.; Wolgemuth, D.J. Cyclin A2 Mediates Cardiomyocyte Mitosis in the Postmitotic Myocardium. J. Biol. Chem. 2004, 279, 35858–35866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Huang, Z.-P.; Seok, H.Y.; Ding, J.; Kataoka, M.; Zhang, Z.; Hu, X.; Wang, G.; Lin, Z.; Wang, S.; et al. mir-17–92 Cluster Is Required for and Sufficient to Induce Cardiomyocyte Proliferation in Postnatal and Adult Hearts. Circ. Res. 2013, 112, 1557–1566. [Google Scholar] [CrossRef] [Green Version]
- Thum, T.; Galuppo, P.; Wolf, C.; Fiedler, J.; Kneitz, S.; van Laake, L.W.; Doevendans, P.A.; Mummery, C.L.; Borlak, J.; Haverich, A.; et al. MicroRNAs in the Human Heart. Circulation 2007, 116, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Liu, Y.; Wang, T.; Zhou, N.; Kong, J.; Chen, L.; Snitow, M.; Morley, M.; Li, D.; Petrenko, N.; et al. A microRNA-Hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Sci. Transl. Med. 2015, 7, 279ra38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.; Nickoloff, E.; Abramova, T.; Johnson, J.; Verma, S.K.; Krishnamurthy, P.; Mackie, A.R.; Vaughan, E.; Garikipati, V.N.S.; Benedict, C.; et al. Embryonic Stem Cell–Derived Exosomes Promote Endogenous Repair Mechanisms and Enhance Cardiac Function Following Myocardial Infarction. Circ. Res. 2015, 117, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manco, R.; Leclercq, I.A.; Clerbaux, L.-A. Liver Regeneration: Different Sub-Populations of Parenchymal Cells at Play Choreographed by an Injury-Specific Microenvironment. Int. J. Mol. Sci. 2018, 19, 4115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlyng, P.; Åbyholm, A.; Grotmol, T.; Erikstein, B.; Huitfeldt, H.S.; Stokke, T.; Seglen, P.O. Binucleation and polyploidization patterns in developmental and regenerative rat liver growth. Cell Prolif. 1993, 26, 557–565. [Google Scholar] [CrossRef]
- Geschwind, I.; Alfert, M.; Schooley, C. Liver regeneration and hepatic polyploidy in the hypophysectomized rat. Exp. Cell Res. 1958, 15, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Maillet, V.; Boussetta, N.; Leclerc, J.; Fauveau, V.; Foretz, M.; Viollet, B.; Couty, J.-P.; Celton-Morizur, S.; Perret, C.; Desdouets, C. LKB1 as a Gatekeeper of Hepatocyte Proliferation and Genomic Integrity during Liver Regeneration. Cell Rep. 2018, 22, 1994–2005. [Google Scholar] [CrossRef] [Green Version]
- Sigal, S.H.; Rajvanshi, P.; Gorla, G.R.; Sokhi, R.P.; Saxena, R.; Gebhard, D.R.; Reid, L.M.; Gupta, S. Partial hepatectomy-induced polyploidy attenuates hepatocyte replication and activates cell aging events. Am. J. Physiol. Liver Physiol. 1999, 276, G1260–G1272. [Google Scholar] [CrossRef]
- Patterson, M.; Barske, L.; Van Handel, B.; Rau, C.D.; Gan, P.; Sharma, A.; Parikh, S.; Denholtz, M.; Huang, Y.; Yamaguchi, Y.; et al. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat. Genet. 2017, 49, 1346–1353. [Google Scholar] [CrossRef]
- Miyaoka, Y.; Ebato, K.; Kato, H.; Arakawa, S.; Shimizu, S.; Miyajima, A. Hypertrophy and Unconventional Cell Division of Hepatocytes Underlie Liver Regeneration. Curr. Biol. 2012, 22, 1166–1175. [Google Scholar] [CrossRef] [Green Version]
- Celton-Morizur, S.; Merlen, G.; Couton, D.; Margall-Ducos, G.; Desdouets, C. The insulin/Akt pathway controls a specific cell division program that leads to generation of binucleated tetraploid liver cells in rodents. J. Clin. Investig. 2009, 119, 1880–1887. [Google Scholar] [CrossRef] [Green Version]
- Gentric, G.; Desdouets, C. Polyploidization in Liver Tissue. Am. J. Pathol. 2014, 184, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Celton-Morizur, S.; Merlen, G.; Couton, D.; Desdouets, C. Polyploidy and liver proliferation: Central role of insulin signaling. Cell Cycle 2010, 9, 460–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anatskaya, O.V.; Vinogradov, A.E. Somatic polyploidy promotes cell function under stress and energy depletion: Evidence from tissue-specific mammal transcriptome. Funct. Integr. Genom. 2010, 10, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.W.; Newell, A.E.H.; Smith, L.; Wilson, E.M.; Olson, S.B.; Thayer, M.J.; Strom, S.C.; Grompe, M. Frequent Aneuploidy Among Normal Human Hepatocytes. Gastroenterology 2012, 142, 25–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, A.W.; Taylor, M.H.; Hickey, R.D.; Newell, A.E.H.; Lenzi, M.L.; Olson, S.B.; Finegold, M.J.; Grompe, M. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 2010, 467, 707–710. [Google Scholar] [CrossRef] [Green Version]
- Minnis-Lyons, S.E.; Ferreira-González, S.; Aleksieva, N.; Man, T.Y.; Gadd, V.L.; Williams, M.J.; Guest, R.V.; Lu, W.-Y.; Dwyer, B.J.; Jamieson, T.; et al. Notch-IGF1 signaling during liver regeneration drives biliary epithelial cell expansion and inhibits hepatocyte differentiation. Sci. Signal. 2021, 14, eaay9185. [Google Scholar] [CrossRef]
- Li, F.; Wang, X.; Gerdes, A. Formation of Binucleated Cardiac Myocytes in Rat Heart: II. Cytoskeletal Organisation. J. Mol. Cell. Cardiol. 1997, 29, 1553–1565. [Google Scholar] [CrossRef]
- Li, F.; Wang, X.; Bunger, P.C.; Gerdes, A. Formation of Binucleated Cardiac Myocytes in Rat Heart: I. Role of Actin–myosin Contractile Ring. J. Mol. Cell. Cardiol. 1997, 29, 1541–1551. [Google Scholar] [CrossRef]
- Soonpaa, M.H.; Kim, K.K.; Pajak, L.; Franklin, M.; Field, L.J. Cardiomyocyte DNA synthesis and binucleation during murine development. Am. J. Physiol. Circ. Physiol. 1996, 271, H2183–H2189. [Google Scholar] [CrossRef]
- Liu, Z.; Yue, S.; Chen, X.; Kubin, T.; Braun, T. Regulation of Cardiomyocyte Polyploidy and Multinucleation by CyclinG1. Circ. Res. 2010, 106, 1498–1506. [Google Scholar] [CrossRef]
- Clubb, F.J.; Bishop, S.P. Formation of binucleated myocardial cells in the neonatal rat. An index for growth hypertrophy. Lab. Investig. 1984, 50, 571–577. [Google Scholar] [PubMed]
- Gräbner, W.; Pfitzer, P. Number of nuclei in isolated myocardial cells of pigs. Virchows Arch. B Cell Pathol. 1974, 15, 279–294. [Google Scholar] [CrossRef]
- Olivetti, G. Aging, Cardiac Hypertrophy and Ischemic Cardiomyopathy Do Not Affect the Proportion of Mononucleated and Multinucleated Myocytes in the Human Heart. J. Mol. Cell. Cardiol. 1996, 28, 1463–1477. [Google Scholar] [CrossRef]
- Schmid, G.; Pfitzer, P. Mitoses and binucleated cells in perinatal human hearts. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1985, 48, 59–67. [Google Scholar] [CrossRef]
- Koh, K.N.; Kang, M.J.; Frith-Terhune, A.; Park, S.K.; Kim, I.; Lee, C.O.; Koh, G.Y. Persistent and Heterogenous Expression of the Cyclin-dependent Kinase Inhibitor, p27KIP1, in Rat Hearts During Development. J. Mol. Cell. Cardiol. 1998, 30, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Poolman, R.A.; Gilchrist, R.; Brooks, G. Cell cycle profiles and expressions of p21CIP1 and p27KIP1 during myocyte development. Int. J. Cardiol. 1998, 67, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Walsh, K.; Perlman, H. Cell cycle exit upon myogenic differentiation. Curr. Opin. Genet. Dev. 1997, 7, 597–602. [Google Scholar] [CrossRef]
- Wang, J.J.; Nadalginard, B. Regulation of Cyclins and P34CDC2 Expression during Terminal Differentiation of C2C12 Myocytes. Biochem. Biophys. Res. Commun. 1995, 206, 82–88. [Google Scholar] [CrossRef]
- Rumyantsev, P.P.; Borisov, A. DNA-Synthesis in Myocytes from Different Myocardial Compartments of Young-Rats in Norm, after Experimental Infarction and Invitro. Biomed. Biochim. Acta 1987, 46, S610–S615. [Google Scholar]
- Soonpaa, M.H.; Field, L.J. Survey of Studies Examining Mammalian Cardiomyocyte DNA Synthesis. Circ. Res. 1998, 83, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Soonpaa, M.H.; Field, L.J. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am. J. Physiol. Circ. Physiol. 1997, 272, H220–H226. [Google Scholar] [CrossRef] [PubMed]
- Pasumarthi, K.B.; Nakajima, H.; Nakajima, H.O.; Soonpaa, M.H.; Field, L.J. Targeted Expression of Cyclin D2 Results in Cardiomyocyte DNA Synthesis and Infarct Regression in Transgenic Mice. Circ. Res. 2005, 96, 110–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltrami, A.P.; Urbanek, K.; Kajstura, J.; Yan, S.-M.; Finato, N.; Bussani, R.; Nadal-Ginard, B.; Silvestri, F.; Leri, A.; Beltrami, C.A.; et al. Evidence That Human Cardiac Myocytes Divide after Myocardial Infarction. N. Engl. J. Med. 2001, 344, 1750–1757. [Google Scholar] [CrossRef] [PubMed]
- Kajstura, J.; Leri, A.; Finato, N.; Di Loreto, C.; Beltrami, C.A.; Anversa, P. Myocyte proliferation in end-stage cardiac failure in humans. Proc. Natl. Acad. Sci. USA 1998, 95, 8801–8805. [Google Scholar] [CrossRef] [Green Version]
- Herget, G.W.; Neuburger, M.; Plagwitz, R.; Adler, C.P. DNA content, ploidy level and number of nuclei in the human heart after myocardial infarction. Cardiovasc. Res. 1997, 36, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Meckert, P.C.; Rivello, H.G.; Vigliano, C.; González, P.; Favaloro, R.; Laguens, R. Endomitosis and polyploidization of myocardial cells in the periphery of human acute myocardial infarction. Cardiovasc. Res. 2005, 67, 116–123. [Google Scholar] [CrossRef]
- Matz, D.; Oberpriller, J. Comparison of mitosis in binucleated and mononucleated newt cardiac myocytes. Anat. Rec. 1998, 251, 245–255. [Google Scholar] [CrossRef]
- Stukenberg, P.T. Triggering p53 after cytokinesis failure. J. Cell Biol. 2004, 165, 607–608. [Google Scholar] [CrossRef] [Green Version]
- Urbanek, K.; Torella, D.; Sheikh, F.; De Angelis, A.; Nurzynska, D.; Silvestri, F.; Beltrami, C.A.; Bussani, R.; Beltrami, A.P.; Quaini, F.; et al. Myocardial regeneration by activation of multipotent cardiac stem cells in ischemic heart failure. Proc. Natl. Acad. Sci. USA 2005, 102, 8692–8697. [Google Scholar] [CrossRef] [Green Version]
- Xiao, G.; Mao, S.; Baumgarten, G.; Serrano, J.; Jordan, M.C.; Roos, K.P.; Fishbein, M.C.; MacLellan, W.R. Inducible Activation of c-Myc in Adult Myocardium In Vivo Provokes Cardiac Myocyte Hypertrophy and Reactivation of DNA Synthesis. Circ. Res. 2001, 89, 1122–1129. [Google Scholar] [CrossRef] [Green Version]
- Derks, W.; Bergmann, O. Polyploidy in Cardiomyocytes: Roadblock to Heart Regeneration? Circ. Res. 2020, 126, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Bishop, S.P.; Zhou, Y.; Nakada, Y.; Zhang, J. Changes in Cardiomyocyte Cell Cycle and Hypertrophic Growth During Fetal to Adult in Mammals. J. Am. Hear. Assoc. 2021, 10, e017839. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, V.Y.; Arefyeva, A.M.; Gvasava, I.G.; Sarkisov, D.S.; Panova, N.W. Polyploidy in cardiac myocytes of normal and hypertrophic human hearts; range of values. Virchows Arch. 1994, 424, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Olivetti, G.; Ricci, R.; Anversa, P. Hyperplasia of myocyte nuclei in long-term cardiac hypertrophy in rats. J. Clin. Investig. 1987, 80, 1818–1821. [Google Scholar] [CrossRef] [PubMed]
- Vliegen, H.W.; Van Der Laarse, A.; Cornelisse, C.J.; Eulderink, F. Myocardial changes in pressure overload-induced left ventricular hypertrophy. Eur. Hear. J. 1991, 12, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Payer, B.; Cayouette, M.; Harris, W.A. In Vivo Time-Lapse Imaging of Cell Divisions during Neurogenesis in the Developing Zebrafish Retina. Neuron 2003, 37, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Senyo, S.E.; Steinhauser, M.L.; Pizzimenti, C.L.; Yang, V.K.; Cai, L.; Wang, M.; Wu, T.-D.; Guerquin-Kern, J.-L.; Lechene, C.P.; Lee, R.T. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 2013, 493, 433–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, P.; Patterson, M.; Sucov, H.M. Cardiomyocyte Polyploidy and Implications for Heart Regeneration. Annu. Rev. Physiol. 2020, 82, 45–61. [Google Scholar] [CrossRef] [Green Version]
- Patterson, M.; Swift, S.K. Residual Diploidy in Polyploid Tissues: A Cellular State with Enhanced Proliferative Capacity for Tissue Regeneration? Stem Cells Dev. 2019, 28, 1527–1539. [Google Scholar] [CrossRef]
- Hirose, K.; Payumo, A.Y.; Cutie, S.; Hoang, A.; Zhang, H.; Guyot, R.; Lunn, D.; Bigley, R.B.; Yu, H.; Wang, J.; et al. Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science 2019, 364, 184–188. [Google Scholar] [CrossRef]
- González-Rosa, J.M.; Sharpe, M.; Field, D.; Soonpaa, M.H.; Field, L.J.; Burns, C.E.; Burns, C.G. Myocardial Polyploidization Creates a Barrier to Heart Regeneration in Zebrafish. Dev. Cell 2018, 44, 433–446.e437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirillova, A.; Han, L.; Liu, H.; Kühn, B. Polyploid cardiomyocytes: Implications for heart regeneration. Development 2021, 148, dev199401. [Google Scholar] [CrossRef] [PubMed]
- Donne, R.; Saroul-Aïnama, M.; Cordier, P.; Celton-Morizur, S.; Desdouets, C. Polyploidy in liver development, homeostasis and disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 391–405. [Google Scholar] [CrossRef]
- Tiemann, K.; Weyer, D.; Djoufack, P.C.; Ghanem, A.; Lewalter, T.; Dreiner, U.; Meyer, R.; Grohe, C.; Fink, K.B. Increasing myocardial contraction and blood pressure in C57BL/6 mice during early postnatal development. Am. J. Physiol. Circ. Physiol. 2003, 284, H464–H474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkass, K.; Panula, J.; Westman, M.; Wu, T.-D.; Guerquin-Kern, J.-L.; Bergmann, O. No Evidence for Cardiomyocyte Number Expansion in Preadolescent Mice. Cell 2015, 163, 1026–1036. [Google Scholar] [CrossRef] [Green Version]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. Correction: Puente et al. The Oxygen-Rich Postnatal Environment Induces Cardiomyocyte Cell-Cycle Arrest through DNA Damage Response. Cell 2014, 157, 1243. [Google Scholar] [CrossRef] [Green Version]
- Armengaud, J.; Yzydorczyk, C.; Siddeek, B.; Peyter, A.; Simeoni, U. Intrauterine growth restriction: Clinical consequences on health and disease at adulthood. Reprod. Toxicol. 2020, 99, 168–176. [Google Scholar] [CrossRef]
- Barker, D.J.; Gelow, J.; Thornburg, K.; Osmond, C.; Kajantie, E.; Eriksson, J.G. The early origins of chronic heart failure: Impaired placental growth and initiation of insulin resistance in childhood. Eur. J. Hear. Fail. 2010, 12, 819–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, D.J.; Osmond, C.; Golding, J.; Kuh, D.; Wadsworth, M.E. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 1989, 298, 564–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corstius, H.B.; Zimanyi, M.A.; Maka, N.; Herath, T.; Thomas, W.; van der Laarse, A.; Wreford, N.G.; Black, M.J. Effect of Intrauterine Growth Restriction on the Number of Cardiomyocytes in Rat Hearts. Pediatr. Res. 2005, 57, 796–800. [Google Scholar] [CrossRef] [Green Version]
- Schipke, J.; Gonzalez-Tendero, A.; Cornejo, L.; Willführ, A.; Bijnens, B.; Crispi, F.; Mühlfeld, C.; Gratacós, E. Experimentally induced intrauterine growth restriction in rabbits leads to differential remodelling of left versus right ventricular myocardial microstructure. Histochem. Cell Biol. 2017, 148, 557–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botting, K.J.; Loke, X.Y.; Zhang, S.; Andersen, J.B.; Nyengaard, J.R.; Morrison, J.L. IUGR decreases cardiomyocyte endowment and alters cardiac metabolism in a sex- and cause-of-IUGR-specific manner. Am. J. Physiol. Integr. Comp. Physiol. 2018, 315, R48–R67. [Google Scholar] [CrossRef] [PubMed]
- Vranas, S.; Heinemann, G.K.; Liu, H.; De Blasio, M.J.; Owens, J.A.; Gatford, K.L.; Black, M.J. Small size at birth predicts decreased cardiomyocyte number in the adult ovine heart. J. Dev. Orig. Health Dis. 2017, 8, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Mühlfeld, C.; Schipke, J. Methodological Progress of Stereology in Cardiac Research and Its Application to Normal and Pathological Heart Development. Cells 2022, 11, 2032. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-F.; Chen, Y.-C.; Yeh, H.-I.; Chen, S.-A. Mononucleated and binucleated cardiomyocytes in left atrium and pulmonary vein have different electrical activity and calcium dynamics. Prog. Biophys. Mol. Biol. 2012, 108, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Raulf, A.; Horder, H.; Tarnawski, L.; Geisen, C.; Ottersbach, A.; Röll, W.; Jovinge, S.; Fleischmann, B.K.; Hesse, M. Transgenic systems for unequivocal identification of cardiac myocyte nuclei and analysis of cardiomyocyte cell cycle status. Basic Res. Cardiol. 2015, 110, 33. [Google Scholar] [CrossRef] [Green Version]
- Engel, F.B.; Schebesta, M.; Keating, M.T. Anillin localization defect in cardiomyocyte binucleation. J. Mol. Cell. Cardiol. 2006, 41, 601–612. [Google Scholar] [CrossRef]
- Adler, C.P.; Friedburg, H. Myocardial DNA content, ploidy level and cell number in geriatric hearts: Post-mortem examinations of human myocardium in old age. J. Mol. Cell. Cardiol. 1986, 18, 39–53. [Google Scholar] [CrossRef]
- Vliegen, H.W.; Eulderink, F.; Bruschke, A.V.; Van Der Laarse, A.; Cornelisse, C.J. Polyploidy of myocyte nuclei in pressure overloaded human hearts: A flow cytometric study in left and right ventricular myocardium. Am. J. Cardiovasc. Pathol. 1995, 5, 27–31. [Google Scholar]
- Sukhacheva, T.V.; Serov, R.A.; Nizyaeva, N.V.; Burov, A.A.; Pavlovich, S.V.; Podurovskaya, Y.L.; Samsonova, M.V.; Chernyaev, A.L.; Shchegolev, A.I.; Kim, A.I.; et al. Accelerated Growth, Differentiation, and Ploidy with Reduced Proliferation of Right Ventricular Cardiomyocytes in Children with Congenital Heart Defect Tetralogy of Fallot. Cells 2022, 11, 175. [Google Scholar] [CrossRef]
- Gilsbach, R.; Schwaderer, M.; Preissl, S.; Grüning, B.A.; Kranzhöfer, D.; Schneider, P.; Nührenberg, T.G.; Mulero-Navarro, S.; Weichenhan, D.; Braun, C.; et al. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun. 2018, 9, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, M.; Wang, Z.; Bassel-Duby, R.; Olson, E.N. Genetic and epigenetic regulation of cardiomyocytes in development, regeneration and disease. Development 2018, 145, dev171983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillette, T.G.; Hill, J.A. Readers, Writers, and Erasers. Circ. Res. 2015, 116, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-J.; Tamadon, A.; Kim, Y.-Y.; Kang, B.-C.; Ku, S.-Y. Epigenetic Regulation of Cardiomyocyte Differentiation from Embryonic and Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2021, 22, 8599. [Google Scholar] [CrossRef]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, L. Targeting Epigenetic Regulation of Cardiomyocytes through Development for Therapeutic Cardiac Regeneration after Heart Failure. Int. J. Mol. Sci. 2022, 23, 11878. [Google Scholar] [CrossRef]
- Heineke, J.; Molkentin, J. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Hixon, M.L.; Muro-Cacho, C.; Wagner, M.W.; Obejero-Paz, C.; Millie, E.; Fujio, Y.; Kureishi, Y.; Hassold, T.; Walsh, K.; Gualberto, A. Akt1/PKB upregulation leads to vascular smooth muscle cell hypertrophy and polyploidization. J. Clin. Investig. 2000, 106, 1011–1020. [Google Scholar] [CrossRef] [Green Version]
- Schneider, R.; Pfitzer, P. Number of nuclei in isolated human myocardial cells. Virchows Arch. B Cell Pathol. 1973, 12, 238–258. [Google Scholar] [CrossRef]
- Li, F.; Wang, X.; Capasso, J.M.; Gerdes, A.M. Rapid Transition of Cardiac Myocytes from Hyperplasia to Hypertrophy During Postnatal Development. J. Mol. Cell. Cardiol. 1996, 28, 1737–1746. [Google Scholar] [CrossRef]
- Ascuitto, R.J.; Ross-Ascuitto, N.T. Substrate metabolism in the developing heart. Semin. Perinatol. 1996, 20, 542–563. [Google Scholar] [CrossRef] [PubMed]
- Quaife-Ryan, G.A.; Sim, C.B.; Ziemann, M.; Kaspi, A.; Rafehi, H.; Ramialison, M.; El-Osta, A.; Hudson, J.E.; Porrello, E.R. Multicellular Transcriptional Analysis of Mammalian Heart Regeneration. Circulation 2017, 136, 1123–1139. [Google Scholar] [CrossRef] [PubMed]
- Padula, S.L.; Velayutham, N.; Yutzey, K.E. Transcriptional Regulation of Postnatal Cardiomyocyte Maturation and Regeneration. Int. J. Mol. Sci. 2021, 22, 3288. [Google Scholar] [CrossRef] [PubMed]
- Vujic, A.; Lerchenmuller, C.; Wu, T.D.; Guillermier, C.; Rabolli, C.P.; Gonzalez, E.; Senyo, S.E.; Liu, X.J.; Guerquin-Kern, J.L.; Steinhauser, M.L.; et al. Exercise induces new cardiomyocyte generation in the adult mammalian heart. Nat. Commun. 2018, 9, 1659. [Google Scholar] [CrossRef] [Green Version]
- Levkau, B.; Schäfers, M.; Wohlschlaeger, J.; Lipinski, K.V.W.; Keul, P.; Hermann, S.; Kawaguchi, N.; Kirchhof, P.; Fabritz, L.; Stypmann, J.; et al. Survivin Determines Cardiac Function by Controlling Total Cardiomyocyte Number. Circulation 2008, 117, 1583–1593. [Google Scholar] [CrossRef] [Green Version]
- Soonpaa, M.H.; Koh, G.Y.; Pajak, L.; Jing, S.; Wang, H.; Franklin, M.T.; Kim, K.K.; Field, L.J. Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J. Clin. Investig. 1997, 99, 2644–2654. [Google Scholar] [CrossRef] [Green Version]
- Rivello, H.G.; Meckert, P.C.; Vigliano, C.; Favaloro, R.; Laguens, R.P. Cardiac myocyte nuclear size and ploidy status decrease after mechanical support. Cardiovasc. Pathol. 2001, 10, 53–57. [Google Scholar] [CrossRef]
- Wohlschlaeger, J.; Levkau, B.; Brockhoff, G.; Schmitz, K.J.; Von Winterfeld, M.; Takeda, A.; Takeda, N.; Stypmann, J.; Vahlhaus, C.; Schmid, C.; et al. Hemodynamic Support by Left Ventricular Assist Devices Reduces Cardiomyocyte DNA Content in the Failing Human Heart. Circulation 2010, 121, 989–996. [Google Scholar] [CrossRef]
- Tamamori-Adachi, M.; Ito, H.; Nobori, K.; Hayashida, K.; Kawauchi, J.; Adachi, S.; Ikeda, M.-A.; Kitajima, S. Expression of cyclin D1 and CDK4 causes hypertrophic growth of cardiomyocytes in culture: A possible implication for cardiac hypertrophy. Biochem. Biophys. Res. Commun. 2002, 296, 274–280. [Google Scholar] [CrossRef]
- Malumbres, M.; Sotillo, R.; Santamaría, D.; Galán, J.; Cerezo, A.; Ortega, S.; Dubus, P.; Barbacid, M. Mammalian Cells Cycle without the D-Type Cyclin-Dependent Kinases Cdk4 and Cdk6. Cell 2004, 118, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Sherr, C.J. G1 phase progression: Cycling on cue. Cell 1994, 79, 551–555. [Google Scholar] [CrossRef]
- Gogiraju, R.; Xu, X.; Bochenek, M.L.; Steinbrecher, J.H.; Lehnart, S.E.; Wenzel, P.; Kessel, M.; Zeisberg, E.M.; Dobbelstein, M.; Schäfer, K. Endothelial p53 Deletion Improves Angiogenesis and Prevents Cardiac Fibrosis and Heart Failure Induced by Pressure Overload in Mice. J. Am. Hear. Assoc. 2015, 4, e001770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoffner, A.; Cigliola, V.; Lee, N.; Ou, J.; Poss, K.D. Tp53 Suppression Promotes Cardiomyocyte Proliferation during Zebrafish Heart Regeneration. Cell Rep. 2020, 32, 108089. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Abe, K.; Tanno, M.; Miki, T.; Kuno, A.; Miura, T.; Steenbergen, C. Does p53 Inhibition Suppress Myocardial Ischemia–Reperfusion Injury? J. Cardiovasc. Pharmacol. Ther. 2018, 23, 350–357. [Google Scholar] [CrossRef]
- Lin, Z.Q.; von Gise, A.; Zhou, P.Z.; Ma, Q.; Chen, J.H.; Jiang, J.M.; Seidman, J.; Wang, D.Z.; Pu, W.T. Cardiac-specific Yap Activation Improve Cardiac Function and Survival in an Experimental Murine Mi Model. Circ. Res. 2014, 115, 354–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Liu, S.; Heallen, T.; Martin, J.F. The Hippo pathway in the heart: Pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, K.; Florencio, L.W.; Tang, L.; Heallen, T.R.; Leach, J.P.; Wang, Y.; Grisanti, F.; Willerson, J.T.; Perin, E.C.; et al. Gene therapy knockdown of Hippo signaling induces cardiomyocyte renewal in pigs after myocardial infarction. Sci. Transl. Med. 2021, 13, eabd6892. [Google Scholar] [CrossRef]
- Heallen, T.; Morikawa, Y.; Leach, J.; Tao, G.; Willerson, J.T.; Johnson, R.L.; Martin, J.F. Hippo signaling impedes adult heart regeneration. Development 2013, 140, 4683–4690. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.Q.; Li, Y.L.; Luo, J.D.; Hou, N. Molecular Mechanism of Hippo-YAP1/TAZ Pathway in Heart Development, Disease, and Regeneration. Front. Physiol. 2020, 11, 389. [Google Scholar] [CrossRef] [Green Version]
- Bak, S.T.; Harvald, E.B.; Ellman, D.G.; Mathiesen, S.B.; Chen, T.; Fang, S.; Andersen, K.S.; Fenger, C.D.; Burton, M.; Thomassen, M.; et al. Ploidy-stratified single cardiomyocyte transcriptomics map Zinc Finger E-Box Binding Homeobox 1 to underly cardiomyocyte proliferation before birth. Basic Res. Cardiol. 2023, 118, 8. [Google Scholar] [CrossRef]
- Rigaud, V.O.C.; Khan, M. Aging in reverse: Reactivating developmental signaling for cardiomyocyte proliferation. J. Mol. Cell. Cardiol. 2021, 154, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Borden, A.; Kurian, J.; Nickoloff, E.; Yang, Y.; Troupes, C.D.; Ibetti, J.; Lucchese, A.M.; Gao, E.; Mohsin, S.; Koch, W.J.; et al. Transient Introduction of miR-294 in the Heart Promotes Cardiomyocyte Cell Cycle Reentry After Injury. Circ. Res. 2019, 125, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Rigaud, V.O.C.; Hoy, R.C.; Kurian, J.; Zarka, C.; Behanan, M.; Brosious, I.; Pennise, J.; Patel, T.; Wang, T.; Johnson, J.; et al. RNA-Binding Protein LIN28a Regulates New Myocyte Formation in the Heart Through Long Noncoding RNA-H19. Circulation 2023, 147, 324–337. [Google Scholar] [CrossRef]
- Dong, Y.; Liu, C.; Zhao, Y.; Ponnusamy, M.; Li, P.; Wang, K. Role of noncoding RNAs in regulation of cardiac cell death and cardiovascular diseases. Cell. Mol. Life Sci. 2017, 75, 291–300. [Google Scholar] [CrossRef]
- Kansakar, U.; Varzideh, F.; Mone, P.; Jankauskas, S.S.; Santulli, G. Functional Role of microRNAs in Regulating Cardiomyocyte Death. Cells 2022, 11, 983. [Google Scholar] [CrossRef]
- Wang, K.; An, T.; Zhou, L.-Y.; Liu, C.-Y.; Zhang, X.-J.; Feng, C.; Li, P.-F. E2F1-regulated miR-30b suppresses Cyclophilin D and protects heart from ischemia/reperfusion injury and necrotic cell death. Cell Death Differ. 2014, 22, 743–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Cardiac-specific miRNA in cardiogenesis, heart function, and cardiac pathology (with focus on myocardial infarction). J. Mol. Cell. Cardiol. 2016, 94, 107–121. [Google Scholar] [CrossRef]
- Stopp, S.; Gründl, M.; Fackler, M.; Malkmus, J.; Leone, M.; Naumann, R.; Frantz, S.; Wolf, E.; von Eyss, B.; Engel, F.B.; et al. Deletion of Gas2l3 in mice leads to specific defects in cardiomyocyte cytokinesis during development. Proc. Natl. Acad. Sci. USA 2017, 114, 8029–8034. [Google Scholar] [CrossRef] [Green Version]
- Green, R.A.; Paluch, E.; Oegema, K. Cytokinesis in Animal Cells. Annu. Rev. Cell Dev. Biol. 2012, 28, 29–58. [Google Scholar] [CrossRef] [Green Version]
- Gan, P.; Patterson, M.; Velasquez, A.; Wang, K.; Tian, D.; Windle, J.J.; Tao, G.; Judge, D.P.; Makita, T.; Park, T.J.; et al. Tnni3k alleles influence ventricular mononuclear diploid cardiomyocyte frequency. PLOS Genet. 2019, 15, e1008354. [Google Scholar] [CrossRef] [Green Version]
- Lai, Z.F.; Chen, Y.Z.; Feng, L.P.; Meng, X.M.; Ding, J.F.; Monzen, K.; Komuro, I. A novel cardiac-specific MAP kinase, TNNI3K, promotes cardiac differentiation and myogenesis in a pluripotent P19CL6 cell model. J. Pharmacol. Sci. 2005, 97, 128p. [Google Scholar]
- Pham, C.; Muñoz-Martín, N.; Lodder, E.M. The Diverse Roles of TNNI3K in Cardiac Disease and Potential for Treatment. Int. J. Mol. Sci. 2021, 22, 6422. [Google Scholar] [CrossRef]
- Yekelchyk, M.; Guenther, S.; Preussner, J.; Braun, T. Mono- and multi-nucleated ventricular cardiomyocytes constitute a transcriptionally homogenous cell population. Basic Res. Cardiol. 2019, 114, 36. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhang, C.-H.; Ammanamanchi, N.; Suresh, S.; Lewarchik, C.; Rao, K.; Uys, G.M.; Han, L.; Abrial, M.; Yimlamai, D.; et al. Control of cytokinesis by β-adrenergic receptors indicates an approach for regulating cardiomyocyte endowment. Sci. Transl. Med. 2019, 11, eaaw6419. [Google Scholar] [CrossRef] [PubMed]
- Mollova, M.; Bersell, K.; Walsh, S.; Savla, J.; Das, L.T.; Park, S.-Y.; Silberstein, L.E.; Dos Remedios, C.G.; Graham, D.; Colan, S.; et al. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc. Natl. Acad. Sci. USA 2013, 110, 1446–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velayutham, N.; Alfieri, C.M.; Agnew, E.J.; Riggs, K.W.; Baker, R.S.; Ponny, S.R.; Zafar, F.; Yutzey, K.E. Cardiomyocyte cell cycling, maturation, and growth by multinucleation in postnatal swine. J. Mol. Cell. Cardiol. 2020, 146, 95–108. [Google Scholar] [CrossRef]
- Swift, S.K.; Purdy, A.L.; Kolell, M.E.; Andresen, K.G.; Lahue, C.; Buddell, T.; Akins, K.A.; Rau, C.D.; O’Meara, C.C.; Patterson, M. Cardiomyocyte ploidy is dynamic during postnatal development and varies across genetic backgrounds. Development 2023, 150, dev201318. [Google Scholar] [CrossRef]
- Jonker, S.S.; Zhang, L.; Louey, S.; Giraud, G.D.; Thornburg, K.L.; Faber, J.J. Myocyte enlargement, differentiation, and proliferation kinetics in the fetal sheep heart. J. Appl. Physiol. 2007, 102, 1130–1142. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.Y.; Eiby, Y.A.; Lumbers, E.R.; Wright, L.L.; Gibson, K.J.; Barnett, A.C.; Lingwood, B.E. Effects of Glucocorticoid Exposure on Growth and Structural Maturation of the Heart of the Preterm Piglet. PLoS ONE 2014, 9, e93407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltrami, C.A.; Di Loreto, C.; Finato, N.; Yan, S.M. DNA Content in End-Stage Heart Failure. Adv. Clin. Pathol. Off. J. Adriat. Soc. Pathol. 1997, 1, 59–73. [Google Scholar]
- Swynghedauw, B.; Delcayre, C. Biology of cardiac overload. Pathobiol. Annu. 1982, 12, 137–183. [Google Scholar] [PubMed]
- Stukenberg, P.T.; Burke, D.J. Connecting the microtubule attachment status of each kinetochore to cell cycle arrest through the spindle assembly checkpoint. Chromosoma 2015, 124, 463–480. [Google Scholar] [CrossRef]
- Vitale, I.; Galluzzi, L.; Castedo, M.; Kroemer, G. Mitotic catastrophe: A mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 2011, 12, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Zebrowski, D.C.; Vergarajauregui, S.; Wu, C.-C.; Piatkowski, T.; Becker, R.; Leone, M.; Hirth, S.; Ricciardi, F.; Falk, N.; Giessl, A.; et al. Developmental alterations in centrosome integrity contribute to the post-mitotic state of mammalian cardiomyocytes. Elife 2015, 4, e05563. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ahmad, F.; Parikh, S.; Hoffman, N.E.; Rajan, S.; Verma, V.; Song, J.; Yuan, A.; Shanmughapriya, S.; Guo, Y.; et al. Loss of Adult Cardiac Myocyte GSK-3 Leads to Mitotic Catastrophe Resulting in Fatal Dilated Cardiomyopathy. Circ. Res. 2016, 118, 1208–1222. [Google Scholar] [CrossRef]
- Mohamed, T.M.; Ang, Y.-S.; Radzinsky, E.; Zhou, P.; Huang, Y.; Elfenbein, A.; Foley, A.; Magnitsky, S.; Srivastava, D. Regulation of Cell Cycle to Stimulate Adult Cardiomyocyte Proliferation and Cardiac Regeneration. Cell 2018, 173, 104–116.e12. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, A.C. The Regulation of Cell Size. Cell 2013, 154, 1194–1205. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, T.P.; Pessa, H.K.; Caldez, M.J.; Fuhrer, T.; Diril, M.K.; Sauer, U.; Kaldis, P.; Björklund, M. Identification of Transcriptional and Metabolic Programs Related to Mammalian Cell Size. Curr. Biol. 2014, 24, 598–608. [Google Scholar] [CrossRef] [Green Version]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The Oxygen-Rich Postnatal Environment Induces Cardiomyocyte Cell-Cycle Arrest through DNA Damage Response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Ashrafian, H.; Redwood, C.; Blair, E.; Watkins, H. Hypertrophic cardiomyopathy:a paradigm for myocardial energy depletion. Trends Genet. 2003, 19, 263–268. [Google Scholar] [CrossRef]
- Bischof, C.; Mirtschink, P.; Yuan, T.; Wu, M.; Zhu, C.; Kaur, J.; Pham, M.D.; Gonzalez-Gonoggia, S.; Hammer, M.; Rogg, E.-M.; et al. Mitochondrial–cell cycle cross-talk drives endoreplication in heart disease. Sci. Transl. Med. 2021, 13, eabi7964. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Ventura-Clapier, R. Maturation of Cardiac Energy Metabolism During Perinatal Development. Front. Physiol. 2018, 9, 959. [Google Scholar] [CrossRef] [Green Version]
- Garbern, J.C.; Lee, R.T. Mitochondria and metabolic transitions in cardiomyocytes: Lessons from development for stem cell-derived cardiomyocytes. Stem Cell Res. Ther. 2021, 12, 177. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Liccardo, D.; Lacanna, R.; Zhang, X.; Lu, R.; Finck, B.N.; Leigh, T.; Chen, X.; Drosatos, K.; Tian, Y. Fatty Acid Oxidation Promotes Cardiomyocyte Proliferation Rate but Does Not Change Cardiomyocyte Number in Infant Mice. Front. Cell Dev. Biol. 2019, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Murray, T.V.; Ahmad, A.; Brewer, A.C. Reactive oxygen at the heart of metabolism. Trends Cardiovasc. Med. 2014, 24, 113–120. [Google Scholar] [CrossRef]
- Kimura, W.; Xiao, F.; Canseco, D.C.; Muralidhar, S.; Thet, S.; Zhang, H.M.; Abderrahman, Y.; Chen, R.; Garcia, J.A.; Shelton, J.M.; et al. Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nature 2015, 523, 226–230. [Google Scholar] [CrossRef]
- Nakada, Y.; Canseco, D.C.; Thet, S.; Abdisalaam, S.; Asaithamby, A.; Santos, C.X.; Shah, A.M.; Zhang, H.; Faber, J.E.; Kinter, M.T.; et al. Hypoxia induces heart regeneration in adult mice. Nature 2017, 541, 222–227. [Google Scholar] [CrossRef]
- Kulisz, A.; Chen, N.; Chandel, N.S.; Shao, Z.; Schumacker, P.T. Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am. J. Physiol. Cell. Mol. Physiol. 2002, 282, L1324–L1329. [Google Scholar] [CrossRef] [Green Version]
- Matsuyama, D.; Kawahara, K. Oxidative stress-induced formation of a positive-feedback loop for the sustained activation of p38 MAPK leading to the loss of cell division in cardiomyocytes soon after birth. Basic Res. Cardiol. 2011, 106, 815–828. [Google Scholar] [CrossRef] [Green Version]
- Sayre, N.L.; Lechleiter, J.D. Fatty acid metabolism and thyroid hormones. Curr. Trends Endocrinol. 2012, 6, 65–76. [Google Scholar]
- Cui, M.; Wang, Z.; Chen, K.; Shah, A.M.; Tan, W.; Duan, L.; Sanchez-Ortiz, E.; Li, H.; Xu, L.; Liu, N.; et al. Dynamic Transcriptional Responses to Injury of Regenerative and Non-regenerative Cardiomyocytes Revealed by Single-Nucleus RNA Sequencing. Dev. Cell 2020, 55, 665–667. [Google Scholar] [CrossRef]
- Rigaud, V.O.; Zarka, C.; Kurian, J.; Harlamova, D.; Elia, A.; Kasatkin, N.; Johnson, J.; Behanan, M.; Kraus, L.; Pepper, H.; et al. UCP2 modulates cardiomyocyte cell cycle activity, acetyl-CoA, and histone acetylation in response to moderate hypoxia. J. Clin. Investig. 2022, 7, 155475. [Google Scholar] [CrossRef]
- Honkoop, H.; de Bakker, D.E.; Aharonov, A.; Kruse, F.; Shakked, A.; Nguyen, P.D.; de Heus, C.; Garric, L.; Muraro, M.J.; Shoffner, A.; et al. Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. Elife 2019, 8, e50163. [Google Scholar] [CrossRef]
- Jopling, C.; Sleep, E.; Raya, M.; Martí, M.; Raya, A.; Belmonte, J.C.I. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 2010, 464, 606–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef]
- Marín-Sedeño, E.; de Morentin, X.M.; Pérez-Pomares, J.M.; Gómez-Cabrero, D.; Ruiz-Villalba, A. Understanding the Adult Mammalian Heart at Single-Cell RNA-Seq Resolution. Front. Cell Dev. Biol. 2021, 9, 645276. [Google Scholar] [CrossRef] [PubMed]
- Galow, A.-M.; Wolfien, M.; Müller, P.; Bartsch, M.; Brunner, R.M.; Hoeflich, A.; Wolkenhauer, O.; David, R.; Goldammer, T. Integrative Cluster Analysis of Whole Hearts Reveals Proliferative Cardiomyocytes in Adult Mice. Cells 2020, 9, 1144. [Google Scholar] [CrossRef] [PubMed]
- Tabula Muris Consortium. Overall Coordination; Logistical Coordination; Organ Collection and Processing; Library Preparation and Sequencing; Computational Data Analysis; Cell Type Annotation; Writing Group; Supplemental Text Writing Group; Principal Investigators. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar] [CrossRef]
- Shen, H.; Gan, P.; Wang, K.; Darehzereshki, A.; Wang, K.; Kumar, S.R.; Lien, C.-L.; Patterson, M.; Tao, G.; Sucov, H.M.; et al. Mononuclear diploid cardiomyocytes support neonatal mouse heart regeneration in response to paracrine IGF2 signaling. Elife 2020, 9, e53071. [Google Scholar] [CrossRef]
- Peng, S.; Chen, L.-L.; Lei, X.-X.; Yang, L.; Lin, H.; Carmichael, G.G.; Huang, Y. Genome-Wide Studies Reveal That Lin28 Enhances the Translation of Genes Important for Growth and Survival of Human Embryonic Stem Cells. Stem Cells 2011, 29, 496–504. [Google Scholar] [CrossRef]
- Zhu, H.; Ng, S.-C.; Segrè, A.V.; Shinoda, G.; Shah, S.P.; Einhorn, W.S.; Takeuchi, A.; Engreitz, J.M.; Hagan, J.P.; Kharas, M.G.; et al. The Lin28/let-7 Axis Regulates Glucose Metabolism. Cell 2011, 147, 81–94. [Google Scholar] [CrossRef] [Green Version]
- de Carvalho, A.E.T.S.; Bassaneze, V.; Forni, M.F.; Keusseyan, A.A.; Kowaltowski, A.J.; Krieger, J.E. Early Postnatal Cardiomyocyte Proliferation Requires High Oxidative Energy Metabolism. Sci. Rep. 2017, 7, 15434. [Google Scholar] [CrossRef]
- Magadum, A.; Singh, N.; Kurian, A.A.; Munir, I.; Mehmood, T.; Brown, K.; Sharkar, M.T.K.; Chepurko, E.; Sassi, Y.; Oh, J.G.; et al. Pkm2 Regulates Cardiomyocyte Cell Cycle and Promotes Cardiac Regeneration. Circulation 2020, 141, 1249–1265. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Marín-Juez, R.; El-Sammak, H.; Beisaw, A.; Ramadass, R.; Kuenne, C.; Guenther, S.; Konzer, A.; Bhagwat, A.M.; Graumann, J.; et al. Stimulation of glycolysis promotes cardiomyocyte proliferation after injury in adult zebrafish. EMBO Rep. 2020, 21, e49752. [Google Scholar] [CrossRef]
- Fan, C.; Tang, Y.; Wang, J.; Xiong, F.; Guo, C.; Wang, Y.; Zhang, S.; Gong, Z.; Wei, X.; Yang, L.; et al. Role of long non-coding RNAs in glucose metabolism in cancer. Mol. Cancer 2017, 16, 130. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Wang, J.-H.; Fan, W.-J.; Meng, Y.-T.; Li, M.-M.; Li, T.-T.; Cui, B.; Wang, H.-F.; Zhao, Y.; An, F.; et al. Glycolysis gatekeeper PDK1 reprograms breast cancer stem cells under hypoxia. Oncogene 2017, 37, 1062–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Stimulus | Cellular Change | Metabolic Change | Functional Change | Signaling Pathway | References |
|---|---|---|---|---|---|
| Cyclins D1, G1 | ↑ DNA synthesis ↑ Multinucleation | Not measured | ↑ Cardiac hypertrophy ↑ Apoptosis | CDK4, 6 p53 | [83,86] [89,90,91] |
| Hippo/YAP1 | ↑ Cell cycle ↑ Cytokinesis | Not measured | ↑ Cardiac function ↑ Repair and regeneration | Salv, Lats1/2 | [94] |
| Tnni3k | ↑ Cell cycle ↑ Diploid mononuclear | Not measured | ↑ survival ↓ inflammation | Oxidative stress signaling | [101] |
| Thyroid hormone | ↑ Cell cycle ↑ Diploid mononuclear | ↓OxPHOS | ↑ survival ↑ proliferation | Cell cycle, E2F, G2M signaling | [64] |
| Gas2l3 | ↑ Cytokinesis ↓ Binucleation | Not measured | ↑ Proliferation ↓ Cardiac hypertrophy | p53, p21 signaling | [99] |
| microRNA-294 | ↑ Cell cycle ↑ Mononucleation | ↑Glycolysis | ↑ Cardiac function ↑ proliferation | Wee1 | [97] |
| LIN28a | ↑ Cell cycle ↑ Diploid mononuclear | ↑Glycolysis | ↑ Cardiac function ↑ Proliferation ↓ apoptosis | lncRNA-H19 | [98] |
| UCP2 | ↑ Cell cycle ↓ Ploidy | ↑Glycolysis | ↑ Cardiac function | Acetyl-CoA | [115] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elia, A.; Mohsin, S.; Khan, M. Cardiomyocyte Ploidy, Metabolic Reprogramming and Heart Repair. Cells 2023, 12, 1571. https://doi.org/10.3390/cells12121571
Elia A, Mohsin S, Khan M. Cardiomyocyte Ploidy, Metabolic Reprogramming and Heart Repair. Cells. 2023; 12(12):1571. https://doi.org/10.3390/cells12121571
Chicago/Turabian StyleElia, Andrea, Sadia Mohsin, and Mohsin Khan. 2023. "Cardiomyocyte Ploidy, Metabolic Reprogramming and Heart Repair" Cells 12, no. 12: 1571. https://doi.org/10.3390/cells12121571