
Autophagy in Crohn’s Disease: Converging on Dysfunctional Innate Immunity

Abstract
:1. Introduction
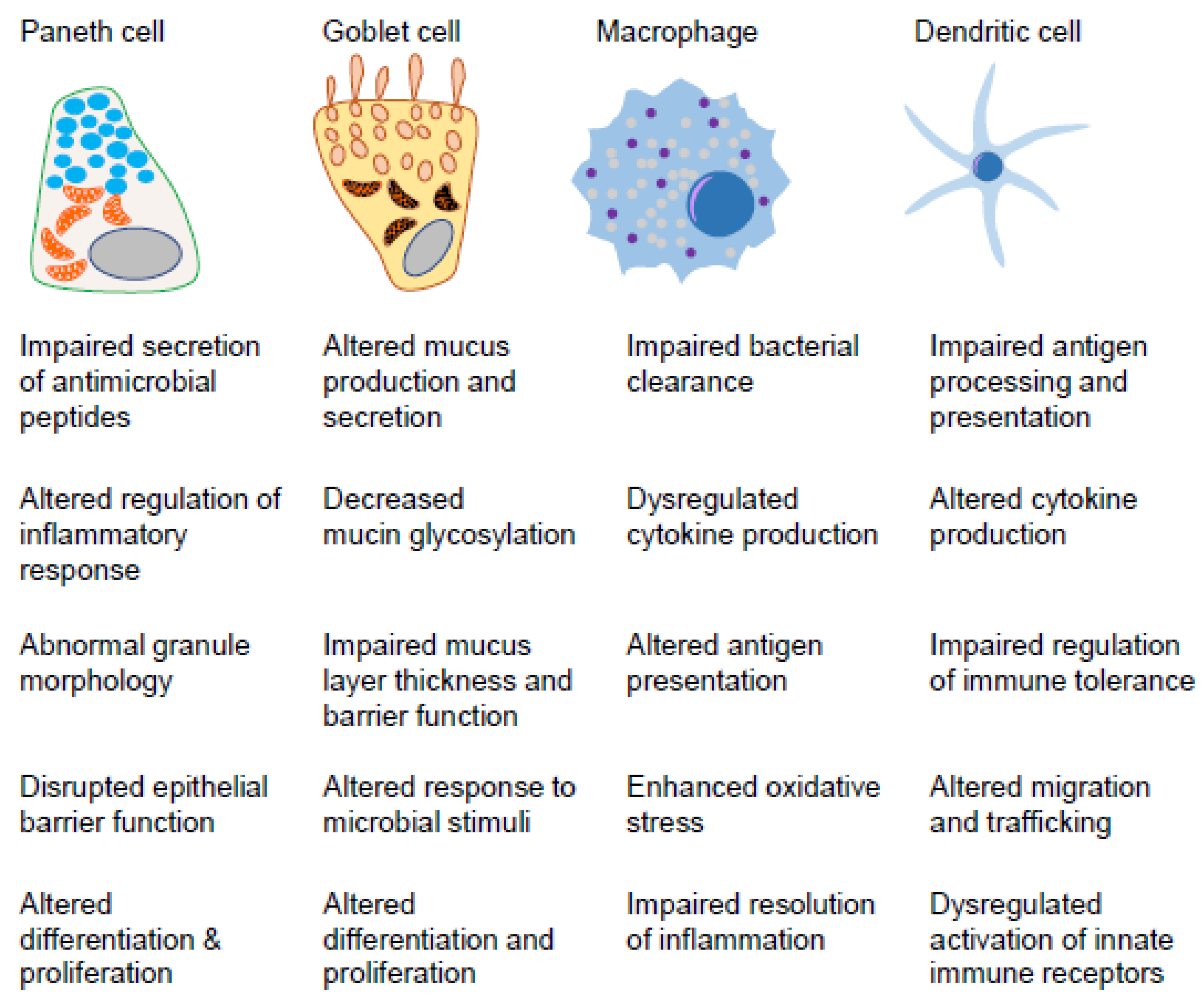
2. Innate Immunity in the Gut Mucosa
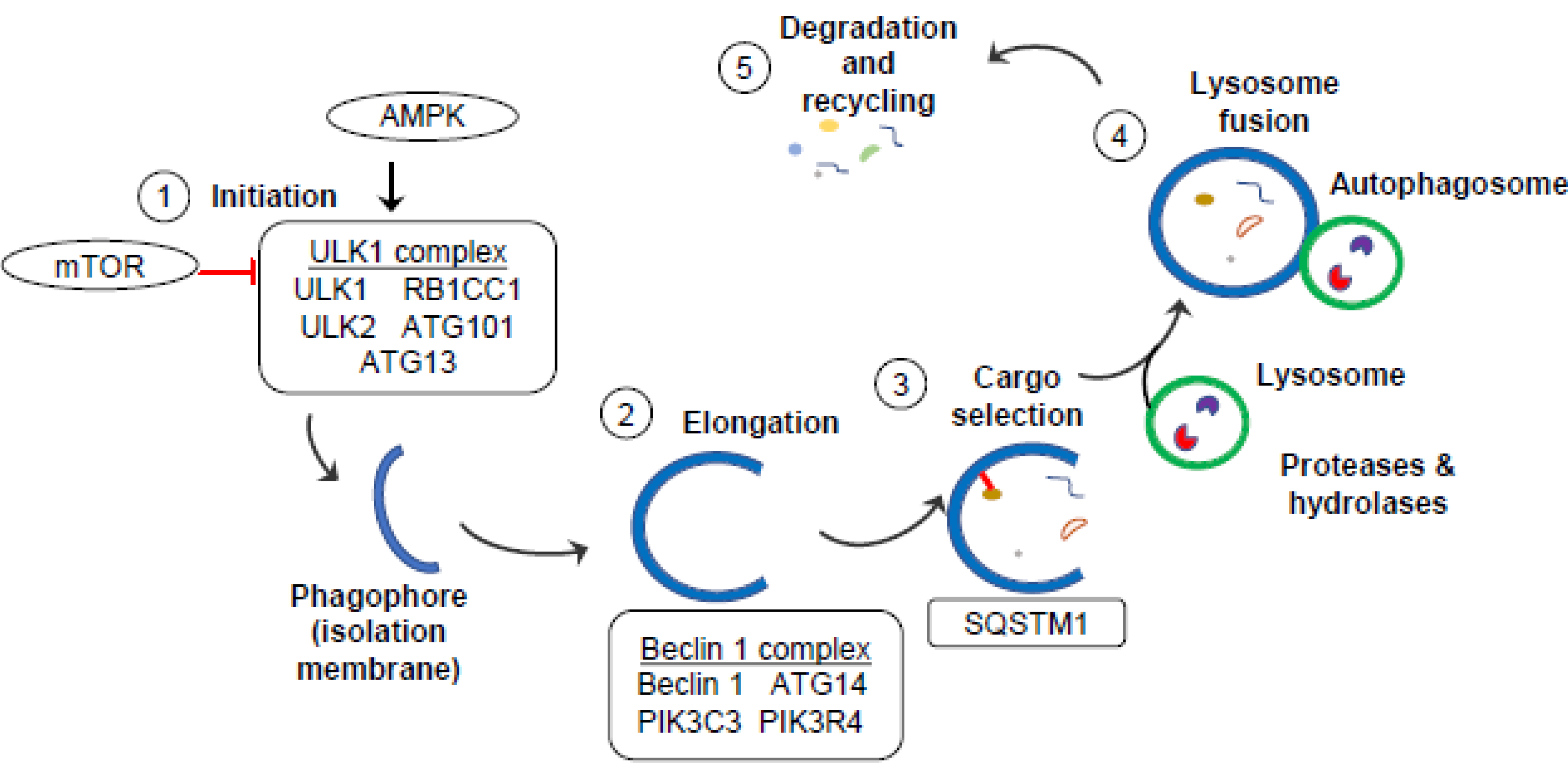
3. Autophagy
4. Autophagy-Related CD Susceptibility Genes
4.1. ATG16L1
4.2. IRGM
4.3. NOD2
4.4. LRRK2
4.5. ULK1
4.6. ATG4
4.7. TCF4
5. Types of Autophagy Linked to CD Pathogenesis
5.1. Xenophagy
5.2. ERphagy
5.3. Mitophagy
6. Targeting Autophagy as Therapy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ananthakrishnan, A.N.; Kaplan, G.G.; Ng, S.C. Changing Global Epidemiology of Inflammatory Bowel Diseases: Sustaining Health Care Delivery Into the 21st Century. Clin. Gastroenterol. Hepatol. 2020, 18, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Bris, R.; Saez, A.; Herrero-Fernandez, B.; Rius, C.; Sanchez-Martinez, H.; Gonzalez-Granado, J.M. CD4 T-Cell Subsets and the Pathophysiology of Inflammatory Bowel Disease. Int. J. Mol. Sci. 2023, 24, 2696. [Google Scholar] [CrossRef] [PubMed]
- Saez, A.; Herrero-Fernandez, B.; Gomez-Bris, R.; Sanchez-Martinez, H.; Gonzalez-Granado, J.M. Pathophysiology of Inflammatory Bowel Disease: Innate Immune System. Int. J. Mol. Sci. 2023, 24, 1526. [Google Scholar] [CrossRef] [PubMed]
- Molnar, T.; Tiszlavicz, L.; Gyulai, C.; Nagy, F.; Lonovics, J. Clinical significance of granuloma in Crohn’s disease. World J. Gastroenterol. 2005, 11, 3118–3121. [Google Scholar] [CrossRef]
- Jackson, D.N.; Theiss, A.L. Gut bacteria signaling to mitochondria in intestinal inflammation and cancer. Gut Microbes 2020, 11, 285–304. [Google Scholar] [CrossRef] [Green Version]
- Roda, G.; Chien Ng, S.; Kotze, P.G.; Argollo, M.; Panaccione, R.; Spinelli, A.; Kaser, A.; Peyrin-Biroulet, L.; Danese, S. Crohn’s disease. Nat. Rev. Dis. Prim. 2020, 6, 22. [Google Scholar] [CrossRef]
- Rieder, F.; Zimmermann, E.M.; Remzi, F.H.; Sandborn, W.J. Crohn’s disease complicated by strictures: A systematic review. Gut 2013, 62, 1072–1084. [Google Scholar] [CrossRef] [Green Version]
- Magro, F.; Moreira, P.L.; Catalano, G.; Alves, C.; Roseira, J.; Estevinho, M.M.; Silva, I.; Dignass, A.; Peyrin-Biroulet, L.; Danese, S.; et al. Has the therapeutical ceiling been reached in Crohn’s disease randomized controlled trials? A systematic review and meta-analysis. United Eur. Gastroenterol. J. 2023, 11, 202–217. [Google Scholar] [CrossRef]
- Sazonovs, A.; Stevens, C.R.; Venkataraman, G.R.; Yuan, K.; Avila, B.; Abreu, M.T.; Ahmad, T.; Allez, M.; Ananthakrishnan, A.N.; Atzmon, G.; et al. Large-scale sequencing identifies multiple genes and rare variants associated with Crohn’s disease susceptibility. Nat. Genet. 2022, 54, 1275–1283. [Google Scholar] [CrossRef]
- Sewell, G.W.; Kaser, A. Interleukin-23 in the Pathogenesis of Inflammatory Bowel Disease and Implications for Therapeutic Intervention. J. Crohn’s Colitis 2022, 16, ii3–ii19. [Google Scholar] [CrossRef]
- Nakata, T.; Creasey, E.A.; Kadoki, M.; Lin, H.; Selig, M.K.; Yao, J.; Lefkovith, A.; Daly, M.J.; Graham, D.B.; Xavier, R.J. A missense variant in SLC39A8 confers risk for Crohn’s disease by disrupting manganese homeostasis and intestinal barrier integrity. Proc. Natl. Acad. Sci. USA 2020, 117, 28930–28938. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Yang, Z.; Chen, B.; Zhong, X. The multifaceted role of CARD9 in inflammatory bowel disease. J. Cell. Mol. Med. 2020, 24, 34–39. [Google Scholar] [CrossRef] [Green Version]
- Prager, M.; Buettner, J.; Buening, C. Genes involved in the regulation of intestinal permeability and their role in ulcerative colitis. J. Dig. Dis. 2015, 16, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Glocker, E.O.; Kotlarz, D.; Boztug, K.; Gertz, E.M.; Schaffer, A.A.; Noyan, F.; Perro, M.; Diestelhorst, J.; Allroth, A.; Murugan, D.; et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 2009, 361, 2033–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aviello, G.; Knaus, U.G. ROS in gastrointestinal inflammation: Rescue Or Sabotage? Br. J. Pharm. 2017, 174, 1704–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.H.; Brant, S.R. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology 2011, 140, 1704–1712. [Google Scholar] [CrossRef] [Green Version]
- Clevers, H.C.; Bevins, C.L. Paneth cells: Maestros of the small intestinal crypts. Annu. Rev. Physiol. 2013, 75, 289–311. [Google Scholar] [CrossRef]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.J.; Bock, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Jackson, D.N.; Panopoulos, M.; Neumann, W.L.; Turner, K.; Cantarel, B.L.; Thompson-Snipes, L.; Dassopoulos, T.; Feagins, L.A.; Souza, R.F.; Mills, J.C.; et al. Mitochondrial dysfunction during loss of prohibitin 1 triggers Paneth cell defects and ileitis. Gut 2020, 69, 1928–1938. [Google Scholar] [CrossRef] [Green Version]
- Deuring, J.J.; Fuhler, G.M.; Konstantinov, S.R.; Peppelenbosch, M.P.; Kuipers, E.J.; de Haar, C.; van der Woude, C.J. Genomic ATG16L1 risk allele-restricted Paneth cell ER stress in quiescent Crohn’s disease. Gut 2014, 63, 1081–1091. [Google Scholar] [CrossRef]
- Lassen, K.G.; Kuballa, P.; Conway, K.L.; Patel, K.K.; Becker, C.E.; Peloquin, J.M.; Villablanca, E.J.; Norman, J.M.; Liu, T.C.; Heath, R.J.; et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl. Acad. Sci. USA 2014, 111, 7741–7746. [Google Scholar] [CrossRef] [PubMed]
- VanDussen, K.L.; Liu, T.C.; Li, D.; Towfic, F.; Modiano, N.; Winter, R.; Haritunians, T.; Taylor, K.D.; Dhall, D.; Targan, S.R.; et al. Genetic variants synthesize to produce paneth cell phenotypes that define subtypes of Crohn’s disease. Gastroenterology 2014, 146, 200–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eshleman, E.M.; Shao, T.Y.; Woo, V.; Rice, T.; Engleman, L.; Didriksen, B.J.; Whitt, J.; Haslam, D.B.; Way, S.S.; Alenghat, T. Intestinal epithelial HDAC3 and MHC class II coordinate microbiota-specific immunity. J. Clin. Investig. 2023, 133, e162190. [Google Scholar] [CrossRef] [PubMed]
- Farache, J.; Koren, I.; Milo, I.; Gurevich, I.; Kim, K.W.; Zigmond, E.; Furtado, G.C.; Lira, S.A.; Shakhar, G. Luminal bacteria recruit CD103+ dendritic cells into the intestinal epithelium to sample bacterial antigens for presentation. Immunity 2013, 38, 581–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagg, A.J. Intestinal Dendritic Cells in Health and Gut Inflammation. Front. Immunol. 2018, 9, 2883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDole, J.R.; Wheeler, L.W.; McDonald, K.G.; Wang, B.; Konjufca, V.; Knoop, K.A.; Newberry, R.D.; Miller, M.J. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature 2012, 483, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Flannigan, K.L.; Geem, D.; Harusato, A.; Denning, T.L. Intestinal Antigen-Presenting Cells: Key Regulators of Immune Homeostasis and Inflammation. Am. J. Pathol. 2015, 185, 1809–1819. [Google Scholar] [CrossRef]
- Jeong, J.; Choi, Y.J.; Lee, H.K. The Role of Autophagy in the Function of CD4+ T Cells and the Development of Chronic Inflammatory Diseases. Front. Pharm. 2022, 13, 860146. [Google Scholar] [CrossRef]
- Metur, S.P.; Klionsky, D.J. Adaptive immunity at the crossroads of autophagy and metabolism. Cell. Mol. Immunol. 2021, 18, 1096–1105. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Henderson, P.; Stevens, C. The role of autophagy in Crohn’s disease. Cells 2012, 1, 492–519. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Su, T.; Li, X.; Yang, M.; Shao, Q.; Zhao, Y.; Ma, C.; Wang, P. Autophagy: An Intracellular Degradation Pathway Regulating Plant Survival and Stress Response. Front. Plant Sci. 2020, 11, 164. [Google Scholar] [CrossRef] [Green Version]
- Hooper, K.M.; Barlow, P.G.; Stevens, C.; Henderson, P. Inflammatory Bowel Disease Drugs: A Focus on Autophagy. J. Crohn’s Colitis 2017, 11, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Simonsen, A.; Tooze, S.A. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J. Cell. Biol. 2009, 186, 773–782. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martin, P.; Komatsu, M. p62/SQSTM1—Steering the cell through health and disease. J. Cell. Sci. 2018, 131, jcs222836. [Google Scholar] [CrossRef] [Green Version]
- Shao, B.Z.; Yao, Y.; Zhai, J.S.; Zhu, J.H.; Li, J.P.; Wu, K. The Role of Autophagy in Inflammatory Bowel Disease. Front. Physiol. 2021, 12, 621132. [Google Scholar] [CrossRef]
- Deretic, V. Autophagy in inflammation, infection, and immunometabolism. Immunity 2021, 54, 437–453. [Google Scholar] [CrossRef]
- Ajayi, T.A.; Innes, C.L.; Grimm, S.A.; Rai, P.; Finethy, R.; Coers, J.; Wang, X.; Bell, D.A.; McGrath, J.A.; Schurman, S.H.; et al. Crohn’s disease IRGM risk alleles are associated with altered gene expression in human tissues. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G95–G105. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, S.; Mandell, M.A.; Deretic, V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol. Cell. 2015, 58, 507–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, G.A.; Huang, H.I.; Fee, B.E.; Youssef, N.; Jewell, M.L.; Cantillana, V.; Schoenborn, A.A.; Rogala, A.R.; Buckley, A.F.; Feng, C.G.; et al. Irgm1-deficiency leads to myeloid dysfunction in colon lamina propria and susceptibility to the intestinal pathogen Citrobacter rodentium. PLoS Pathog. 2020, 16, e1008553. [Google Scholar] [CrossRef] [PubMed]
- Mehto, S.; Jena, K.K.; Nath, P.; Chauhan, S.; Kolapalli, S.P.; Das, S.K.; Sahoo, P.K.; Jain, A.; Taylor, G.A.; Chauhan, S. The Crohn’s Disease Risk Factor IRGM Limits NLRP3 Inflammasome Activation by Impeding Its Assembly and by Mediating Its Selective Autophagy. Mol. Cell 2019, 73, 429–445.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Gulati, A.S.; Cantillana, V.; Henry, S.C.; Schmidt, E.A.; Daniell, X.; Grossniklaus, E.; Schoenborn, A.A.; Sartor, R.B.; Taylor, G.A. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G573–G584. [Google Scholar] [CrossRef] [Green Version]
- Baskaran, K.; Pugazhendhi, S.; Ramakrishna, B.S. Association of IRGM gene mutations with inflammatory bowel disease in the Indian population. PLoS ONE 2014, 9, e106863. [Google Scholar] [CrossRef]
- Al Nabhani, Z.; Dietrich, G.; Hugot, J.P.; Barreau, F. Nod2: The intestinal gate keeper. PLoS Pathog. 2017, 13, e1006177. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhang, Y.; Jin, T.; Yi, C.; Ocansey, D.K.W.; Mao, F. The role of NOD2 in intestinal immune response and microbiota modulation: A therapeutic target in inflammatory bowel disease. Int. Immunopharmacol. 2022, 113, 109466. [Google Scholar] [CrossRef]
- Hall, L.J.; Watson, A.J. Role of autophagy in NOD2-induced inflammation in Crohn’s disease. Gastroenterology 2012, 142, 1032–1034. [Google Scholar] [CrossRef]
- Fritz, T.; Niederreiter, L.; Adolph, T.; Blumberg, R.S.; Kaser, A. Crohn’s disease: NOD2, autophagy and ER stress converge. Gut 2011, 60, 1580–1588. [Google Scholar] [CrossRef] [Green Version]
- Lesage, S.; Zouali, H.; Cezard, J.P.; Colombel, J.F.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.; Gassull, M.; Binder, V.; et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am. J. Hum. Genet. 2002, 70, 845–857. [Google Scholar] [CrossRef] [Green Version]
- Sidiq, T.; Yoshihama, S.; Downs, I.; Kobayashi, K.S. Nod2: A Critical Regulator of Ileal Microbiota and Crohn’s Disease. Front. Immunol. 2016, 7, 367. [Google Scholar] [CrossRef] [Green Version]
- Travassos, L.H.; Carneiro, L.A.; Ramjeet, M.; Hussey, S.; Kim, Y.G.; Magalhaes, J.G.; Yuan, L.; Soares, F.; Chea, E.; Le Bourhis, L.; et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar] [CrossRef]
- Ogura, Y.; Lala, S.; Xin, W.; Smith, E.; Dowds, T.A.; Chen, F.F.; Zimmermann, E.; Tretiakova, M.; Cho, J.H.; Hart, J.; et al. Expression of NOD2 in Paneth cells: A possible link to Crohn’s ileitis. Gut 2003, 52, 1591–1597. [Google Scholar] [CrossRef]
- Liu, Z.; Lenardo, M.J. The role of LRRK2 in inflammatory bowel disease. Cell. Res. 2012, 22, 1092–1094. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Zhi, L.; Zhang, H. LRRK2 and mitochondria: Recent advances and current views. Brain Res. 2019, 1702, 96–104. [Google Scholar] [CrossRef]
- Yang, E.; Shen, J. The roles and functions of Paneth cells in Crohn’s disease: A critical review. Cell. Prolif. 2021, 54, e12958. [Google Scholar] [CrossRef]
- Boecker, C.A.; Goldsmith, J.; Dou, D.; Cajka, G.G.; Holzbaur, E.L.F. Increased LRRK2 kinase activity alters neuronal autophagy by disrupting the axonal transport of autophagosomes. Curr. Biol. 2021, 31, 2140–2154.e6. [Google Scholar] [CrossRef]
- Manzoni, C.; Mamais, A.; Dihanich, S.; Soutar, M.P.M.; Plun-Favreau, H.; Bandopadhyay, R.; Abeti, R.; Giunti, P.; Hardy, J.; Cookson, M.R.; et al. mTOR independent alteration in ULK1 Ser758 phosphorylation following chronic LRRK2 kinase inhibition. Biosci. Rep. 2018, 38, BSR20171669. [Google Scholar] [CrossRef] [Green Version]
- Rocha, E.M.; Keeney, M.T.; Di Maio, R.; De Miranda, B.R.; Greenamyre, J.T. LRRK2 and idiopathic Parkinson’s disease. Trends Neurosci. 2022, 45, 224–236. [Google Scholar] [CrossRef]
- Wallings, R.L.; Tansey, M.G. LRRK2 regulation of immune-pathways and inflammatory disease. Biochem. Soc. Trans. 2019, 47, 1581–1595. [Google Scholar] [CrossRef]
- Ahmadi Rastegar, D.; Hughes, L.P.; Perera, G.; Keshiya, S.; Zhong, S.; Gao, J.; Halliday, G.M.; Schule, B.; Dzamko, N. Effect of LRRK2 protein and activity on stimulated cytokines in human monocytes and macrophages. NPJ Park. Dis. 2022, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Ikezu, T.; Koro, L.; Wolozin, B.; Farraye, F.A.; Strongosky, A.J.; Wszolek, Z.K. Crohn’s and Parkinson’s Disease-Associated LRRK2 Mutations Alter Type II Interferon Responses in Human CD14+ Blood Monocytes Ex Vivo. J. Neuroimmune Pharm. 2020, 15, 794–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Lee, J.W.; Cooper, S.C.; Broxmeyer, H.E.; Cannon, J.R.; Kim, C.H. Parkinson disease-associated LRRK2 G2019S transgene disrupts marrow myelopoiesis and peripheral Th17 response. J. Leukoc. Biol. 2017, 102, 1093–1102. [Google Scholar] [CrossRef] [Green Version]
- Herrick, M.K.; Tansey, M.G. Is LRRK2 the missing link between inflammatory bowel disease and Parkinson’s disease? NPJ Park. Dis. 2021, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Shutinoski, B.; Hakimi, M.; Harmsen, I.E.; Lunn, M.; Rocha, J.; Lengacher, N.; Zhou, Y.Y.; Khan, J.; Nguyen, A.; Hake-Volling, Q.; et al. Lrrk2 alleles modulate inflammation during microbial infection of mice in a sex-dependent manner. Sci. Transl. Med. 2019, 11, eaas9292. [Google Scholar] [CrossRef]
- Hui, K.Y.; Fernandez-Hernandez, H.; Hu, J.; Schaffner, A.; Pankratz, N.; Hsu, N.Y.; Chuang, L.S.; Carmi, S.; Villaverde, N.; Li, X.; et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaai7795. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.L.; Matikainen-Ankney, B.A.; Hussein, A.; Huntley, G.W. Functional and behavioral consequences of Parkinson’s disease-associated LRRK2-G2019S mutation. Biochem. Soc. Trans. 2018, 46, 1697–1705. [Google Scholar] [CrossRef]
- Usmani, A.; Shavarebi, F.; Hiniker, A. The Cell Biology of LRRK2 in Parkinson’s Disease. Mol. Cell. Biol. 2021, 41, e00660-20. [Google Scholar] [CrossRef]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Ianniciello, A.; Zarou, M.M.; Rattigan, K.M.; Scott, M.; Dawson, A.; Dunn, K.; Brabcova, Z.; Kalkman, E.R.; Nixon, C.; Michie, A.M.; et al. ULK1 inhibition promotes oxidative stress-induced differentiation and sensitizes leukemic stem cells to targeted therapy. Sci. Transl. Med. 2021, 13, eabd5016. [Google Scholar] [CrossRef]
- Ro, S.H.; Jung, C.H.; Hahn, W.S.; Xu, X.; Kim, Y.M.; Yun, Y.S.; Park, J.M.; Kim, K.H.; Seo, M.; Ha, T.Y.; et al. Distinct functions of Ulk1 and Ulk2 in the regulation of lipid metabolism in adipocytes. Autophagy 2013, 9, 2103–2114. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.H.; Seo, M.; Otto, N.M.; Kim, D.H. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy 2011, 7, 1212–1221. [Google Scholar] [CrossRef] [Green Version]
- Morgan, A.R.; Lam, W.J.; Han, D.Y.; Fraser, A.G.; Ferguson, L.R. Association Analysis of ULK1 with Crohn’s Disease in a New Zealand Population. Gastroenterol. Res. Pract. 2012, 2012, 715309. [Google Scholar] [CrossRef] [Green Version]
- Henckaerts, L.; Cleynen, I.; Brinar, M.; John, J.M.; Van Steen, K.; Rutgeerts, P.; Vermeire, S. Genetic variation in the autophagy gene ULK1 and risk of Crohn’s disease. Inflamm. Bowel Dis. 2011, 17, 1392–1397. [Google Scholar] [CrossRef]
- Radhi, O.A.; Davidson, S.; Scott, F.; Zeng, R.X.; Jones, D.H.; Tomkinson, N.C.O.; Yu, J.; Chan, E.Y.W. Inhibition of the ULK1 protein complex suppresses Staphylococcus-induced autophagy and cell death. J. Biol. Chem. 2019, 294, 14289–14307. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Pantoom, S.; Wu, Y.W. Distinct Mechanisms for Processing Autophagy Protein LC3-PE by RavZ and ATG4B. ChemBioChem 2020, 21, 3377–3382. [Google Scholar] [CrossRef]
- Fernandez, A.F.; Lopez-Otin, C. The functional and pathologic relevance of autophagy proteases. J. Clin. Investig. 2015, 125, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wang, G.; Feng, D.; Zu, G.; Li, Y.; Shi, X.; Zhao, Y.; Jing, H.; Ning, S.; Le, W.; et al. Targeting the miR-665-3p-ATG4B-autophagy axis relieves inflammation and apoptosis in intestinal ischemia/reperfusion. Cell. Death Dis. 2018, 9, 483. [Google Scholar] [CrossRef] [Green Version]
- Papes, F.; Camargo, A.P.; de Souza, J.S.; Carvalho, V.M.A.; Szeto, R.A.; LaMontagne, E.; Teixeira, J.R.; Avansini, S.H.; Sanchez-Sanchez, S.M.; Nakahara, T.S.; et al. Transcription Factor 4 loss-of-function is associated with deficits in progenitor proliferation and cortical neuron content. Nat. Commun. 2022, 13, 2387. [Google Scholar] [CrossRef]
- Forrest, M.P.; Waite, A.J.; Martin-Rendon, E.; Blake, D.J. Knockdown of human TCF4 affects multiple signaling pathways involved in cell survival, epithelial to mesenchymal transition and neuronal differentiation. PLoS ONE 2013, 8, e73169. [Google Scholar] [CrossRef] [Green Version]
- Wehkamp, J.; Wang, G.; Kubler, I.; Nuding, S.; Gregorieff, A.; Schnabel, A.; Kays, R.J.; Fellermann, K.; Burk, O.; Schwab, M.; et al. The Paneth cell alpha-defensin deficiency of ileal Crohn’s disease is linked to Wnt/Tcf-4. J. Immunol. 2007, 179, 3109–3118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gersemann, M.; Wehkamp, J.; Fellermann, K.; Stange, E.F. Crohn’s disease—Defect in innate defence. World J. Gastroenterol. 2008, 14, 5499–5503. [Google Scholar] [CrossRef] [PubMed]
- Koslowski, M.J.; Kubler, I.; Chamaillard, M.; Schaeffeler, E.; Reinisch, W.; Wang, G.; Beisner, J.; Teml, A.; Peyrin-Biroulet, L.; Winter, S.; et al. Genetic variants of Wnt transcription factor TCF-4 (TCF7L2) putative promoter region are associated with small intestinal Crohn’s disease. PLoS ONE 2009, 4, e4496. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Heden, T.D.; Chow, L.S.; Hughey, C.C.; Mashek, D.G. Regulation and role of glycophagy in skeletal muscle energy metabolism. Autophagy 2022, 18, 1078–1089. [Google Scholar] [CrossRef]
- Farre, J.C.; Subramani, S. Peroxisome turnover by micropexophagy: An autophagy-related process. Trends Cell Biol. 2004, 14, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Dunn, W.A., Jr.; Cregg, J.M.; Kiel, J.A.; van der Klei, I.J.; Oku, M.; Sakai, Y.; Sibirny, A.A.; Stasyk, O.V.; Veenhuis, M. Pexophagy: The selective autophagy of peroxisomes. Autophagy 2005, 1, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Krick, R.; Muehe, Y.; Prick, T.; Bremer, S.; Schlotterhose, P.; Eskelinen, E.L.; Millen, J.; Goldfarb, D.S.; Thumm, M. Piecemeal microautophagy of the nucleus requires the core macroautophagy genes. Mol. Biol. Cell 2008, 19, 4492–4505. [Google Scholar] [CrossRef] [Green Version]
- Beese, C.J.; Brynjolfsdottir, S.H.; Frankel, L.B. Selective Autophagy of the Protein Homeostasis Machinery: Ribophagy, Proteaphagy and ER-Phagy. Front. Cell Dev. Biol. 2019, 7, 373. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Juste, Y.R.; Cuervo, A.M. Analysis of Chaperone-Mediated Autophagy. Methods Mol. Biol. 2019, 1880, 703–727. [Google Scholar] [CrossRef]
- Koga, H.; Martinez-Vicente, M.; Arias, E.; Kaushik, S.; Sulzer, D.; Cuervo, A.M. Constitutive upregulation of chaperone-mediated autophagy in Huntington’s disease. J. Neurosci. 2011, 31, 18492–18505. [Google Scholar] [CrossRef] [Green Version]
- Tekirdag, K.; Cuervo, A.M. Chaperone-mediated autophagy and endosomal microautophagy: Joint by a chaperone. J. Biol. Chem. 2018, 293, 5414–5424. [Google Scholar] [CrossRef] [Green Version]
- Orenstein, S.J.; Cuervo, A.M. Chaperone-mediated autophagy: Molecular mechanisms and physiological relevance. Semin. Cell Dev. Biol. 2010, 21, 719–726. [Google Scholar] [CrossRef] [Green Version]
- Assaye, M.A.; Gizaw, S.T. Chaperone-Mediated Autophagy and Its Implications for Neurodegeneration and Cancer. Int. J. Gen. Med. 2022, 15, 5635–5649. [Google Scholar] [CrossRef]
- Wang, L.; Klionsky, D.J.; Shen, H.M. The emerging mechanisms and functions of microautophagy. Nat. Rev. Mol. Cell Biol. 2023, 24, 186–203. [Google Scholar] [CrossRef]
- Jin, M.; Liu, X.; Klionsky, D.J. SnapShot: Selective autophagy. Cell 2013, 152, 368–368.e2. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, A.; Miller, S.I. Xenophagy: Pathogen-Containing Vacuoles Are Hard to Digest. Curr. Biol. 2019, 29, R1086–R1088. [Google Scholar] [CrossRef]
- Bauckman, K.A.; Owusu-Boaitey, N.; Mysorekar, I.U. Selective autophagy: Xenophagy. Methods 2015, 75, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Knodler, L.A.; Celli, J. Eating the strangers within: Host control of intracellular bacteria via xenophagy. Cell. Microbiol. 2011, 13, 1319–1327. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Tu, S.; Ding, L.; Jin, M.; Chen, H.; Zhou, H. The role of autophagy in viral infections. J. Biomed. Sci. 2023, 30, 5. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Wang, Z.; Chen, K.; Li, D.; Lv, Z.; Zhang, C.; Zhang, W.; Li, C. Xenophagy of invasive bacteria is differentially activated and modulated via a TLR-TRAF6-Beclin1 axis in echinoderms. J. Biol. Chem. 2022, 298, 101667. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.H.; Kamada, N.; Warner, N.; Kim, Y.G.; Nunez, G. The ever-expanding function of NOD2: Autophagy, viral recognition, and T cell activation. Trends Immunol. 2011, 32, 73–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brain, O.; Allan, P.; Simmons, A. NOD2-mediated autophagy and Crohn disease. Autophagy 2010, 6, 412–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay Between NLRP3 Inflammasome and Autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef] [PubMed]
- Paulus, G.L.; Xavier, R.J. Autophagy and checkpoints for intracellular pathogen defense. Curr. Opin. Gastroenterol. 2015, 31, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Verma, S.; Seranova, E.; Sarkar, S.; Kumar, D. Selective Autophagy and Xenophagy in Infection and Disease. Front. Cell Dev. Biol. 2018, 6, 147. [Google Scholar] [CrossRef] [Green Version]
- Lapaquette, P.; Bringer, M.A.; Darfeuille-Michaud, A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell. Microbiol. 2012, 14, 791–807. [Google Scholar] [CrossRef] [Green Version]
- Silwal, P.; Kim, J.K.; Jeon, S.M.; Lee, J.Y.; Kim, Y.J.; Kim, Y.S.; Seo, Y.; Kim, J.; Kim, S.Y.; Lee, M.J.; et al. Mitofusin-2 boosts innate immunity through the maintenance of aerobic glycolysis and activation of xenophagy in mice. Commun. Biol. 2021, 4, 548. [Google Scholar] [CrossRef]
- Bah, A.; Vergne, I. Macrophage Autophagy and Bacterial Infections. Front. Immunol. 2017, 8, 1483. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, H.; Nguyen, T.; Goins, W.F.; Chiocca, E.A. Interferon-stimulated gene 15 (ISG15) and ISG15-linked proteins can associate with members of the selective autophagic process, histone deacetylase 6 (HDAC6) and SQSTM1/p62. J. Biol. Chem. 2015, 290, 1485–1495. [Google Scholar] [CrossRef] [Green Version]
- Bhushan, J.; Radke, J.B.; Perng, Y.C.; McAllaster, M.; Lenschow, D.J.; Virgin, H.W.; Sibley, L.D. ISG15 Connects Autophagy and IFN-gamma-Dependent Control of Toxoplasma gondii Infection in Human Cells. mBio 2020, 11, e00852-20. [Google Scholar] [CrossRef]
- Germic, N.; Frangez, Z.; Yousefi, S.; Simon, H.U. Regulation of the innate immune system by autophagy: Monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ. 2019, 26, 715–727. [Google Scholar] [CrossRef]
- Germic, N.; Frangez, Z.; Yousefi, S.; Simon, H.U. Regulation of the innate immune system by autophagy: Neutrophils, eosinophils, mast cells, NK cells. Cell Death Differ. 2019, 26, 703–714. [Google Scholar] [CrossRef]
- Gradzka, S.; Thomas, O.S.; Kretz, O.; Haimovici, A.; Vasilikos, L.; Wong, W.W.; Hacker, G.; Gentle, I.E. Inhibitor of apoptosis proteins are required for effective fusion of autophagosomes with lysosomes. Cell Death Dis. 2018, 9, 529. [Google Scholar] [CrossRef]
- Cabrera, S.; Fernandez, A.F.; Marino, G.; Aguirre, A.; Suarez, M.F.; Espanol, Y.; Vega, J.A.; Laura, R.; Fueyo, A.; Fernandez-Garcia, M.S.; et al. ATG4B/autophagin-1 regulates intestinal homeostasis and protects mice from experimental colitis. Autophagy 2013, 9, 1188–1200. [Google Scholar] [CrossRef] [Green Version]
- Alsaadi, R.M.; Losier, T.T.; Tian, W.; Jackson, A.; Guo, Z.; Rubinsztein, D.C.; Russell, R.C. ULK1-mediated phosphorylation of ATG16L1 promotes xenophagy, but destabilizes the ATG16L1 Crohn’s mutant. EMBO Rep. 2019, 20, e46885. [Google Scholar] [CrossRef]
- Marchiando, A.M.; Ramanan, D.; Ding, Y.; Gomez, L.E.; Hubbard-Lucey, V.M.; Maurer, K.; Wang, C.; Ziel, J.W.; van Rooijen, N.; Nunez, G.; et al. A deficiency in the autophagy gene Atg16L1 enhances resistance to enteric bacterial infection. Cell Host Microbe 2013, 14, 216–224. [Google Scholar] [CrossRef] [Green Version]
- Brest, P.; Lapaquette, P.; Souidi, M.; Lebrigand, K.; Cesaro, A.; Vouret-Craviari, V.; Mari, B.; Barbry, P.; Mosnier, J.F.; Hebuterne, X.; et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat. Genet. 2011, 43, 242–245. [Google Scholar] [CrossRef]
- Rogala, A.R.; Schoenborn, A.A.; Fee, B.E.; Cantillana, V.A.; Joyce, M.J.; Gharaibeh, R.Z.; Roy, S.; Fodor, A.A.; Sartor, R.B.; Taylor, G.A.; et al. Environmental factors regulate Paneth cell phenotype and host susceptibility to intestinal inflammation in Irgm1-deficient mice. Dis. Model. Mech. 2018, 11, dmm031070. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Eun, H.S.; Jo, E.K. Roles of Autophagy-Related Genes in the Pathogenesis of Inflammatory Bowel Disease. Cells 2019, 8, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochida, K.; Nakatogawa, H. ER-phagy: Selective autophagy of the endoplasmic reticulum. EMBO Rep. 2022, 23, e55192. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef] [PubMed]
- Hosomi, S.; Kaser, A.; Blumberg, R.S. Role of endoplasmic reticulum stress and autophagy as interlinking pathways in the pathogenesis of inflammatory bowel disease. Curr. Opin. Gastroenterol. 2015, 31, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Kaser, A.; Blumberg, R.S. Endoplasmic reticulum stress and intestinal inflammation. Mucosal Immunol. 2010, 3, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Naama, M.; Telpaz, S.; Awad, A.; Ben-Simon, S.; Harshuk-Shabso, S.; Modilevsky, S.; Rubin, E.; Sawaed, J.; Zelik, L.; Zigdon, M.; et al. Autophagy controls mucus secretion from intestinal goblet cells by alleviating ER stress. Cell Host Microbe 2023, 31, 433–446.e4. [Google Scholar] [CrossRef]
- Amen, O.M.; Sarker, S.D.; Ghildyal, R.; Arya, A. Endoplasmic Reticulum Stress Activates Unfolded Protein Response Signaling and Mediates Inflammation, Obesity, and Cardiac Dysfunction: Therapeutic and Molecular Approach. Front. Pharm. 2019, 10, 977. [Google Scholar] [CrossRef] [Green Version]
- Tschurtschenthaler, M.; Adolph, T.E. The Selective Autophagy Receptor Optineurin in Crohn’s Disease. Front. Immunol. 2018, 9, 766. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Qian, X.; Cui, Y. Advances in ER-Phagy and Its Diseases Relevance. Cells 2021, 10, 2328. [Google Scholar] [CrossRef]
- Aden, K.; Tran, F.; Ito, G.; Sheibani-Tezerji, R.; Lipinski, S.; Kuiper, J.W.; Tschurtschenthaler, M.; Saveljeva, S.; Bhattacharyya, J.; Hasler, R.; et al. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS-STING. J. Exp. Med. 2018, 215, 2868–2886. [Google Scholar] [CrossRef] [Green Version]
- Stengel, S.T.; Fazio, A.; Lipinski, S.; Jahn, M.T.; Aden, K.; Ito, G.; Wottawa, F.; Kuiper, J.W.P.; Coleman, O.I.; Tran, F.; et al. Activating Transcription Factor 6 Mediates Inflammatory Signals in Intestinal Epithelial Cells Upon Endoplasmic Reticulum Stress. Gastroenterology 2020, 159, 1357–1374.e10. [Google Scholar] [CrossRef]
- Chipurupalli, S.; Samavedam, U.; Robinson, N. Crosstalk Between ER Stress, Autophagy and Inflammation. Front. Med. 2021, 8, 758311. [Google Scholar] [CrossRef]
- Kamimura, D.; Bevan, M.J. Endoplasmic reticulum stress regulator XBP-1 contributes to effector CD8+ T cell differentiation during acute infection. J. Immunol. 2008, 181, 5433–5441. [Google Scholar] [CrossRef] [Green Version]
- Kaser, A.; Adolph, T.E.; Blumberg, R.S. The unfolded protein response and gastrointestinal disease. Semin. Immunopathol. 2013, 35, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Margariti, A.; Li, H.; Chen, T.; Martin, D.; Vizcay-Barrena, G.; Alam, S.; Karamariti, E.; Xiao, Q.; Zampetaki, A.; Zhang, Z.; et al. XBP1 mRNA splicing triggers an autophagic response in endothelial cells through BECLIN-1 transcriptional activation. J. Biol. Chem. 2013, 288, 859–872. [Google Scholar] [CrossRef] [Green Version]
- Tschurtschenthaler, M.; Adolph, T.E.; Ashcroft, J.W.; Niederreiter, L.; Bharti, R.; Saveljeva, S.; Bhattacharyya, J.; Flak, M.B.; Shih, D.Q.; Fuhler, G.M.; et al. Defective ATG16L1-mediated removal of IRE1alpha drives Crohn’s disease-like ileitis. J. Exp. Med. 2017, 214, 401–422. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Wang, K.; Wang, M.; Zhao, W.; Zhang, C.; Cai, M.; Qiu, Y.; Zhang, T.; Shao, R.; Zhao, W. ER-phagy in the Occurrence and Development of Cancer. Biomedicines 2022, 10, 707. [Google Scholar] [CrossRef]
- Matsuzawa-Ishimoto, Y.; Shono, Y.; Gomez, L.E.; Hubbard-Lucey, V.M.; Cammer, M.; Neil, J.; Dewan, M.Z.; Lieberman, S.R.; Lazrak, A.; Marinis, J.M.; et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J. Exp. Med. 2017, 214, 3687–3705. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Doblado, L.; Lueck, C.; Rey, C.; Samhan-Arias, A.K.; Prieto, I.; Stacchiotti, A.; Monsalve, M. Mitophagy in Human Diseases. Int. J. Mol. Sci. 2021, 22, 3903. [Google Scholar] [CrossRef]
- Ho, G.T.; Theiss, A.L. Mitochondria and Inflammatory Bowel Diseases: Toward a Stratified Therapeutic Intervention. Annu. Rev. Physiol. 2022, 84, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dai, L.; Li, D. Mitophagy in neurological disorders. J. Neuroinflammation 2021, 18, 297. [Google Scholar] [CrossRef] [PubMed]
- Alula, K.M.; Delgado-Deida, Y.; Callahan, R.; Till, A.; Underwood, L.; Thompson, W.E.; Souza, R.F.; Dassopoulos, T.; Onyiah, J.; Venuprasad, K.; et al. Inner mitochondrial membrane protein Prohibitin 1 mediates Nix-induced, Parkin-independent mitophagy. Sci. Rep. 2023, 13, 18. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.C.; Gao, F.; McGovern, D.P.; Stappenbeck, T.S. Spatial and temporal stability of paneth cell phenotypes in Crohn’s disease: Implications for prognostic cellular biomarker development. Inflamm. Bowel Dis. 2014, 20, 646–651. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.C.; Gurram, B.; Baldridge, M.T.; Head, R.; Lam, V.; Luo, C.; Cao, Y.; Simpson, P.; Hayward, M.; Holtz, M.L.; et al. Paneth cell defects in Crohn’s disease patients promote dysbiosis. JCI Insight 2016, 1, e86907. [Google Scholar] [CrossRef]
- Liu, T.C.; Kern, J.T.; VanDussen, K.L.; Xiong, S.; Kaiko, G.E.; Wilen, C.B.; Rajala, M.W.; Caruso, R.; Holtzman, M.J.; Gao, F.; et al. Interaction between smoking and ATG16L1T300A triggers Paneth cell defects in Crohn’s disease. J. Clin. Investig. 2018, 128, 5110–5122. [Google Scholar] [CrossRef] [Green Version]
- Alula, K.M.; Jackson, D.N.; Smith, A.D.; Kim, D.S.; Turner, K.; Odstrcil, E.; Kaipparettu, B.A.; Dassopoulos, T.; Venuprasad, K.; Feagins, L.A.; et al. Targeting Mitochondrial Damage as a Therapeutic for Ileal Crohn’s Disease. Cells 2021, 10, 1349. [Google Scholar] [CrossRef]
- Mao, H.; Chen, W.; Chen, L.; Li, L. Potential role of mitochondria-associated endoplasmic reticulum membrane proteins in diseases. Biochem. Pharm. 2022, 199, 115011. [Google Scholar] [CrossRef]
- Levin, A.D.; Koelink, P.J.; Bloemendaal, F.M.; Vos, A.C.; D’Haens, G.R.; van den Brink, G.R.; Wildenberg, M.E. Autophagy Contributes to the Induction of Anti-TNF Induced Macrophages. J. Crohn’s Colitis 2016, 10, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Prins, M.M.C.; Giugliano, F.P.; van Roest, M.; van de Graaf, S.F.J.; Koelink, P.J.; Wildenberg, M.E. Thiopurines correct the effects of autophagy impairment on intestinal healing—A potential role for ARHGAP18/RhoA. Dis. Model. Mech. 2021, 14, dmm047233. [Google Scholar] [CrossRef]
- Thellung, S.; Corsaro, A.; Nizzari, M.; Barbieri, F.; Florio, T. Autophagy Activator Drugs: A New Opportunity in Neuroprotection from Misfolded Protein Toxicity. Int. J. Mol. Sci. 2019, 20, 901. [Google Scholar] [CrossRef] [Green Version]
- Azzman, N. Crohn’s Disease: Potential Drugs for Modulation of Autophagy. Medicina 2019, 55, 224. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Gene | Functional Relation to Autophagy |
|---|---|
| ATG16L1 | Assembly of autophagy machinery, autophagosome formation, and bacterial defense by Paneth cells |
| IRGM | Promotion of autophagosome formation, dampening inflammation via NLRP3, and maintenance of Paneth cell health |
| NOD2 | Regulation of immune system, maintenance of intestinal barrier integrity, elimination of invading bacteria via activation of NFκB, and interaction with ATG16L1 and IRGM to form autophagosomes |
| LRRK2 | Regulation of autophagosome formation via Rab29, ATG9, and ULK1 interactions, maintenance of functional Paneth cells, and mediation of the immune response through TLRs |
| ULK1 | Initiation of autophagy by forming ULK1 complex, regulation of immune cell growth and differentiation, and maintenance of intestinal barrier function |
| ATG4 | Regulation of autophagy via processing of LC3 and reduction of inflammation |
| TCF4 | Contribution to functional autophagy, regulation of gene expression and survival, and ensuring Paneth cell health |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alula, K.M.; Theiss, A.L. Autophagy in Crohn’s Disease: Converging on Dysfunctional Innate Immunity. Cells 2023, 12, 1779. https://doi.org/10.3390/cells12131779
Alula KM, Theiss AL. Autophagy in Crohn’s Disease: Converging on Dysfunctional Innate Immunity. Cells. 2023; 12(13):1779. https://doi.org/10.3390/cells12131779
Chicago/Turabian StyleAlula, Kibrom M., and Arianne L. Theiss. 2023. "Autophagy in Crohn’s Disease: Converging on Dysfunctional Innate Immunity" Cells 12, no. 13: 1779. https://doi.org/10.3390/cells12131779