
Genetic Primary Microcephalies: When Centrosome Dysfunction Dictates Brain and Body Size

Abstract
:1. Introduction
2. Primary Microcephaly: Small Brain Size or Small Brain and Body Sizes?
2.1. Primary Microcephaly: An Early Defect in Brain Growth with or without Cortical Malformations
2.2. Primary Microcephaly Is an Early Defect in Brain Growth with or without Defects in Body Growth (Microcephalic Primordial Dwarfism)
2.3. Primary Microcephaly: An Early Defect in Brain Growth with or without Neurosensory Disorders
2.4. Primary Microcephaly: An Early Defect in Brain Growth with or without Intellectual Disability
3. ASPM, WDR62, and Dynein: Three Emblematic PM Genes Implicated in PMs with or without MCDs
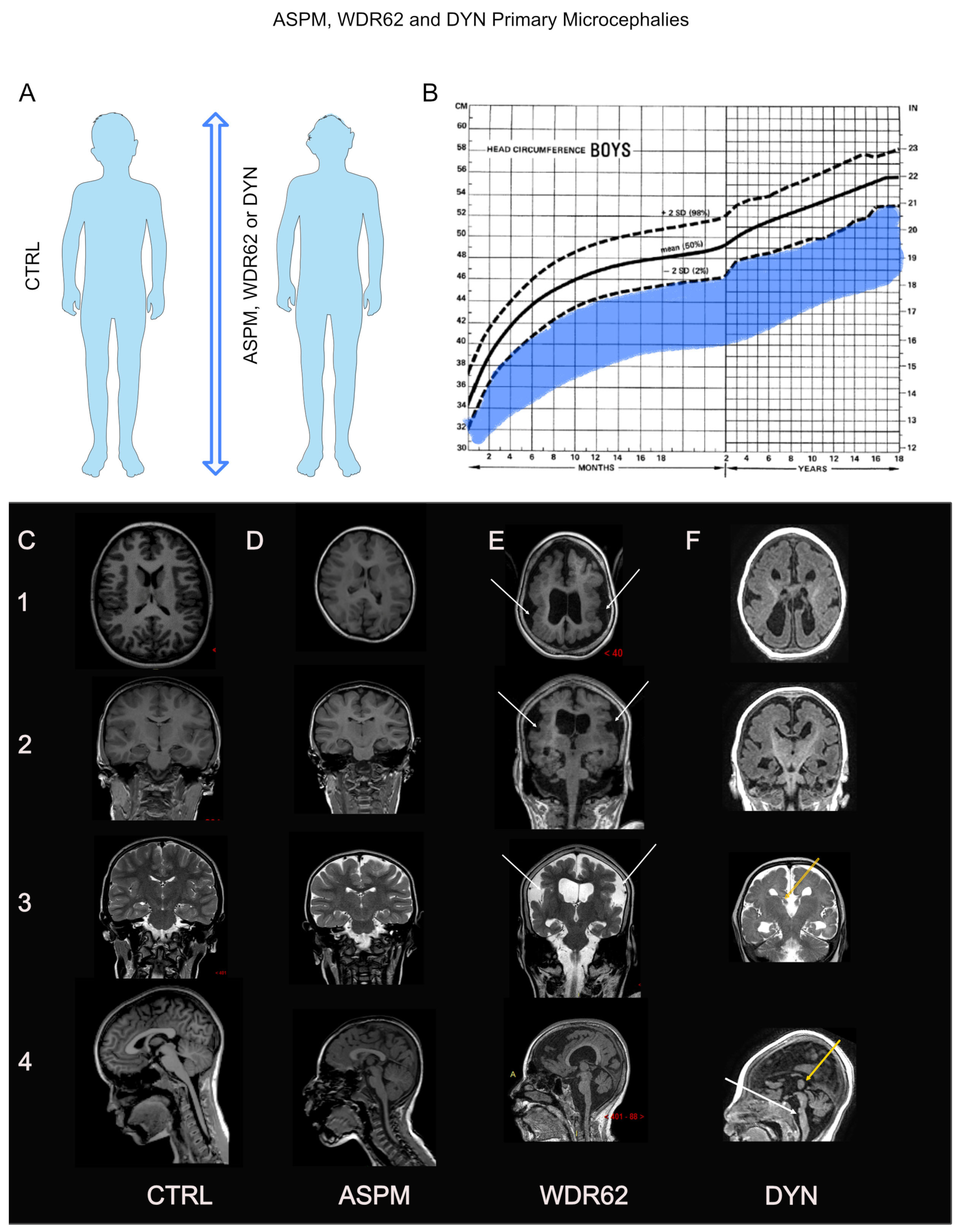
3.1. ASPM: Phenotype–Gene Relationships
3.1.1. Genetics
3.1.2. Growth
3.1.3. Brain Development and Cognition
3.2. WDR62: Phenotype–Gene Relationships
3.2.1. Genetics
3.2.2. Growth
3.2.3. Brain Development and Cognition
3.3. Dynein: Phenotype–Gene Relationships
3.3.1. Genetics
3.3.2. Growth
3.3.3. Brain Development and Cognition
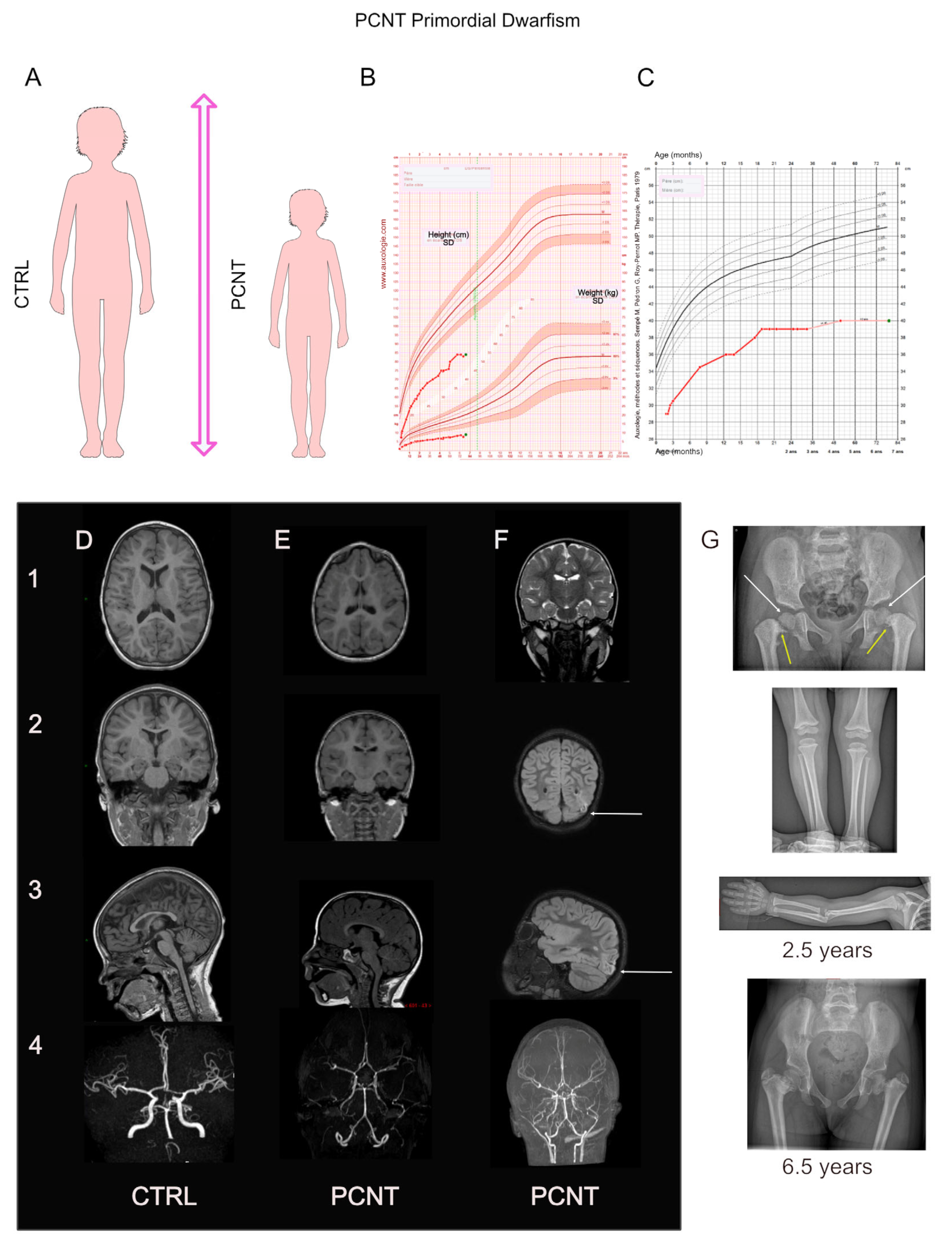
4. PCNT: The Major Microcephalic Primordial Dwarfism-Causing Gene
4.1. PCNT: Phenotype–Gene Relationships
4.1.1. Genetics
4.1.2. Growth
4.1.3. Brain Development and Cognition
4.1.4. Associated Features or Comorbidities
5. CDK5RAP2, CEP152, and PLK4: Three Emblematic PM Genes Associated with Short Stature or Chorioretinopathy or Both
5.1. CDK5RAP2: Phenotype–Gene Relationships
5.1.1. Genetics
5.1.2. Growth
5.1.3. Brain Development and Cognition
5.1.4. Neurosensory Impairment
5.2. CEP152: Phenotype–Gene Relationships
5.2.1. Genetics
5.2.2. Growth
5.2.3. Brain Development and Cognition
5.2.4. Neurosensory Impairment
5.3. PLK4: Phenotype–Gene Relationships
5.3.1. Genetics
5.3.2. Growth
5.3.3. Brain Development and Cognition
5.3.4. Neurosensory Impairment
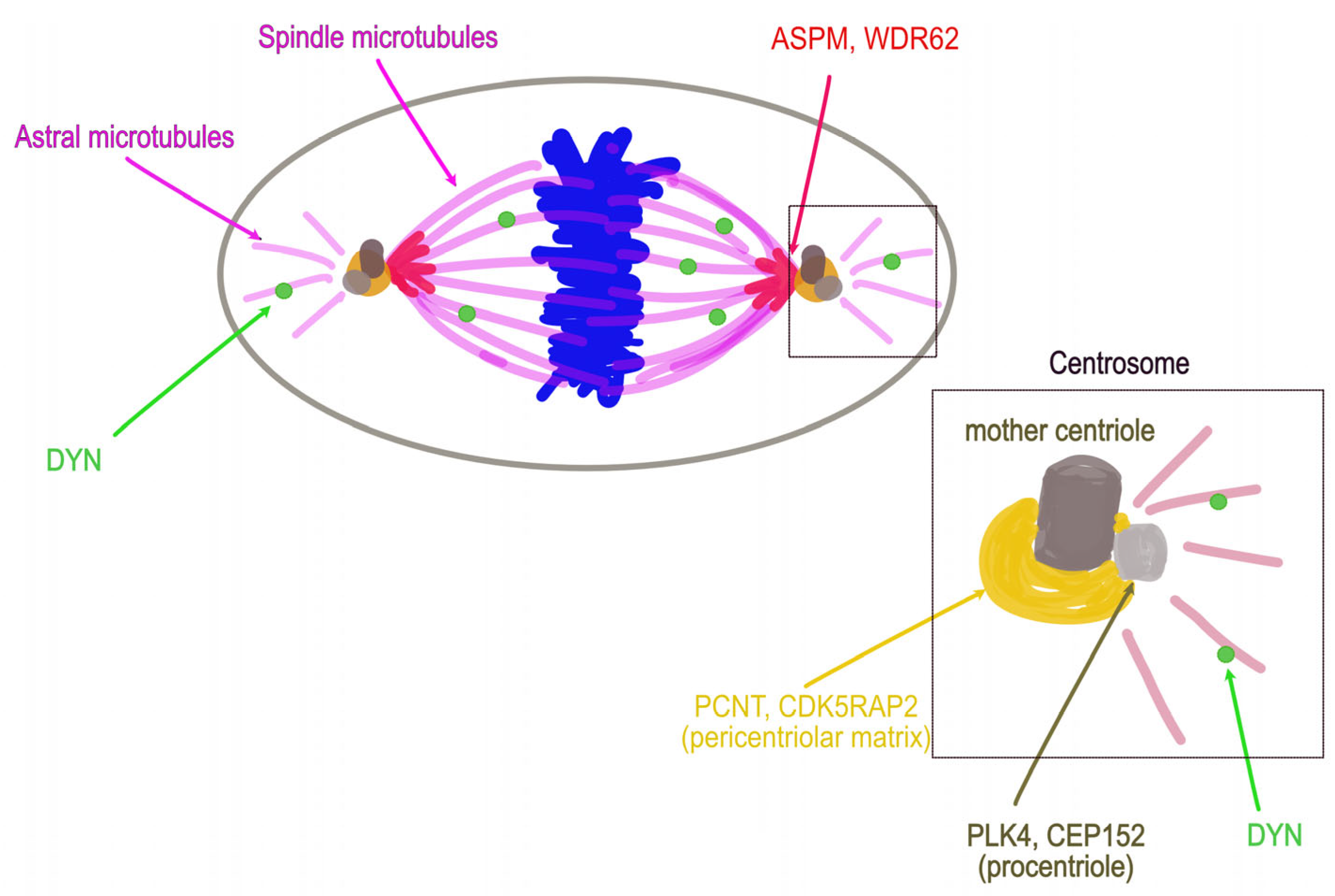
6. Emblematic PM Genes Encode Centrosome or Spindle Pole Proteins
7. Unresolved Issues and Clinical Pitfalls
7.1. Mutations in Emblematic PM Genes Playing a Role in Spindle Pole Structure and Function Are Responsible for Different Diseases with Partial Overlap
7.2. Why Is the Brain Relatively More Vulnerable than Other Organs?
7.3. Do Single Nucleotide Polymorphisms, Not Pathogenic, or Heterozygous Pathogenic Variants in PM Genes Affect Cognitive Functions, Brain Size, or Both?
7.4. Do Mutations in One or More PM Genes Allow the Establishment of Prognosis?
7.5. Understanding What Occurred during Neurogenesis in the Brain of Each Individual Affected by PM
7.6. Identifying the Temporal Window during Which Defects Occur and during Which Intervention Will Be Possible to Improve the Conditions of Patients
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Locus | Gene/Protein | Subcellular Location | Main Molecular Functions | Functional Partners Involved in the Same Pathway | Syndrome Related to Mutations | Affected Organs | References |
|---|---|---|---|---|---|---|---|
| MCPH1 | MCPH1/ MICROCEPHALIN | Nucleus, mitochondria | Chromosome condensation, DNA replication, DNA damage response | CDC27, H2AX upon DNA damage | PM/MPD (AR) | Brain, skeleton | [2,210,211,212,213,214,215] |
| MCPH2 | WD62/WDR62 | Minus end of microtubules | Neuronal proliferation and migration | ASPM, AURORA A, CEP152, CEP63, CDK5RAP2 | PM + MCD (AR) | Brain | [3,10,11,216,217] |
| MCPH3 | CDK5RAP2/ CDK5RAP2 | PCM | Microtubule nucleation, centriole disengagement after duplication | PCNT, GAMMA TUBULIN, WDR62, CEP152, CEP63 | PM (AR) | Brain, eye, ear | [4,162,216,218,219,220,221] |
| MCPH4 | CASC5/KLN1 | Kinetochore | Attachment of centromeres to spindle microtubules, spindle-assembly checkpoint signaling | BUB1, BUB1B | PM (AR) | Brain | [222,223,224,225,226] |
| MCPH5 | ASPM/ASPM | Minus end of microtubules | Spindle pole organization, microtubule dynamics at spindle pole | KATNA1, KATNB1, WDR62, CIT | PM (AR) | Brain | [6,56,76,217] |
| MCPH6 & SCKL4 | CENPJ (CPAP)/ CENPJ (CPAP) | Centriole | Centriole duplication/biogenesis | CEP152, STIL, SAS6, GAMMA TUBULIN, CEP135, microtubules | PM/MPD (AR) | Brain, skeleton | [4,221,227,228,229,230,231] |
| MCPH7 | STIL/STIL | Centriole cartwheel | Centriole duplication/ biogenesis, primary cilium | PLK4, CENPJ/CPAP, SAS6 | PM/MPD (AR) | Brain | [229,232,233,234,235,236] |
| MCPH8 | CEP135/CEP135 | Centriole | Centriole biogenesis & cohesion | CEP152, CENPJ/CPAP, SAS6, GAMMA TUBULIN, Microtubules | PM/MPD (AR) | Brain | [231,237,238,239,240] |
| MCPH9 & SCKL5 | CEP152/CEP152 | Centriole | Centriole duplication/ Biogenesis | PLK4, CENPJ/CPAP, WDR62, CEP192, CEP63, CEP135, CDK5RAP2 | PM/MPD (AR) | Brain, skeleton, teeth | [8,173,174,216,221,228,231,241] |
| MCPH10 | ZNF332/ZNF332 | Nucleus | Transcription, neural progenitor proliferation | CCAR2, EMSY, ASCL2 | PM (AR) | Brain, skeleton, eye, ear | [242,243] |
| MCPH11 | PHC1/PHC1 | Nucleus | Chromatin remodeling | PRC1 complex | PM/MPD (AR) | Brain | [244] |
| MCPH12 | CDK6/CDK6 | Nucleus, centrosome | Cell cycle control (G1/S transition) | CYCLIN D | PM (AR) | Brain | [245,246] |
| MCPH13 | CENPE/CENPE | Plus-ends of microtubules, kinesin-like motor protein | Microtubule attachment at the kinetochore, chromosome congression | CENPF, BUB1B | PM/MPD (AR) | Brain, skeleton, heart | [247,248,249,250] |
| MCPH14 | SAS6/SAS6 | Centriole cartwheel | Centriole duplication/ Biogenesis | CENPJ/CPAP, STIL, CEP135, CEP152 | PM (AR) | Brain | [174,229,231,251,252,253] |
| MCPH15 | MSFD2A/ MSFD2A | Cell membrane | Blood-brain barrier formation | PM (AR) | Brain | [254,255,256] | |
| MCPH16 | ANKLE2/ANKLE2 | Endoplasmic reticulum | Nuclear envelope reassembly | BAF/BANF1 | PM/MPD (AR) | Brain, eye, skin, bone marrow, skeleton, heart | [148,257,258] |
| MCPH17 | CIT/CIT | Midbody | Cytokinesis | ASPM, KIF14 | PM/MPD (AR) | Brain | [259,260,261,262,263] |
| MCPH18 | WDFY3/WDFY3 | Nucleus | Autophagy | ATG5 | PM (AD) | Brain | [264,265] |
| MCPH19 | COPB2/COPB2 | Golgi, cytoplasmic vesicle | ER-to-Golgi and Golgi-to-ER transport. Coatomer complex protein | alpha, beta, gamma, delta, epsilon and zeta COP-subunits | PM (AR) | Brain, skeleton | [266] |
| MCPH20 | KIF14/KIF14 | Mitotic spindle, midbody | Cell cycle progression, cytokinesis | CIT (AR) | PM/MPD (AR) | Brain, +/− kidney and eye | [262,267,268,269] |
| MCPH21 | NCAPD2/NCAPD2 | Nucleus, chromosomes | Mitotic chromosome condensation/integrity | Component of the condensing complex NCAPD3/H/G | MPD (AR) | Brain | [270,271] |
| MCPH22 | NCAPD3/NCAPD3 | Nucleus, chromosomes | Mitotic chromosome condensation/integrity | Component of the condensing complex NCAPD2/H/G | PM/MPD (AR) | Brain | [270,271] |
| MCPH23 | NCAPH/NCAPH | Nucleus, chromosomes | Mitotic chromosome condensation/integrity | Component of the condensing complex NCAPD2/D3/G | PM (AR) | Brain | [270,271] |
| MCPH24 | NUP37/NUP37 | Nucleus, nuclear pore complex | Kinetochore microtubule attachment, mitotic progression | Component of the nuclear pore complex (NUP) | PM (AR) | Brain | [272,273] |
| MCPH25 | MAP11/MAP11 | Mitotic spindle, midbody | Mitotic spindle dynamics, cytokinesis | TUBA1A | PM (AR) | Brain | [274] |
| MCPH26 | LMNB1/LMNB1 | Nucleus, inner nuclear membrane | Nuclear shape, spindle assembly | Lamin-associated polypeptides | PM (AD) | Brain | [275,276,277,278] |
| MCPH27 | LMNB2/LMNB2 | Nucleus, inner nuclear membrane | Nuclear shape, spindle assembly | Lamin-associated polypeptides | PM (AD) | Brain | [275,276,277,278] |
| MCPH28 | RRP7A/RRP7A | Nucleus, cilium, microtubules | Ribosome biogenesis, cilium resorption | Small subunits of the processosome | PM (AR) | Brain | [279,280] |
| MCPH29 | PDCD6IP/ PDCD6IP | Cytoplasmic vesicle membrane | Cytokinesis, apoptosis | ESCRT-III components | PM (AR) | Brain | [281] |
| MCPH30 | BUB1/BUB1 | Chromosomes, kinetochore | spindle-assembly checkpoint signaling, chromosome segregation | MAD1L1, BUB3, CASC5/KNL1, CENPE | PM/MPD (AR) | Brain, skeleton, heart | [282,283,284,285] |
| MOPD2 | PCNT/PCNT | PCM | PCM formation and centrosome cohesion | GAMMA TUBULIN, CDK5RAP2 | MPD, MOPD2 and Seckel syndromes (AR) | Brain, cerebral arteries, heart, kidney, bone marrow, skin, pituitary gland, pancreas, skeleton, teeth | [35,36,286,287,288,289] |
| SCKL1 | ATR/ATR | Nucleus, chromosomes | DNA damage response | ATRIP, RAD17 | MPD (AR) | Brain, skeleton, bone marrow, teeth | [36,290,291,292] |
| SCKL2 | RBBP8 (CTIP)/RBBP8 | Nucleus, chromosomes | Double-stand break repair, homologous recombination | BRCA1, MRN complex, RAD50 | MPD (AR) | Brain, skeleton, eye, teeth | [293,294,295,296] |
| SCKL6 | CEP63/CEP63 | Mother Centriole | Centriole integrity/assembly | CEP152, CDK5RAP2, WDR62 | MPD (AR) | Brain | [216,297,298] |
| SCKL7 | NIN/NINEIN | Sub-distal appendage of mother centriole | Microtubule anchoring at mother centriole | CCDC120 | MPD (AR) | Brain, pituitary gland, skeleton | [299,300,301] |
| SCKL8 | DNA2/DNA2 | Nucleus, mitochondria | DNA replication/repair | BLM | MPD (AR) | Brain | [302,303,304,305] |
| SCKL9 | TRAIP/TRAIP | Nucleus | Replication stress response | PCNA | MPD (AR) | Brain, gonads, skeleton, lung, kidney | [306,307,308,309] |
| SCKL10 | NSMCE2/ NSMCE2 | Nucleus | DNA double-strand break repair–homologous recombination | Component of the SMC5-SMC6 complex | MPD ‘AR) | Brain, eye, pancreas, gonads | [310,311,312] |
| MCCRP1 | TUBGCP6/ TUBGCP6 | PCM/MTOC | Microtubule nucleation | TUGCP2/3/4/5, GAMMA TUBULIN | PM + neurosensory disorder (AR) | Brain, eye | [46,313] |
| MCCRP2 | PLK4/PLK4 | Centriole | Centriole duplication/ Biogenesis | CEP152, CEP192, STIL | MPD + neurosensory disorder (AR) | Brain, eye, ear, skin | [46,174,209,237,314,315] |
| MCCRP3 | TUBGCP4/ TUBGCP4 | PCM/MTOC | Microtubule nucleation | TUGCP2/3/4/5, GAMMA TUBULIN | PM + neurosensory disorder (AR) | Brain, eye | [47,316] |
| MCLRM | KIF11/KIF11 | Plus end of microtubules | Bipolar spindle establishment | Microtubules | PM + neurosensory disorder (AD) | Brain, eye | [317,318] |
| CDCBM/SMELED | DYNC1H1 | Microtubules | Transporter of cargo towards the minus ends of microtubules | DYNC1LI1/2, DYNACTIN, LIS1, NDEL1 | PM + MCD (AD) | Brain, muscle | [100,319,320,321] |
| Bloom (BLM) syndrome | RECQL3/RECQL3 | Nucleus | DNA replication/repair | RAD51, FANCD2, DNA2 | MPD (AR) | Brain, skin, pituitary gland, bone marrow, immune system, gonads, pancreas | [322,323,324,325] |
| LIG4 syndrome | LIG4/LIG4 | Nucleus | DNA repair | XRCC4/5/6, NHEJ1/XLF | MPD (AR) | Brain, immune system, skin | [39,326,327,328] |
| SSMED | XRCC4/XRCC4 | Nucleus | DNA repair | XRCC5/6, LIG4, | MPD (AR) | Brain, eye, ear, pituitary gland, gonads, pancreas, immune system, kidney, +/− heart | [40,328,329] |
| Meyer Gorlin syndrome 1 | ORC1/ORC1 | Nucleus | DNA replication | ORC2/3/4/5/6, CDC6 | MPD (AR) | Brain, skeleton | [37,330,331] |
| Meyer Gorlin syndrome 2 | ORC4/ORC4 | Nucleus | DNA replication | ORC1/2/3/5/6, CDC6 | MPD (AR) | Brain, skeleton | [241] |
| Meyer Gorlin syndrome 3 | ORC6/ORC6 | Nucleus | DNA replication | ORC1/2/3/4/5, CDC6 | MPD (AR) | Brain, skeleton | [37] |
| Meyer Gorlin syndrome 4 | CDT1/CDT1 | Nucleus, chromosomes, kinetochore | DNA replication, mitosis | CDC6, PCNA, GMMN | MPD (AR) | Brain, skeleton | [37,241,332,333] |
| Meyer Gorlin syndrome 5 | CDC6/CDC6 | Nucleus | DNA replication | ORC1, CDT1, PCNA | MPD (AR) | Brain, skeleton, teeth | [37,334,335] |
| Meyer Gorlin syndrome 6 | GMNN/GMNN | Nucleus | DNA replication | CDT1 | MPD (AD) | Brain, skeleton, ear | [336,337] |
References
- Wright, C.M.; Emond, A. Head Growth and Neurocognitive Outcomes. Pediatrics 2015, 135, e1393–e1398. [Google Scholar] [CrossRef] [Green Version]
- Jackson, A.P.; Eastwood, H.; Bell, S.M.; Adu, J.; Toomes, C.; Carr, I.M.; Roberts, E.; Hampshire, D.J.; Crow, Y.J.; Mighell, A.J.; et al. Identification of Microcephalin, a Protein Implicated in Determining the Size of the Human Brain. Am. J. Hum. Genet. 2002, 71, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholas, A.K.; Khurshid, M.; Desir, J.; Carvalho, O.P.; Cox, J.J.; Thornton, G.; Kausar, R.; Ansar, M.; Ahmad, W.; Verloes, A.; et al. WDR62 Is Associated with the Spindle Pole and Is Mutated in Human Microcephaly. Nat. Genet. 2010, 42, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.; Roberts, E.; Springell, K.; Lizarraga, S.B.; Scott, S.; Higgins, J.; Hampshire, D.J.; Morrison, E.E.; Leal, G.F.; Silva, E.O.; et al. A Centrosomal Mechanism Involving CDK5RAP2 and CENPJ Controls Brain Size. Nat. Genet. 2005, 37, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Genin, A.; Desir, J.; Lambert, N.; Biervliet, M.; Van Der Aa, N.; Pierquin, G.; Killian, A.; Tosi, M.; Urbina, M.; Lefort, A.; et al. Kinetochore KMN Network Gene CASC5 Mutated in Primary Microcephaly. Hum. Mol. Genet. 2012, 21, 5306–5317. [Google Scholar] [CrossRef] [Green Version]
- Bond, J.; Roberts, E.; Mochida, G.H.; Hampshire, D.J.; Scott, S.; Askham, J.M.; Springell, K.; Mahadevan, M.; Crow, Y.J.; Markham, A.F.; et al. ASPM Is a Major Determinant of Cerebral Cortical Size. Nat. Genet. 2002, 32, 316–320. [Google Scholar] [CrossRef]
- Al-Dosari, M.S.; Shaheen, R.; Colak, D.; Alkuraya, F.S. Novel CENPJ Mutation Causes Seckel Syndrome. J. Med. Genet. 2010, 47, 411–414. [Google Scholar] [CrossRef]
- Kalay, E.; Yigit, G.; Aslan, Y.; Brown, K.E.; Pohl, E.; Bicknell, L.S.; Kayserili, H.; Li, Y.; Tuysuz, B.; Nurnberg, G.; et al. CEP152 Is a Genome Maintenance Protein Disrupted in Seckel Syndrome. Nat. Genet. 2011, 43, 23–26. [Google Scholar] [CrossRef] [Green Version]
- Guernsey, D.L.; Jiang, H.; Hussin, J.; Arnold, M.; Bouyakdan, K.; Perry, S.; Babineau-Sturk, T.; Beis, J.; Dumas, N.; Evans, S.C.; et al. Mutations in Centrosomal Protein CEP152 in Primary Microcephaly Families Linked to MCPH4. Am. J. Hum. Genet. 2010, 87, 40–51. [Google Scholar] [CrossRef] [Green Version]
- Bilgüvar, K.; Oztürk, A.K.; Louvi, A.; Kwan, K.Y.; Choi, M.; Tatli, B.; Yalnizoğlu, D.; Tüysüz, B.; Cağlayan, A.O.; Gökben, S.; et al. Whole-Exome Sequencing Identifies Recessive WDR62 Mutations in Severe Brain Malformations. Nature 2010, 467, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.W.; Mochida, G.H.; Tischfield, D.J.; Sgaier, S.K.; Flores-Sarnat, L.; Sergi, C.M.; Topcu, M.; McDonald, M.T.; Barry, B.J.; Felie, J.M.; et al. Mutations in WDR62, Encoding a Centrosome-Associated Protein, Cause Microcephaly with Simplified Gyri and Abnormal Cortical Architecture. Nat. Genet. 2010, 42, 1015–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasser, H.; Vera, L.; Elmaleh-Bergès, M.; Steindl, K.; Letard, P.; Teissier, N.; Ernault, A.; Guimiot, F.; Afenjar, A.; Moutard, M.L.; et al. CDK5RAP2 Primary Microcephaly Is Associated with Hypothalamic, Retinal and Cochlear Developmental Defects. J. Med. Genet. 2020, 57, 389–399. [Google Scholar] [CrossRef]
- Gotz, M.; Huttner, W.B. The Cell Biology of Neurogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Lui, J.H.; Hansen, D.V.; Kriegstein, A.R. Development and Evolution of the Human Neocortex. Cell 2011, 146, 18–36. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, D.; Bae, B.I.; Walsh, C.A. The Genetics of Primary Microcephaly. Annu. Rev. Genom. Hum. Genet. 2018, 19, 177–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conduit, P.T.; Wainman, A.; Raff, J.W. Centrosome Function and Assembly in Animal Cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A.; Holland, A.J. Once and Only Once: Mechanisms of Centriole Duplication and Their Deregulation in Disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 297–312. [Google Scholar] [CrossRef]
- Iwata, R.; Vanderhaeghen, P. Regulatory Roles of Mitochondria and Metabolism in Neurogenesis. Curr. Opin. Neurobiol. 2021, 69, 231–240. [Google Scholar] [CrossRef]
- Saade, M.; Blanco-Ameijeiras, J.; Gonzalez-Gobartt, E.; Martí, E. A Centrosomal View of CNS Growth. Development 2018, 145, dev170613. [Google Scholar] [CrossRef] [Green Version]
- Chavali, P.L.; Putz, M.; Gergely, F. Small Organelle, Big Responsibility: The Role of Centrosomes in Development and Disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130468. [Google Scholar] [CrossRef] [Green Version]
- Marthiens, V.; Basto, R. Centrosomes: The Good and the Bad for Brain Development. Biol. Cell 2020, 112, 153–172. [Google Scholar] [CrossRef]
- Yang, J.; Hu, X.; Ma, J.; Shi, S.-H. Centrosome Regulation and Function in Mammalian Cortical Neurogenesis. Curr. Opin. Neurobiol. 2021, 69, 256–266. [Google Scholar] [CrossRef]
- Anjur-Dietrich, M.I.; Kelleher, C.P.; Needleman, D.J. Mechanical Mechanisms of Chromosome Segregation. Cells 2021, 10, 465. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Limeta, A.; Loncarek, J. Human Centrosome Organization and Function in Interphase and Mitosis. Semin. Cell Dev. Biol. 2021, 117, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Barisic, M.; Rajendraprasad, G.; Steblyanko, Y. The Metaphase Spindle at Steady State–Mechanism and Functions of Microtubule Poleward Flux. Semin. Cell Dev. Biol. 2021, 117, 99–117. [Google Scholar] [CrossRef] [PubMed]
- Prosser, S.L.; Pelletier, L. Mitotic Spindle Assembly in Animal Cells: A Fine Balancing Act. Nat. Rev. Mol. Cell Biol. 2017, 18, 187–201. [Google Scholar] [CrossRef]
- Storchova, Z. Consequences of Mitotic Failure–The Penalties and the Rewards. Semin. Cell Dev. Biol. 2021, 117, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Pan, Y.; Auger, N.; Sun, W.; Dai, L.; Li, S.; Xie, S.; Wen, S.W.; Chen, D. Small Head Circumference at Birth: An 8-Year Retrospective Cohort Study in China. BMJ Paediatr. Open 2019, 3, e000470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passemard, S.; Perez, F.; Colin-Lemesre, E.; Rasika, S.; Gressens, P.; El Ghouzzi, V. Golgi Trafficking Defects in Postnatal Microcephaly: The Evidence for “Golgipathies”. Prog. Neurobiol. 2017, 153, 46–63. [Google Scholar] [CrossRef] [Green Version]
- Gilmore, E.C.; Walsh, C.A. Genetic Causes of Microcephaly and Lessons for Neuronal Development. Wiley Interdiscip. Rev. Dev. Biol. 2013, 2, 461–478. [Google Scholar] [CrossRef]
- Oegema, R.; Barakat, T.S.; Wilke, M.; Stouffs, K.; Amrom, D.; Aronica, E.; Bahi-Buisson, N.; Conti, V.; Fry, A.E.; Geis, T.; et al. International Consensus Recommendations on the Diagnostic Work-up for Malformations of Cortical Development. Nat. Rev. Neurol. 2020, 16, 618–635. [Google Scholar] [CrossRef]
- Helmut, P.G. Seckel Bird-Headed Dwarfs: Studies in Developemental Anthropology Including Human Proportions; S. Karger AG: Basel, Switzerland, 1960. [Google Scholar]
- Majewski, F.; Ranke, M.; Schinzel, A. Studies of Microcephalic Primordial Dwarfism II: The Osteodysplastic Type II of Primordial Dwarfism. Am. J. Med. Genet. 1982, 12, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Boles, R.G.; Teebi, A.S.; Schwartz, D.; Harper, J.F. Further Delineation of the Ear, Patella, Short Stature Syndrome (Meier-Gorlin Syndrome). Clin. Dysmorphol. 1994, 3, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Rauch, A.; Thiel, C.T.; Schindler, D.; Wick, U.; Crow, Y.J.; Ekici, A.B.; van Essen, A.J.; Goecke, T.O.; Al-Gazali, L.; Chrzanowska, K.H.; et al. Mutations in the Pericentrin (PCNT) Gene Cause Primordial Dwarfism. Science 2008, 319, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Griffith, E.; Walker, S.; Martin, C.A.; Vagnarelli, P.; Stiff, T.; Vernay, B.; Al Sanna, N.; Saggar, A.; Hamel, B.; Earnshaw, W.C.; et al. Mutations in Pericentrin Cause Seckel Syndrome with Defective ATR-Dependent DNA Damage Signaling. Nat. Genet. 2008, 40, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bicknell, L.S.; Bongers, E.M.H.F.; Leitch, A.; Brown, S.; Schoots, J.; Harley, M.E.; Aftimos, S.; Al-Aama, J.Y.; Bober, M.; Brown, P.A.J.; et al. Mutations in the Pre-Replication Complex Cause Meier-Gorlin Syndrome. Nat. Genet. 2011, 43, 356–359. [Google Scholar] [CrossRef] [Green Version]
- Ellis, N.A.; German, J. Molecular Genetics of Bloom’s Syndrome. Hum. Mol. Genet. 1996, 5, 1457–1463. [Google Scholar] [CrossRef] [Green Version]
- O’Driscoll, M.; Cerosaletti, K.M.; Girard, P.M.; Dai, Y.; Stumm, M.; Kysela, B.; Hirsch, B.; Gennery, A.; Palmer, S.E.; Seidel, J.; et al. DNA Ligase IV Mutations Identified in Patients Exhibiting Developmental Delay and Immunodeficiency. Mol. Cell 2001, 8, 1175–1185. [Google Scholar] [CrossRef]
- Murray, J.E.; van der Burg, M.; IJspeert, H.; Carroll, P.; Wu, Q.; Ochi, T.; Leitch, A.; Miller, E.S.; Kysela, B.; Jawad, A.; et al. Mutations in the NHEJ Component XRCC4 Cause Primordial Dwarfism. Am. J. Hum. Genet. 2015, 96, 412–424. [Google Scholar] [CrossRef] [Green Version]
- Shurygina, M.F.; Simonett, J.M.; Parker, M.A.; Mitchell, A.; Grigorian, F.; Lifton, J.; Nagiel, A.; Shpak, A.A.; Dadali, E.L.; Mishina, I.A.; et al. Genotype Phenotype Correlation and Variability in Microcephaly Associated With Chorioretinopathy or Familial Exudative Vitreoretinopathy. Investig. Ophthalmol. Vis. Sci. 2020, 61, 2. [Google Scholar] [CrossRef]
- Morton, C.C.; Nance, W.E. Newborn Hearing Screening—A Silent Revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Wentland, C.J.; Ronner, E.A.; Basonbul, R.A.; Pinnapureddy, S.; Mankarious, L.; Keamy, D.; Lee, D.J.; Cohen, M.S. Utilization of Diagnostic Testing for Pediatric Sensorineural Hearing Loss. Int. J. Pediatr. Otorhinolaryngol. 2018, 111, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Teissier, N.; Van Den Abbeele, T.; Sebag, G.; Elmaleh-Berges, M. Computed Tomography Measurements of the Normal and the Pathologic Cochlea in Children. Pediatr. Radiol. 2010, 40, 275–283. [Google Scholar] [CrossRef]
- Birtel, J.; Gliem, M.; Mangold, E.; Tebbe, L.; Spier, I.; Müller, P.L.; Holz, F.G.; Neuhaus, C.; Wolfrum, U.; Bolz, H.J.; et al. Novel Insights Into the Phenotypical Spectrum of KIF11-Associated Retinopathy, Including a New Form of Retinal Ciliopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3950–3959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, C.A.; Ahmad, I.; Klingseisen, A.; Hussain, M.S.; Bicknell, L.S.; Leitch, A.; Nurnberg, G.; Toliat, M.R.; Murray, J.E.; Hunt, D.; et al. Mutations in PLK4, Encoding a Master Regulator of Centriole Biogenesis, Cause Microcephaly, Growth Failure and Retinopathy. Nat. Genet. 2014, 46, 1283–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheidecker, S.; Etard, C.; Haren, L.; Stoetzel, C.; Hull, S.; Arno, G.; Plagnol, V.; Drunat, S.; Passemard, S.; Toutain, A.; et al. Mutations in TUBGCP4 Alter Microtubule Organization via the Gamma-Tubulin Ring Complex in Autosomal-Recessive Microcephaly with Chorioretinopathy. Am. J. Hum. Genet. 2015, 96, 666–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaheen, R.; Al Tala, S.; Almoisheer, A.; Alkuraya, F.S. Mutation in PLK4, Encoding a Master Regulator of Centriole Formation, Defines a Novel Locus for Primordial Dwarfism. J. Med. Genet. 2014, 51, 814–816. [Google Scholar] [CrossRef]
- Duerinckx, S.; Désir, J.; Perazzolo, C.; Badoer, C.; Jacquemin, V.; Soblet, J.; Maystadt, I.; Tunca, Y.; Blaumeiser, B.; Ceulemans, B.; et al. Phenotypes and Genotypes in Non-Consanguineous and Consanguineous Primary Microcephaly: High Incidence of Epilepsy. Mol. Genet. Genom. Med. 2021, 9, e1768. [Google Scholar] [CrossRef]
- Verloes, A.; Drunat, S.; Passemard, S. ASPM Primary Microcephaly. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Letard, P.; Drunat, S.; Vial, Y.; Duerinckx, S.; Ernault, A.; Amram, D.; Arpin, S.; Bertoli, M.; Busa, T.; Ceulemans, B.; et al. Autosomal Recessive Primary Microcephaly Due to ASPM Mutations: An Update. Hum. Mutat. 2018, 39, 319–332. [Google Scholar] [CrossRef]
- Woods, C.G.; Parker, A. Investigating Microcephaly. Arch. Dis. Child. 2013, 98, 707–713. [Google Scholar] [CrossRef]
- Ruaud, L.; Drunat, S.; Elmaleh-Bergès, M.; Ernault, A.; Guilmin Crepon, S.; The MCPH Consortium; El Ghouzzi, V.; Auvin, S.; Verloes, A.; Passemard, S.; et al. Neurological Outcome in WDR62 Primary Microcephaly. Dev. Med. Child Neurol. 2022, 64, 509–517. [Google Scholar] [CrossRef]
- Higgins, J.; Midgley, C.; Bergh, A.M.; Bell, S.M.; Askham, J.M.; Roberts, E.; Binns, R.K.; Sharif, S.M.; Bennett, C.; Glover, D.M.; et al. Human ASPM Participates in Spindle Organisation, Spindle Orientation and Cytokinesis. BMC Cell Biol. 2010, 11, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakefield, J.G.; Bonaccorsi, S.; Gatti, M. The Drosophila Protein Asp Is Involved in Microtubule Organization during Spindle Formation and Cytokinesis. J. Cell Biol. 2001, 153, 637–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, K.; Rezabkova, L.; Hua, S.; Liu, Q.; Capitani, G.; Altelaar, A.F.M.; Heck, A.J.R.; Kammerer, R.A.; Steinmetz, M.O.; Akhmanova, A. Microtubule Minus-End Regulation at Spindle Poles by an ASPM-Katanin Complex. Nat. Cell Biol. 2017, 19, 480–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhargav, D.S.; Sreedevi, N.; Swapna, N.; Vivek, S.; Kovvali, S. Whole Exome Sequencing Identifies a Novel Homozygous Frameshift Mutation in the ASPM Gene, Which Causes Microcephaly 5, Primary, Autosomal Recessive. F1000Res 2017, 6, 2163. [Google Scholar] [CrossRef] [Green Version]
- Weitensteiner, V.; Zhang, R.; Bungenberg, J.; Marks, M.; Gehlen, J.; Ralser, D.J.; Hilger, A.C.; Sharma, A.; Schumacher, J.; Gembruch, U.; et al. Exome Sequencing in Syndromic Brain Malformations Identifies Novel Mutations in ACTB, and SLC9A6, and Suggests BAZ1A as a New Candidate Gene. Birth Defects Res. 2018, 110, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Wang, R.; Han, S.; Ahmad, W.; Zhang, X. Identification of a Novel Nonsense ASPM Mutation in a Large Consanguineous Pakistani Family Using Targeted Next-Generation Sequencing. Genet. Test. Mol. Biomark. 2018, 22, 159–164. [Google Scholar] [CrossRef]
- Marakhonov, A.V.; Konovalov, F.A.; Makaov, A.K.; Vasilyeva, T.A.; Kadyshev, V.V.; Galkina, V.A.; Dadali, E.L.; Kutsev, S.I.; Zinchenko, R.A. Primary Microcephaly Case from the Karachay-Cherkess Republic Poses an Additional Support for Microcephaly and Seckel Syndrome Spectrum Disorders. BMC Med. Genom. 2018, 11, 8. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, N.; Kohmoto, T.; Naruto, T.; Masuda, K.; Imoto, I. Primary Microcephaly Caused by Novel Compound Heterozygous Mutations in ASPM. Hum. Genome Var. 2018, 5, 18015. [Google Scholar] [CrossRef] [Green Version]
- Duerinckx, S.; Jacquemin, V.; Drunat, S.; Vial, Y.; Passemard, S.; Perazzolo, C.; Massart, A.; Soblet, J.; Racapé, J.; Desmyter, L.; et al. Digenic Inheritance of Human Primary Microcephaly Delineates Centrosomal and Non-Centrosomal Pathways. Hum. Mutat. 2019, 41, 512–524. [Google Scholar] [CrossRef] [Green Version]
- Bazgir, A.; Agha Gholizadeh, M.; Sarvar, F.; Pakzad, Z. A Novel Frameshift Mutation in Abnormal Spindle-Like Microcephaly (ASPM) Gene in an Iranian Patient with Primary Microcephaly: A Case Report. Iran. J. Public Health 2019, 48, 2074–2078. [Google Scholar] [CrossRef] [PubMed]
- Rasool, S.; Baig, J.M.; Moawia, A.; Ahmad, I.; Iqbal, M.; Waseem, S.S.; Asif, M.; Abdullah, U.; Makhdoom, E.U.H.; Kaygusuz, E.; et al. An Update of Pathogenic Variants in ASPM, WDR62, CDK5RAP2, STIL, CENPJ, and CEP135 Underlying Autosomal Recessive Primary Microcephaly in 32 Consanguineous Families from Pakistan. Mol. Genet. Genom. Med. 2020, 8, e1408. [Google Scholar] [CrossRef]
- Naseer, M.I.; Abdulkareem, A.A.; Muthaffar, O.Y.; Sogaty, S.; Alkhatabi, H.; Almaghrabi, S.; Chaudhary, A.G. Whole Exome Sequencing Identifies Three Novel Mutations in the ASPM Gene From Saudi Families Leading to Primary Microcephaly. Front. Pediatr. 2020, 8, 627122. [Google Scholar] [CrossRef] [PubMed]
- Makhdoom, E.U.H.; Waseem, S.S.; Iqbal, M.; Abdullah, U.; Hussain, G.; Asif, M.; Budde, B.; Höhne, W.; Tinschert, S.; Saadi, S.M.; et al. Modifier Genes in Microcephaly: A Report on WDR62, CEP63, RAD50 and PCNT Variants Exacerbating Disease Caused by Biallelic Mutations of ASPM and CENPJ. Genes 2021, 12, 731. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.M.; Hussain, B.; Zheng, C.; Khan, A.; Masoud, M.S.; Gu, Q.; Qiu, L.; Malik, N.A.; Qasim, M.; Tariq, M.; et al. Updates on Clinical and Genetic Heterogeneity of ASPM in 12 Autosomal Recessive Primary Microcephaly Families in Pakistani Population. Front. Pediatr. 2021, 9, 695133. [Google Scholar] [CrossRef]
- Batool, T.; Irshad, S.; Mahmood, K. Novel Pathogenic Mutation Mapping of ASPM Gene in Consanguineous Pakistani Families with Primary Microcephaly. Braz. J. Biol. 2021, 83, e246040. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.H.; Diep, Q.M.; Cao, M.H.; Luong, L.H.; Pham, V.A.; Lan Dinh, O.T.; Bui, T.-H.; Van Ta, T.; Tran, V.K. Microcephaly Primary Hereditary (MCPH): Report of Novel ASPM Variants and Prenatal Diagnosis in a Vietnamese Family. Taiwan J. Obstet. Gynecol. 2021, 60, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhang, W.; Zhou, R.; Huang, H.; Chen, W.; Xiang, W.; Liu, L.; Song, J. Two Novel Truncating Variants of the ASPM Gene Identified in a Nonconsanguineous Chinese Family Associated with Primary Microcephaly. Clin. Dysmorphol. 2022, 31, 1–5. [Google Scholar] [CrossRef]
- Hussain, S.; Nawaz, A.; Hamid, M.; Ullah, W.; Khan, I.N.; Afshan, M.; Rehman, A.; Nawaz, H.; Halswick, J.; Rehman, S.-U.; et al. Mutation Screening of Multiple Pakistani MCPH Families Revealed Novel and Recurrent Protein-Truncating Mutations of ASPM. Biotechnol. Appl. Biochem. 2022, 69, 2296–2303. [Google Scholar] [CrossRef]
- Makhdoom, E.U.H.; Anwar, H.; Baig, S.M.; Hussain, G. Whole Exome Sequencing Identifies a Novel Mutation in ASPM and Ultra-Rare Mutation in CDK5RAP2 Causing Primary Microcephaly in Consanguineous Pakistani Families. Pak. J. Med. Sci. 2022, 38, 84–89. [Google Scholar] [CrossRef]
- Naqvi, S.F.; Shabbir, R.M.K.; Tolun, A.; Basit, S.; Malik, S. A Two-Base Pair Deletion in IQ Repeats in ASPM Underlies Microcephaly in a Pakistani Family. Genet. Test. Mol. Biomark. 2022, 26, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Türkyılmaz, A.; Sager, S.G. Two New Cases of Primary Microcephaly with Neuronal Migration Defect Caused by Truncating Mutations in the ASPM Gene. Mol. Syndromol. 2022, 13, 56–63. [Google Scholar] [CrossRef]
- Li, M.; Luo, J.; Yang, Q.; Chen, F.; Chen, J.; Qin, J.; He, W.; Chen, J.; Yi, S.; Qin, Z.; et al. Novel and Recurrent ASPM Mutations of Founder Effect in Chinese Population. Brain Dev. 2022, 44, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Kouprina, N.; Pavlicek, A.; Collins, N.K.; Nakano, M.; Noskov, V.N.; Ohzeki, J.; Mochida, G.H.; Risinger, J.I.; Goldsmith, P.; Gunsior, M.; et al. The Microcephaly ASPM Gene Is Expressed in Proliferating Tissues and Encodes for a Mitotic Spindle Protein. Hum. Mol. Genet. 2005, 14, 2155–2165. [Google Scholar] [CrossRef] [PubMed]
- Passemard, S.; Verloes, A.; Billette de Villemeur, T.; Boespflug-Tanguy, O.; Hernandez, K.; Laurent, M.; Isidor, B.; Alberti, C.; Pouvreau, N.; Drunat, S.; et al. Abnormal Spindle-like Microcephaly-Associated (ASPM) Mutations Strongly Disrupt Neocortical Structure but Spare the Hippocampus and Long-Term Memory. Cortex 2016, 74, 158–176. [Google Scholar] [CrossRef]
- Passemard, S.; Titomanlio, L.; Elmaleh, M.; Afenjar, A.; Alessandri, J.L.; Andria, G.; de Villemeur, T.B.; Boespflug-Tanguy, O.; Burglen, L.; Del Giudice, E.; et al. Expanding the Clinical and Neuroradiologic Phenotype of Primary Microcephaly Due to ASPM Mutations. Neurology 2009, 73, 962–969. [Google Scholar] [CrossRef]
- Hu, H.; Suckow, V.; Musante, L.; Roggenkamp, V.; Kraemer, N.; Ropers, H.H.; Hubner, C.; Wienker, T.F.; Kaindl, A.M. Previously Reported New Type of Autosomal Recessive Primary Microcephaly Is Caused by Compound Heterozygous ASPM Gene Mutations. Cell Cycle 2014, 13, 1650–1651. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Liang, Z.; Guan, C.; Hua, S.; Jiang, K. WDR62 Regulates Spindle Dynamics as an Adaptor Protein between TPX2/Aurora A and Katanin. J. Cell Biol. 2021, 220, e202007167. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, S.-L.; Yang, M.; Herrlinger, S.; Shao, Q.; Collar, J.L.; Fierro, E.; Shi, Y.; Liu, A.; Lu, H.; et al. Modeling Microcephaly with Cerebral Organoids Reveals a WDR62-CEP170-KIF2A Pathway Promoting Cilium Disassembly in Neural Progenitors. Nat. Commun. 2019, 10, 2612. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-F.; Zhang, Y.; Wilde, J.; Hansen, K.C.; Lai, F.; Niswander, L. Microcephaly Disease Gene Wdr62 Regulates Mitotic Progression of Embryonic Neural Stem Cells and Brain Size. Nat. Commun. 2014, 5, 3885. [Google Scholar] [CrossRef] [Green Version]
- Sgourdou, P.; Mishra-Gorur, K.; Saotome, I.; Henagariu, O.; Tuysuz, B.; Campos, C.; Ishigame, K.; Giannikou, K.; Quon, J.L.; Sestan, N.; et al. Disruptions in Asymmetric Centrosome Inheritance and WDR62-Aurora Kinase B Interactions in Primary Microcephaly. Sci. Rep. 2017, 7, 43708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verloes, A.; Ruaud, L.; Drunat, S.; Passemard, S. WDR62 Primary Microcephaly. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington, Seattle: Seattle WA, USA, 1993. [Google Scholar]
- Aryan, H.; Zokaei, S.; Farhud, D.; Keykhaei, M.; Ashrafi, M.R.; Rasulinezhad, M.; Hosseini, S.M.M.; Razmara, E.; Tavasoli, A.R. Novel Phenotype and Genotype Spectrum of WDR62 in Two Patients with Associated Primary Autosomal Recessive Microcephaly. Ir. J. Med. Sci. 2022, 191, 2733–2741. [Google Scholar] [CrossRef]
- Bhat, V.; Girimaji, S.C.; Mohan, G.; Arvinda, H.R.; Singhmar, P.; Duvvari, M.R.; Kumar, A. Mutations in WDR62, Encoding a Centrosomal and Nuclear Protein, in Indian Primary Microcephaly Families with Cortical Malformations. Clin. Genet. 2011, 80, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Murdock, D.R.; Clark, G.D.; Bainbridge, M.N.; Newsham, I.; Wu, Y.-Q.; Muzny, D.M.; Cheung, S.W.; Gibbs, R.A.; Ramocki, M.B. Whole-Exome Sequencing Identifies Compound Heterozygous Mutations in WDR62 in Siblings with Recurrent Polymicrogyria. Am. J. Med. Genet. A 2011, 155A, 2071–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kousar, R.; Hassan, M.J.; Khan, B.; Basit, S.; Mahmood, S.; Mir, A.; Ahmad, W.; Ansar, M. Mutations in WDR62 Gene in Pakistani Families with Autosomal Recessive Primary Microcephaly. BMC Neurol. 2011, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Poulton, C.J.; Schot, R.; Seufert, K.; Lequin, M.H.; Accogli, A.; Annunzio, G.D.; Villard, L.; Philip, N.; de Coo, R.; Catsman-Berrevoets, C.; et al. Severe Presentation of WDR62 Mutation: Is There a Role for Modifying Genetic Factors? Am. J. Med. Genet. A 2014, 164A, 2161–2171. [Google Scholar] [CrossRef]
- Bastaki, F.; Mohamed, M.; Nair, P.; Saif, F.; Tawfiq, N.; Aithala, G.; El-Halik, M.; Al-Ali, M.; Hamzeh, A.R. Novel Splice-Site Mutation in WDR62 Revealed by Whole-Exome Sequencing in a Sudanese Family with Primary Microcephaly. Congenit. Anom. 2016, 56, 135–137. [Google Scholar] [CrossRef] [Green Version]
- Kvarnung, M.; Taylan, F.; Nilsson, D.; Anderlid, B.-M.; Malmgren, H.; Lagerstedt-Robinson, K.; Holmberg, E.; Burstedt, M.; Nordenskjöld, M.; Nordgren, A.; et al. Genomic Screening in Rare Disorders: New Mutations and Phenotypes, Highlighting ALG14 as a Novel Cause of Severe Intellectual Disability. Clin. Genet. 2018, 94, 528–537. [Google Scholar] [CrossRef]
- Zombor, M.; Kalmár, T.; Nagy, N.; Berényi, M.; Telcs, B.; Maróti, Z.; Brandau, O.; Sztriha, L. A Novel WDR62 Missense Mutation in Microcephaly with Abnormal Cortical Architecture and Review of the Literature. J. Appl. Genet. 2019, 60, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Sajid Hussain, M.; Marriam Bakhtiar, S.; Farooq, M.; Anjum, I.; Janzen, E.; Reza Toliat, M.; Eiberg, H.; Kjaer, K.W.; Tommerup, N.; Noegel, A.A.; et al. Genetic Heterogeneity in Pakistani Microcephaly Families. Clin. Genet. 2013, 83, 446–451. [Google Scholar] [CrossRef]
- Memon, M.M.; Raza, S.I.; Basit, S.; Kousar, R.; Ahmad, W.; Ansar, M. A Novel WDR62 Mutation Causes Primary Microcephaly in a Pakistani Family. Mol. Biol. Rep. 2013, 40, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Farag, H.G.; Froehler, S.; Oexle, K.; Ravindran, E.; Schindler, D.; Staab, T.; Huebner, A.; Kraemer, N.; Chen, W.; Kaindl, A.M. Abnormal Centrosome and Spindle Morphology in a Patient with Autosomal Recessive Primary Microcephaly Type 2 Due to Compound Heterozygous WDR62 Gene Mutation. Orphanet J. Rare Dis. 2013, 8, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonell, L.M.; Warman Chardon, J.; Schwartzentruber, J.; Foster, D.; Beaulieu, C.L.; FORGE Canada Consortium; Majewski, J.; Bulman, D.E.; Boycott, K.M. The Utility of Exome Sequencing for Genetic Diagnosis in a Familial Microcephaly Epilepsy Syndrome. BMC Neurol. 2014, 14, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Khan, A.; Han, S.; Zhang, X. Molecular Analysis of 23 Pakistani Families with Autosomal Recessive Primary Microcephaly Using Targeted Next-Generation Sequencing. J. Hum. Genet. 2017, 62, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Nardello, R.; Fontana, A.; Antona, V.; Beninati, A.; Mangano, G.D.; Stallone, M.C.; Mangano, S. A Novel Mutation of WDR62 Gene Associated with Severe Phenotype Including Infantile Spasm, Microcephaly, and Intellectual Disability. Brain Dev. 2018, 40, 58–64. [Google Scholar] [CrossRef]
- Poirier, K.; Lebrun, N.; Broix, L.; Tian, G.; Saillour, Y.; Boscheron, C.; Parrini, E.; Valence, S.; Pierre, B.S.; Oger, M.; et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A Cause Malformations of Cortical Development and Microcephaly. Nat. Genet. 2013, 45, 639–647. [Google Scholar] [CrossRef]
- Strickland, A.V.; Schabhüttl, M.; Offenbacher, H.; Synofzik, M.; Hauser, N.S.; Brunner-Krainz, M.; Gruber-Sedlmayr, U.; Moore, S.A.; Windhager, R.; Bender, B.; et al. Mutation Screen Reveals Novel Variants and Expands the Phenotypes Associated with DYNC1H1. J. Neurol. 2015, 262, 2124–2134. [Google Scholar] [CrossRef] [Green Version]
- Weedon, M.N.; Hastings, R.; Caswell, R.; Xie, W.; Paszkiewicz, K.; Antoniadi, T.; Williams, M.; King, C.; Greenhalgh, L.; Newbury-Ecob, R.; et al. Exome Sequencing Identifies a DYNC1H1 Mutation in a Large Pedigree with Dominant Axonal Charcot-Marie-Tooth Disease. Am. J. Hum. Genet. 2011, 89, 308–312. [Google Scholar] [CrossRef] [Green Version]
- Willemsen, M.H.; Vissers, L.E.L.; Willemsen, M.A.A.P.; van Bon, B.W.M.; Kroes, T.; de Ligt, J.; de Vries, B.B.; Schoots, J.; Lugtenberg, D.; Hamel, B.C.J.; et al. Mutations in DYNC1H1 Cause Severe Intellectual Disability with Neuronal Migration Defects. J. Med. Genet. 2012, 49, 179–183. [Google Scholar] [CrossRef]
- Beecroft, S.J.; McLean, C.A.; Delatycki, M.B.; Koshy, K.; Yiu, E.; Haliloglu, G.; Orhan, D.; Lamont, P.J.; Davis, M.R.; Laing, N.G.; et al. Expanding the Phenotypic Spectrum Associated with Mutations of DYNC1H1. Neuromuscul. Disord. 2017, 27, 607–615. [Google Scholar] [CrossRef]
- Niu, Q.; Wang, X.; Shi, M.; Jin, Q. A Novel DYNC1H1 Mutation Causing Spinal Muscular Atrophy with Lower Extremity Predominance. Neurol. Genet. 2015, 1, e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, D.; Chen, Z.; Li, K.; Long, Z.; Ye, W.; Tang, Z.; Xia, K.; Qiu, R.; Tang, B.; Jiang, H. Identification of a de Novo DYNC1H1 Mutation via WES According to Published Guidelines. Sci. Rep. 2016, 6, 20423. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Liu, Z.; Li, X.; Li, F.; Hu, Y.; Chen, B.; Wang, Z.; Liu, Y. Whole-Exome Sequencing Identifies a Novel de Novo Mutation in DYNC1H1 in Epileptic Encephalopathies. Sci. Rep. 2017, 7, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.H.S.; van Alfen, N.; Thuestad, I.J.; Ip, J.; Chan, A.O.-K.; Mak, C.; Chung, B.H.-Y.; Verrips, A.; Kamsteeg, E.-J. A Recurrent de Novo DYNC1H1 Tail Domain Mutation Causes Spinal Muscular Atrophy with Lower Extremity Predominance, Learning Difficulties and Mild Brain Abnormality. Neuromuscul. Disord. 2018, 28, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Gelineau-Morel, R.; Lukacs, M.; Weaver, K.N.; Hufnagel, R.B.; Gilbert, D.L.; Stottmann, R.W. Congenital Cataracts and Gut Dysmotility in a DYNC1H1 Dyneinopathy Patient. Genes 2016, 7, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Xu, Y.; Li, G.; Li, N.; Yu, T.; Yao, R.-E.; Wang, X.; Shen, Y.; Wang, J. Exome Sequencing Identifies De Novo DYNC1H1 Mutations Associated With Distal Spinal Muscular Atrophy and Malformations of Cortical Development. J. Child Neurol. 2017, 32, 379–386. [Google Scholar] [CrossRef]
- Laquerriere, A.; Maillard, C.; Cavallin, M.; Chapon, F.; Marguet, F.; Molin, A.; Sigaudy, S.; Blouet, M.; Benoist, G.; Fernandez, C.; et al. Neuropathological Hallmarks of Brain Malformations in Extreme Phenotypes Related to DYNC1H1 Mutations. J. Neuropathol. Exp. Neurol. 2017, 76, 195–205. [Google Scholar] [CrossRef]
- Hertecant, J.; Komara, M.; Nagi, A.; Suleiman, J.; Al-Gazali, L.; Ali, B.R. A Novel de Novo Mutation in DYNC1H1 Gene Underlying Malformation of Cortical Development and Cataract. Meta Gene 2016, 9, 124–127. [Google Scholar] [CrossRef]
- Peeters, K.; Bervoets, S.; Chamova, T.; Litvinenko, I.; De Vriendt, E.; Bichev, S.; Kancheva, D.; Mitev, V.; Kennerson, M.; Timmerman, V.; et al. Novel Mutations in the DYNC1H1 Tail Domain Refine the Genetic and Clinical Spectrum of Dyneinopathies. Hum. Mutat. 2015, 36, 287–291. [Google Scholar] [CrossRef]
- Tsurusaki, Y.; Saitoh, S.; Tomizawa, K.; Sudo, A.; Asahina, N.; Shiraishi, H.; Ito, J.-I.; Tanaka, H.; Doi, H.; Saitsu, H.; et al. A DYNC1H1 Mutation Causes a Dominant Spinal Muscular Atrophy with Lower Extremity Predominance. Neurogenetics 2012, 13, 327–332. [Google Scholar] [CrossRef]
- Das, J.; Lilleker, J.B.; Jabbal, K.; Ealing, J. A Missense Mutation in DYNC1H1 Gene Causing Spinal Muscular Atrophy–Lower Extremity, Dominant. Neurol. Neurochir. Pol. 2018, 52, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Punetha, J.; Monges, S.; Franchi, M.E.; Hoffman, E.P.; Cirak, S.; Tesi-Rocha, C. Exome Sequencing Identifies DYNC1H1 Variant Associated With Vertebral Abnormality and Spinal Muscular Atrophy With Lower Extremity Predominance. Pediatr. Neurol. 2015, 52, 239–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, M.B.; Ori-McKenney, K.M.; Scoto, M.; Tuck, E.P.; Bell, S.; Ma, D.; Masi, S.; Allred, P.; Al-Lozi, M.; Reilly, M.M.; et al. Mutations in the Tail Domain of DYNC1H1 Cause Dominant Spinal Muscular Atrophy. Neurology 2012, 78, 1714–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zillhardt, J.L.; Poirier, K.; Broix, L.; Lebrun, N.; Elmorjani, A.; Martinovic, J.; Saillour, Y.; Muraca, G.; Nectoux, J.; Bessieres, B.; et al. Mosaic Parental Germline Mutations Causing Recurrent Forms of Malformations of Cortical Development. Eur. J. Hum. Genet. 2016, 24, 611–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorillo, C.; Moro, F.; Yi, J.; Weil, S.; Brisca, G.; Astrea, G.; Severino, M.; Romano, A.; Battini, R.; Rossi, A.; et al. Novel Dynein DYNC1H1 Neck and Motor Domain Mutations Link Distal Spinal Muscular Atrophy and Abnormal Cortical Development. Hum. Mutat. 2014, 35, 298–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scoto, M.; Rossor, A.M.; Harms, M.B.; Cirak, S.; Calissano, M.; Robb, S.; Manzur, A.Y.; Martínez Arroyo, A.; Rodriguez Sanz, A.; Mansour, S.; et al. Novel Mutations Expand the Clinical Spectrum of DYNC1H1-Associated Spinal Muscular Atrophy. Neurology 2015, 84, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Jamuar, S.S.; Lam, A.-T.N.; Kircher, M.; D’Gama, A.M.; Wang, J.; Barry, B.J.; Zhang, X.; Hill, R.S.; Partlow, J.N.; Rozzo, A.; et al. Somatic Mutations in Cerebral Cortical Malformations. N. Engl. J. Med. 2014, 371, 733–743. [Google Scholar] [CrossRef] [Green Version]
- Becker, L.-L.; Dafsari, H.S.; Schallner, J.; Abdin, D.; Seifert, M.; Petit, F.; Smol, T.; Bok, L.; Rodan, L.; Krapels, I.; et al. The Clinical-Phenotype Continuum in DYNC1H1-Related Disorders-Genomic Profiling and Proposal for a Novel Classification. J. Hum. Genet. 2020, 65, 1003–1017. [Google Scholar] [CrossRef]
- Harms, M.B.; Allred, P.; Gardner, R.; Fernandes Filho, J.A.; Florence, J.; Pestronk, A.; Al-Lozi, M.; Baloh, R.H. Dominant Spinal Muscular Atrophy with Lower Extremity Predominance: Linkage to 14q32. Neurology 2010, 75, 539–546. [Google Scholar] [CrossRef]
- Hoang, H.T.; Schlager, M.A.; Carter, A.P.; Bullock, S.L. DYNC1H1 Mutations Associated with Neurological Diseases Compromise Processivity of Dynein-Dynactin-Cargo Adaptor Complexes. Proc. Natl. Acad. Sci. USA 2017, 114, E1597–E1606. [Google Scholar] [CrossRef]
- Lawo, S.; Hasegan, M.; Gupta, G.D.; Pelletier, L. Subdiffraction Imaging of Centrosomes Reveals Higher-Order Organizational Features of Pericentriolar Material. Nat. Cell Biol. 2012, 14, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, J.B.; Wueseke, O.; Hyman, A.A. Pericentriolar Material Structure and Dynamics. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Pelletier, L. Pericentrin: Critical for Spindle Orientation. Curr. Biol. 2014, 24, R962–R964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-T.; Hehnly, H.; Yu, Q.; Farkas, D.; Zheng, G.; Redick, S.D.; Hung, H.-F.; Samtani, R.; Jurczyk, A.; Akbarian, S.; et al. A Unique Set of Centrosome Proteins Requires Pericentrin for Spindle-Pole Localization and Spindle Orientation. Curr. Biol. 2014, 24, 2327–2334. [Google Scholar] [CrossRef] [Green Version]
- Gavilan, M.P.; Gandolfo, P.; Balestra, F.R.; Arias, F.; Bornens, M.; Rios, R.M. The Dual Role of the Centrosome in Organizing the Microtubule Network in Interphase. EMBO Rep. 2018, 19, e45942. [Google Scholar] [CrossRef]
- Pimenta-Marques, A.; Bettencourt-Dias, M. Pericentriolar Material. Curr. Biol. 2020, 30, R687–R689. [Google Scholar] [CrossRef]
- Waich, S.; Janecke, A.R.; Parson, W.; Greber-Platzer, S.; Müller, T.; Huber, L.A.; Valovka, T.; Vodopiutz, J. Novel PCNT Variants in MOPDII with Attenuated Growth Restriction and Pachygyria. Clin. Genet. 2020, 98, 282–287. [Google Scholar] [CrossRef]
- Lorenzo-Betancor, O.; Blackburn, P.R.; Edwards, E.; Vázquez-do-Campo, R.; Klee, E.W.; Labbé, C.; Hodges, K.; Glover, P.; Sigafoos, A.N.; Soto, A.I.; et al. PCNT Point Mutations and Familial Intracranial Aneurysms. Neurology 2018, 91, e2170–e2181. [Google Scholar] [CrossRef]
- Liu, H.; Tao, N.; Wang, Y.; Yang, Y.; He, X.; Zhang, Y.; Zhou, Y.; Liu, X.; Feng, X.; Sun, M.; et al. A Novel Homozygous Mutation of the PCNT Gene in a Chinese Patient with Microcephalic Osteodysplastic Primordial Dwarfism Type II. Mol. Genet. Genom. Med. 2021, 9, e1761. [Google Scholar] [CrossRef]
- Kantaputra, P.; Tanpaiboon, P.; Porntaveetus, T.; Ohazama, A.; Sharpe, P.; Rauch, A.; Hussadaloy, A.; Thiel, C.T. The Smallest Teeth in the World Are Caused by Mutations in the PCNT Gene. Am. J. Med. Genet. A 2011, 155A, 1398–1403. [Google Scholar] [CrossRef]
- Dehghan Tezerjani, M.; Vahidi Mehrjardi, M.Y.; Hozhabri, H.; Rahmanian, M. A Novel PCNT Frame Shift Variant (c.7511delA) Causing Osteodysplastic Primordial Dwarfism of Majewski Type 2 (MOPD II). Front. Pediatr. 2020, 8, 340. [Google Scholar] [CrossRef] [PubMed]
- Willems, M.; Geneviève, D.; Borck, G.; Baumann, C.; Baujat, G.; Bieth, E.; Edery, P.; Farra, C.; Gerard, M.; Héron, D.; et al. Molecular Analysis of Pericentrin Gene (PCNT) in a Series of 24 Seckel/Microcephalic Osteodysplastic Primordial Dwarfism Type II (MOPD II) Families. J. Med. Genet. 2010, 47, 797–802. [Google Scholar] [CrossRef] [Green Version]
- Piane, M.; Della Monica, M.; Piatelli, G.; Lulli, P.; Lonardo, F.; Chessa, L.; Scarano, G. Majewski Osteodysplastic Primordial Dwarfism Type II (MOPD II) Syndrome Previously Diagnosed as Seckel Syndrome: Report of a Novel Mutation of the PCNT Gene. Am. J. Med. Genet. A 2009, 149A, 2452–2456. [Google Scholar] [CrossRef] [PubMed]
- Pachajoa, H.; Ruiz-Botero, F.; Isaza, C. A New Mutation of the PCNT Gene in a Colombian Patient with Microcephalic Osteodysplastic Primordial Dwarfism Type II: A Case Report. J. Med. Case Rep. 2014, 8, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, E.; Dunstheimer, D.; Klammt, J.; Friebe, D.; Kiess, W.; Kratzsch, J.; Kruis, T.; Laue, S.; Pfäffle, R.; Wallborn, T.; et al. Clinical and Functional Characterization of a Patient Carrying a Compound Heterozygous Pericentrin Mutation and a Heterozygous IGF1 Receptor Mutation. PLoS ONE 2012, 7, e38220. [Google Scholar] [CrossRef]
- Bober, M.B.; Niiler, T.; Duker, A.L.; Murray, J.E.; Ketterer, T.; Harley, M.E.; Alvi, S.; Flora, C.; Rustad, C.; Bongers, E.M.H.F.; et al. Growth in Individuals with Majewski Osteodysplastic Primordial Dwarfism Type II Caused by Pericentrin Mutations. Am. J. Med. Genet. A 2012, 158A, 2719–2725. [Google Scholar] [CrossRef]
- Unal, S.; Alanay, Y.; Cetin, M.; Boduroglu, K.; Utine, E.; Cormier-Daire, V.; Huber, C.; Ozsurekci, Y.; Kilic, E.; Simsek Kiper, O.P.; et al. Striking Hematological Abnormalities in Patients with Microcephalic Osteodysplastic Primordial Dwarfism Type II (MOPD II): A Potential Role of Pericentrin in Hematopoiesis. Pediatr. Blood Cancer 2014, 61, 302–305. [Google Scholar] [CrossRef]
- Dieks, J.-K.; Baumer, A.; Wilichowski, E.; Rauch, A.; Sigler, M. Microcephalic Osteodysplastic Primordial Dwarfism Type II (MOPD II) with Multiple Vascular Complications Misdiagnosed as Dubowitz Syndrome. Eur. J. Pediatr. 2014, 173, 1253–1256. [Google Scholar] [CrossRef]
- Li, F.-F.; Wang, X.-D.; Zhu, M.-W.; Lou, Z.-H.; Zhang, Q.; Zhu, C.-Y.; Feng, H.-L.; Lin, Z.-G.; Liu, S.-L. Identification of Two Novel Critical Mutations in PCNT Gene Resulting in Microcephalic Osteodysplastic Primordial Dwarfism Type II Associated with Multiple Intracranial Aneurysms. Metab. Brain Dis. 2015, 30, 1387–1394. [Google Scholar] [CrossRef]
- Weiss, K.; Ekhilevitch, N.; Cohen, L.; Bratman-Morag, S.; Bello, R.; Martinez, A.F.; Hadid, Y.; Shlush, L.I.; Kurolap, A.; Paperna, T.; et al. Identification of a Novel PCNT Founder Pathogenic Variant in the Israeli Druze Population. Eur. J. Med. Genet. 2020, 63, 103643. [Google Scholar] [CrossRef]
- Abdel-Salam, G.M.H.; Sayed, I.S.M.; Afifi, H.H.; Abdel-Ghafar, S.F.; Abouzaid, M.R.; Ismail, S.I.; Aglan, M.S.; Issa, M.Y.; El-Bassyouni, H.T.; El-Kamah, G.; et al. Microcephalic Osteodysplastic Primordial Dwarfism Type II: Additional Nine Patients with Implications on Phenotype and Genotype Correlation. Am. J. Med. Genet. A 2020, 182, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Rossi-Espagnet, M.C.; Dentici, M.L.; Pasquini, L.; Carducci, C.; Lucignani, M.; Longo, D.; Agolini, E.; Novelli, A.; Gonfiantini, M.V.; Digilio, M.C.; et al. Microcephalic Osteodysplastic Primordial Dwarfism Type II and Pachygyria: Morphometric Analysis in a 2-Year-Old Girl. Am. J. Med. Genet. A 2020, 182, 2372–2376. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Xu, Z.; Zhao, J.; Shen, H. Novel Compound Heterozygous Mutations of PCNT Gene in MOPD Type II with Central Precocious Puberty. Gynecol. Endocrinol. 2021, 37, 190–192. [Google Scholar] [CrossRef]
- Shaheen, R.; Maddirevula, S.; Ewida, N.; Alsahli, S.; Abdel-Salam, G.M.H.; Zaki, M.S.; Tala, S.A.; Alhashem, A.; Softah, A.; Al-Owain, M.; et al. Genomic and Phenotypic Delineation of Congenital Microcephaly. Genet. Med. 2019, 21, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Alrajhi, H.; Alallah, J.; Shawli, A.; Alghamdi, K.; Hakami, F. Majewski Dwarfism Type II: An Atypical Neuroradiological Presentation with a Novel Variant in the PCNT Gene. BMJ Case Rep. 2019, 12, e224197. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Tu, C.; Lu, G.; Lin, G.; Tan, Y. Novel Biallelic PCNT Deletion Causing Microcephalic Osteodysplastic Primordial Dwarfism Type II with Congenital Heart Defect. Sci. China Life Sci. 2019, 62, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Huang-Doran, I.; Bicknell, L.S.; Finucane, F.M.; Rocha, N.; Porter, K.M.; Tung, Y.C.L.; Szekeres, F.; Krook, A.; Nolan, J.J.; O’Driscoll, M.; et al. Genetic Defects in Human Pericentrin Are Associated with Severe Insulin Resistance and Diabetes. Diabetes 2011, 60, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.; Kim, S.Y.; Kang, J.; Phi, J.H.; Kim, W.-H.; Yang, S.W.; Kwon, H.W.; Lee, S.Y.; Kim, G.B.; Bae, E.J.; et al. Siblings With Familial Dwarfism Presenting With Acute Myocardial Infarction at Adolescence. JACC Case Rep. 2021, 3, 795–800. [Google Scholar] [CrossRef]
- Eslava, A.; Garcia-Puig, M.; Corripio, R. A 10-Year-Old Boy with Short Stature and Microcephaly, Diagnosed with Moyamoya Syndrome and Microcephalic Osteodysplastic Primordial Dwarfism Type II (MOPD II). Am. J. Case Rep. 2021, 22, e933919. [Google Scholar] [CrossRef]
- Hettiarachchi, D.; Subasinghe, S.M.V.; Anandagoda, G.G.; Panchal, H.; Lai, P.S.; Dissanayake, V.H.W. Novel Frameshift Variant in the PCNT Gene Associated with Microcephalic Osteodysplastic Primordial Dwarfism (MOPD) Type II and Small Kidneys. BMC Med. Genom. 2022, 15, 82. [Google Scholar] [CrossRef]
- Rauch, A. The Shortest of the Short: Pericentrin Mutations and Beyond. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Nguyen, N.-L.; Vu, C.D.; Ngoc, C.T.B.; Nguyen, N.K.; Nguyen, H.H. Identification of Three Novel Mutations in PCNT in Vietnamese Patients with Microcephalic Osteodysplastic Primordial Dwarfism Type II. Genes Genom. 2021, 43, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhou, D.; Dong, N.; Ding, C.; Liu, Y.; Li, F. Clinical and Genetic Analysis of a Patient With Coexisting 17a-Hydroxylase/17,20-Lyase Deficiency and Moyamoya Disease. Front. Genet. 2022, 13, 845016. [Google Scholar] [CrossRef] [PubMed]
- Graser, S.; Stierhof, Y.D.; Nigg, E.A. Cep68 and Cep215 (Cdk5rap2) Are Required for Centrosome Cohesion. J. Cell Sci. 2007, 120, 4321–4331. [Google Scholar] [CrossRef] [Green Version]
- Fong, K.W.; Choi, Y.K.; Rattner, J.B.; Qi, R.Z. CDK5RAP2 Is a Pericentriolar Protein That Functions in Centrosomal Attachment of the {gamma}-Tubulin Ring Complex. Mol. Biol. Cell 2008, 19, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Barr, A.R.; Kilmartin, J.V.; Gergely, F. CDK5RAP2 Functions in Centrosome to Spindle Pole Attachment and DNA Damage Response. J. Cell Biol. 2010, 189, 23–39. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.S.; Park, J.-E.; Ahn, J.I.; Zeng, Y. Constructing PCM with Architecturally Distinct Higher-Order Assemblies. Curr. Opin. Struct. Biol. 2021, 66, 66–73. [Google Scholar] [CrossRef]
- Watanabe, S.; Meitinger, F.; Shiau, A.K.; Oegema, K.; Desai, A. Centriole-Independent Mitotic Spindle Assembly Relies on the PCNT-CDK5RAP2 Pericentriolar Matrix. J. Cell Biol. 2020, 219, e202006010. [Google Scholar] [CrossRef]
- Yigit, G.; Brown, K.E.; Kayserili, H.; Pohl, E.; Caliebe, A.; Zahnleiter, D.; Rosser, E.; Bogershausen, N.; Uyguner, Z.O.; Altunoglu, U.; et al. Mutations in CDK5RAP2 Cause Seckel Syndrome. Mol. Genet. Genom. Med. 2015, 3, 467–480. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral Organoids Model Human Brain Development and Microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Alfares, A.; Alhufayti, I.; Alsubaie, L.; Alowain, M.; Almass, R.; Alfadhel, M.; Kaya, N.; Eyaid, W. A New Association between CDK5RAP2 Microcephaly and Congenital Cataracts. Ann. Hum. Genet. 2018, 82, 165–170. [Google Scholar] [CrossRef]
- Moynihan, L.; Jackson, A.P.; Roberts, E.; Karbani, G.; Lewis, I.; Corry, P.; Turner, G.; Mueller, R.F.; Lench, N.J.; Woods, C.G. A Third Novel Locus for Primary Autosomal Recessive Microcephaly Maps to Chromosome 9q34. Am. J. Hum. Genet. 2000, 66, 724–727. [Google Scholar] [CrossRef] [Green Version]
- Hassan, M.J.; Khurshid, M.; Azeem, Z.; John, P.; Ali, G.; Chishti, M.S.; Ahmad, W. Previously Described Sequence Variant in CDK5RAP2 Gene in a Pakistani Family with Autosomal Recessive Primary Microcephaly. BMC Med. Genet. 2007, 8, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issa, L.; Mueller, K.; Seufert, K.; Kraemer, N.; Rosenkotter, H.; Ninnemann, O.; Buob, M.; Kaindl, A.M.; Morris-Rosendahl, D.J. Clinical and Cellular Features in Patients with Primary Autosomal Recessive Microcephaly and a Novel CDK5RAP2 Mutation. Orphanet J. Rare Dis. 2013, 8, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouan, L.; Ouled Amar Bencheikh, B.; Daoud, H.; Dionne-Laporte, A.; Dobrzeniecka, S.; Spiegelman, D.; Rochefort, D.; Hince, P.; Szuto, A.; Lassonde, M.; et al. Exome Sequencing Identifies Recessive CDK5RAP2 Variants in Patients with Isolated Agenesis of Corpus Callosum. Eur. J. Hum. Genet. 2016, 24, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Pagnamenta, A.T.; Murray, J.E.; Yoon, G.; Sadighi Akha, E.; Harrison, V.; Bicknell, L.S.; Ajilogba, K.; Stewart, H.; Kini, U.; Taylor, J.C.; et al. A Novel Nonsense CDK5RAP2 Mutation in a Somali Child with Primary Microcephaly and Sensorineural Hearing Loss. Am. J. Med. Genet. A 2012, 158A, 2577–2582. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.A.; Topper, S.; Ward Melver, C.; Stein, J.; Reeder, A.; Arndt, K.; Das, S. The First Case of CDK5RAP2-Related Primary Microcephaly in a Non-Consanguineous Patient Identified by next Generation Sequencing. Brain Dev. 2014, 36, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Kakar, N.; Ahmad, J.; Morris-Rosendahl, D.J.; Altmüller, J.; Friedrich, K.; Barbi, G.; Nürnberg, P.; Kubisch, C.; Dobyns, W.B.; Borck, G. STIL Mutation Causes Autosomal Recessive Microcephalic Lobar Holoprosencephaly. Hum. Genet. 2015, 134, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Mouden, C.; de Tayrac, M.; Dubourg, C.; Rose, S.; Carré, W.; Hamdi-Rozé, H.; Babron, M.-C.; Akloul, L.; Héron-Longe, B.; Odent, S.; et al. Homozygous STIL Mutation Causes Holoprosencephaly and Microcephaly in Two Siblings. PLoS ONE 2015, 10, e0117418. [Google Scholar] [CrossRef] [PubMed]
- Hatch, E.M.; Kulukian, A.; Holland, A.J.; Cleveland, D.W.; Stearns, T. Cep152 Interacts with Plk4 and Is Required for Centriole Duplication. J. Cell Biol. 2010, 191, 721–729. [Google Scholar] [CrossRef] [Green Version]
- Cizmecioglu, O.; Arnold, M.; Bahtz, R.; Settele, F.; Ehret, L.; Haselmann-Weiss, U.; Antony, C.; Hoffmann, I. Cep152 Acts as a Scaffold for Recruitment of Plk4 and CPAP to the Centrosome. J. Cell Biol. 2010, 191, 731–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gönczy, P.; Hatzopoulos, G.N. Centriole Assembly at a Glance. J. Cell Sci. 2019, 132, jcs228833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnen, K.F.; Gabryjonczyk, A.-M.; Anselm, E.; Stierhof, Y.-D.; Nigg, E.A. Human Cep192 and Cep152 Cooperate in Plk4 Recruitment and Centriole Duplication. J. Cell Sci. 2013, 126, 3223–3233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avidor-Reiss, T.; Gopalakrishnan, J. Building a Centriole. Curr. Opin. Cell Biol. 2013, 25, 72–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loncarek, J.; Bettencourt-Dias, M. Building the Right Centriole for Each Cell Type. J. Cell Biol. 2018, 217, 823–835. [Google Scholar] [CrossRef]
- Breslow, D.K.; Holland, A.J. Mechanism and Regulation of Centriole and Cilium Biogenesis. Annu. Rev. Biochem. 2019, 88, 691–724. [Google Scholar] [CrossRef]
- Zhang, L.; Teng, Y.; Hu, H.; Zhu, H.; Wen, J.; Liang, D.; Li, Z.; Wu, L. Two Novel Variants in CEP152 Caused Seckel Syndrome 5 in a Chinese Family. Front. Genet. 2022, 13, 1052915. [Google Scholar] [CrossRef]
- Tsutsumi, M.; Yokoi, S.; Miya, F.; Miyata, M.; Kato, M.; Okamoto, N.; Tsunoda, T.; Yamasaki, M.; Kanemura, Y.; Kosaki, K.; et al. Novel Compound Heterozygous Variants in PLK4 Identified in a Patient with Autosomal Recessive Microcephaly and Chorioretinopathy. Eur. J. Hum. Genet. 2016, 24, 1702–1706. [Google Scholar] [CrossRef] [Green Version]
- Martín-Rivada, Á.; Pozo-Román, J.; Güemes, M.; Ortiz-Cabrera, N.V.; Pérez-Jurado, L.A.; Argente, J. Primary Dwarfism, Microcephaly, and Chorioretinopathy Due to a PLK4 Mutation in Two Siblings. Horm. Res. Paediatr. 2020, 93, 567–572. [Google Scholar] [CrossRef]
- Dinçer, T.; Yorgancıoğlu-Budak, G.; Ölmez, A.; Er, İ.; Dodurga, Y.; Özdemir, Ö.M.; Toraman, B.; Yıldırım, A.; Sabir, N.; Akarsu, N.A.; et al. Analysis of Centrosome and DNA Damage Response in PLK4 Associated Seckel Syndrome. Eur. J. Hum. Genet. 2017, 25, 1118–1125. [Google Scholar] [CrossRef]
- Neitzel, H.; Varon, R.; Chughtai, S.; Dartsch, J.; Dutrannoy-Tönsing, V.; Nürnberg, P.; Nürnberg, G.; Schweiger, M.; Digweed, M.; Hildebrand, G.; et al. Transmission Ratio Distortion of Mutations in the Master Regulator of Centriole Biogenesis PLK4. Hum. Genet. 2022, 141, 1785–1794. [Google Scholar] [CrossRef]
- Bornens, M. Centrosome Organization and Functions. Curr. Opin. Struct. Biol. 2021, 66, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Akhmanova, A.; Steinmetz, M.O. Microtubule Minus-End Regulation at a Glance. J. Cell Sci. 2019, 132, jcs227850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Akhmanova, A. Microtubule-Organizing Centers. Annu. Rev. Cell Dev. Biol. 2017, 33, 51–75. [Google Scholar] [CrossRef]
- Borgal, L.; Wakefield, J.G. Context-Dependent Spindle Pole Focusing. Essays Biochem. 2018, 62, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Fraschini, R. Factors That Control Mitotic Spindle Dynamics. Adv. Exp. Med. Biol. 2017, 925, 89–101. [Google Scholar] [CrossRef]
- Goundiam, O.; Basto, R. Centrosomes in Disease: How the Same Music Can Sound so Different? Curr. Opin. Struct. Biol. 2021, 66, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.P.; Holland, A.J. Time Is of the Essence: The Molecular Mechanisms of Primary Microcephaly. Genes Dev. 2021, 35, 1551–1578. [Google Scholar] [CrossRef]
- Gavvovidis, I.; Rost, I.; Trimborn, M.; Kaiser, F.J.; Purps, J.; Wiek, C.; Hanenberg, H.; Neitzel, H.; Schindler, D. A Novel MCPH1 Isoform Complements the Defective Chromosome Condensation of Human MCPH1-Deficient Cells. PLoS ONE 2012, 7, e40387. [Google Scholar] [CrossRef] [Green Version]
- Kraemer, N.; Issa-Jahns, L.; Neubert, G.; Ravindran, E.; Mani, S.; Ninnemann, O.; Kaindl, A.M. Novel Alternative Splice Variants of Mouse Cdk5rap2. PLoS ONE 2015, 10, e0136684. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, M.H.; Wu, X.; Kodani, A.; Fan, J.; Doan, R.; Ozawa, M.; Ma, J.; Yoshida, N.; Reiter, J.F.; et al. Cell-Type-Specific Alternative Splicing Governs Cell Fate in the Developing Cerebral Cortex. Cell 2016, 166, 1147–1162.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knouse, K.A.; Lopez, K.E.; Bachofner, M.; Amon, A. Chromosome Segregation Fidelity in Epithelia Requires Tissue Architecture. Cell 2018, 175, 200–211.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, S.L.; Dobyns, W.B.; Lahn, B.T. Genetic Links between Brain Development and Brain Evolution. Nat. Rev. Genet. 2005, 6, 581–590. [Google Scholar] [CrossRef]
- Mekel-Bobrov, N.; Gilbert, S.L.; Evans, P.D.; Vallender, E.J.; Anderson, J.R.; Hudson, R.R.; Tishkoff, S.A.; Lahn, B.T. Ongoing Adaptive Evolution of ASPM, a Brain Size Determinant in Homo Sapiens. Science 2005, 309, 1720–1722. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.D.; Anderson, J.R.; Vallender, E.J.; Gilbert, S.L.; Malcom, C.M.; Dorus, S.; Lahn, B.T. Adaptive Evolution of ASPM, a Major Determinant of Cerebral Cortical Size in Humans. Hum. Mol. Genet. 2004, 13, 489–494. [Google Scholar] [CrossRef] [Green Version]
- Evans, P.D.; Gilbert, S.L.; Mekel-Bobrov, N.; Vallender, E.J.; Anderson, J.R.; Vaez-Azizi, L.M.; Tishkoff, S.A.; Hudson, R.R.; Lahn, B.T. Microcephalin, a Gene Regulating Brain Size, Continues to Evolve Adaptively in Humans. Science 2005, 309, 1717–1720. [Google Scholar] [CrossRef]
- Timpson, N.; Heron, J.; Smith, G.D.; Enard, W. Comment on Papers by Evans et al. and Mekel-Bobrov et al. on Evidence for Positive Selection of MCPH1 and ASPM. Science 2007, 317, 1036, author reply 1036. [Google Scholar] [CrossRef] [Green Version]
- Mekel-Bobrov, N.; Posthuma, D.; Gilbert, S.L.; Lind, P.; Gosso, M.F.; Luciano, M.; Harris, S.E.; Bates, T.C.; Polderman, T.J.C.; Whalley, L.J.; et al. The Ongoing Adaptive Evolution of ASPM and Microcephalin Is Not Explained by Increased Intelligence. Hum. Mol. Genet. 2007, 16, 600–608. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.K.; Li, Y.; Su, B. A Common SNP of MCPH1 Is Associated with Cranial Volume Variation in Chinese Population. Hum. Mol. Genet. 2008, 17, 1329–1335. [Google Scholar] [CrossRef] [Green Version]
- Dobson-Stone, C.; Gatt, J.M.; Kuan, S.A.; Grieve, S.M.; Gordon, E.; Williams, L.M.; Schofield, P.R. Investigation of MCPH1 G37995C and ASPM A44871G Polymorphisms and Brain Size in a Healthy Cohort. NeuroImage 2007, 37, 394–400. [Google Scholar] [CrossRef]
- Johnson, M.B.; Sun, X.; Kodani, A.; Borges-Monroy, R.; Girskis, K.M.; Ryu, S.C.; Wang, P.P.; Patel, K.; Gonzalez, D.M.; Woo, Y.M.; et al. Aspm Knockout Ferret Reveals an Evolutionary Mechanism Governing Cerebral Cortical Size. Nature 2018, 556, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Fish, J.L.; Kosodo, Y.; Enard, W.; Paabo, S.; Huttner, W.B. Aspm Specifically Maintains Symmetric Proliferative Divisions of Neuroepithelial Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 10438–10443. [Google Scholar] [CrossRef] [PubMed]
- Pulvers, J.N.; Bryk, J.; Fish, J.L.; Wilsch-Brauninger, M.; Arai, Y.; Schreier, D.; Naumann, R.; Helppi, J.; Habermann, B.; Vogt, J.; et al. Mutations in Mouse Aspm (Abnormal Spindle-like Microcephaly Associated) Cause Not Only Microcephaly but Also Major Defects in the Germline. Proc. Natl. Acad. Sci. USA 2010, 107, 16595–16600. [Google Scholar] [CrossRef] [PubMed]
- Lizarraga, S.B.; Margossian, S.P.; Harris, M.H.; Campagna, D.R.; Han, A.P.; Blevins, S.; Mudbhary, R.; Barker, J.E.; Walsh, C.A.; Fleming, M.D. Cdk5rap2 Regulates Centrosome Function and Chromosome Segregation in Neuronal Progenitors. Development 2010, 137, 1907–1917. [Google Scholar] [CrossRef]
- Marthiens, V.; Rujano, M.A.; Pennetier, C.; Tessier, S.; Paul-Gilloteaux, P.; Basto, R. Centrosome Amplification Causes Microcephaly. Nat. Cell Biol. 2013, 15, 731–740. [Google Scholar] [CrossRef]
- Nano, M.; Basto, R. Consequences of Centrosome Dysfunction during Brain Development. Adv. Exp. Med. Biol. 2017, 1002, 19–45. [Google Scholar] [CrossRef]
- Lin, S.Y.; Rai, R.; Li, K.; Xu, Z.X.; Elledge, S.J. BRIT1/MCPH1 Is a DNA Damage Responsive Protein That Regulates the Brca1-Chk1 Pathway, Implicating Checkpoint Dysfunction in Microcephaly. Proc. Natl. Acad. Sci. USA 2005, 102, 15105–15109. [Google Scholar] [CrossRef]
- Alderton, G.K.; Galbiati, L.; Griffith, E.; Surinya, K.H.; Neitzel, H.; Jackson, A.P.; Jeggo, P.A.; O’Driscoll, M. Regulation of Mitotic Entry by Microcephalin and Its Overlap with ATR Signalling. Nat. Cell Biol. 2006, 8, 725–733. [Google Scholar] [CrossRef]
- Gruber, R.; Zhou, Z.; Sukchev, M.; Joerss, T.; Frappart, P.O.; Wang, Z.Q. MCPH1 Regulates the Neuroprogenitor Division Mode by Coupling the Centrosomal Cycle with Mitotic Entry through the Chk1-Cdc25 Pathway. Nat. Cell Biol. 2011, 13, 1325–1334. [Google Scholar] [CrossRef]
- Shao, Z.; Li, F.; Sy, S.M.-H.; Yan, W.; Zhang, Z.; Gong, D.; Wen, B.; Huen, M.S.Y.; Gong, Q.; Wu, J.; et al. Specific Recognition of Phosphorylated Tail of H2AX by the Tandem BRCT Domains of MCPH1 Revealed by Complex Structure. J. Struct. Biol. 2012, 177, 459–468. [Google Scholar] [CrossRef]
- Singh, N.; Wiltshire, T.D.; Thompson, J.R.; Mer, G.; Couch, F.J. Molecular Basis for the Association of Microcephalin (MCPH1) Protein with the Cell Division Cycle Protein 27 (Cdc27) Subunit of the Anaphase-Promoting Complex. J. Biol. Chem. 2012, 287, 2854–2862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Journiac, N.; Gilabert-Juan, J.; Cipriani, S.; Benit, P.; Liu, X.; Jacquier, S.; Faivre, V.; Delahaye-Duriez, A.; Csaba, Z.; Hourcade, T.; et al. Cell Metabolic Alterations Due to Mcph1 Mutation in Microcephaly. Cell Rep. 2020, 31, 107506. [Google Scholar] [CrossRef] [PubMed]
- Kodani, A.; Yu, T.W.; Johnson, J.R.; Jayaraman, D.; Johnson, T.L.; Al-Gazali, L.; Sztriha, L.; Partlow, J.N.; Kim, H.; Krup, A.L.; et al. Centriolar Satellites Assemble Centrosomal Microcephaly Proteins to Recruit CDK2 and Promote Centriole Duplication. eLife 2015, 4, e07519. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, D.; Kodani, A.; Gonzalez, D.M.; Mancias, J.D.; Mochida, G.H.; Vagnoni, C.; Johnson, J.; Krogan, N.; Harper, J.W.; Reiter, J.F.; et al. Microcephaly Proteins Wdr62 and Aspm Define a Mother Centriole Complex Regulating Centriole Biogenesis, Apical Complex, and Cell Fate. Neuron 2016, 92, 813–828. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.-K.; Liu, P.; Sze, S.K.; Dai, C.; Qi, R.Z. CDK5RAP2 Stimulates Microtubule Nucleation by the Gamma-Tubulin Ring Complex. J. Cell Biol. 2010, 191, 1089–1095. [Google Scholar] [CrossRef] [Green Version]
- Pagan, J.K.; Marzio, A.; Jones, M.J.K.; Saraf, A.; Jallepalli, P.V.; Florens, L.; Washburn, M.P.; Pagano, M. Degradation of Cep68 and PCNT Cleavage Mediate Cep215 Removal from the PCM to Allow Centriole Separation, Disengagement and Licensing. Nat. Cell Biol. 2015, 17, 31–43. [Google Scholar] [CrossRef] [Green Version]
- Barrera, J.A.; Kao, L.R.; Hammer, R.E.; Seemann, J.; Fuchs, J.L.; Megraw, T.L. CDK5RAP2 Regulates Centriole Engagement and Cohesion in Mice. Dev. Cell 2010, 18, 913–926. [Google Scholar] [CrossRef] [Green Version]
- Firat-Karalar, E.N.; Rauniyar, N.; Yates, J.R.; Stearns, T. Proximity Interactions among Centrosome Components Identify Regulators of Centriole Duplication. Curr. Biol. 2014, 24, 664–670. [Google Scholar] [CrossRef] [Green Version]
- Cheeseman, I.M.; Hori, T.; Fukagawa, T.; Desai, A. KNL1 and the CENP-H/I/K Complex Coordinately Direct Kinetochore Assembly in Vertebrates. Mol. Biol. Cell 2008, 19, 587–594. [Google Scholar] [CrossRef] [Green Version]
- Ghongane, P.; Kapanidou, M.; Asghar, A.; Elowe, S.; Bolanos-Garcia, V.M. The Dynamic Protein Knl1–a Kinetochore Rendezvous. J. Cell Sci. 2014, 127, 3415–3423. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, J.S.; Cross, F.R.; Funabiki, H. KNL1/Spc105 Recruits PP1 to Silence the Spindle Assembly Checkpoint. Curr. Biol. 2011, 21, 942–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyomitsu, T.; Obuse, C.; Yanagida, M. Human Blinkin/AF15q14 Is Required for Chromosome Alignment and the Mitotic Checkpoint through Direct Interaction with Bub1 and BubR1. Dev. Cell 2007, 13, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Kiyomitsu, T.; Murakami, H.; Yanagida, M. Protein Interaction Domain Mapping of Human Kinetochore Protein Blinkin Reveals a Consensus Motif for Binding of Spindle Assembly Checkpoint Proteins Bub1 and BubR1. Mol. Cell. Biol. 2011, 31, 998–1011. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.J.; Fu, R.H.; Wu, K.S.; Hsu, W.B.; Tang, T.K. CPAP Is a Cell-Cycle Regulated Protein That Controls Centriole Length. Nat. Cell Biol. 2009, 11, 825–831. [Google Scholar] [CrossRef]
- Dzhindzhev, N.S.; Yu, Q.D.; Weiskopf, K.; Tzolovsky, G.; Cunha-Ferreira, I.; Riparbelli, M.; Rodrigues-Martins, A.; Bettencourt-Dias, M.; Callaini, G.; Glover, D.M. Asterless Is a Scaffold for the Onset of Centriole Assembly. Nature 2010, 467, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.-J.C.; Lin, S.-Y.; Hsu, W.-B.; Lin, Y.-N.; Wu, C.-T.; Lin, Y.-C.; Chang, C.-W.; Wu, K.-S.; Tang, T.K. The Human Microcephaly Protein STIL Interacts with CPAP and Is Required for Procentriole Formation. EMBO J. 2011, 30, 4790–4804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabriel, E.; Wason, A.; Ramani, A.; Gooi, L.M.; Keller, P.; Pozniakovsky, A.; Poser, I.; Noack, F.; Telugu, N.S.; Calegari, F.; et al. CPAP Promotes Timely Cilium Disassembly to Maintain Neural Progenitor Pool. EMBO J. 2016, 35, 803–819. [Google Scholar] [CrossRef]
- Lin, Y.-C.; Chang, C.-W.; Hsu, W.-B.; Tang, C.-J.C.; Lin, Y.-N.; Chou, E.-J.; Wu, C.-T.; Tang, T.K. Human Microcephaly Protein CEP135 Binds to HSAS-6 and CPAP, and Is Required for Centriole Assembly. EMBO J. 2013, 32, 1141–1154. [Google Scholar] [CrossRef] [Green Version]
- Izraeli, S.; Colaizzo-Anas, T.; Bertness, V.L.; Mani, K.; Aplan, P.D.; Kirsch, I.R. Expression of the SIL Gene Is Correlated with Growth Induction and Cellular Proliferation. Cell Growth Differ. 1997, 8, 1171–1179. [Google Scholar]
- Izraeli, S.; Lowe, L.A.; Bertness, V.L.; Good, D.J.; Dorward, D.W.; Kirsch, I.R.; Kuehn, M.R. The SIL Gene Is Required for Mouse Embryonic Axial Development and Left-Right Specification. Nature 1999, 399, 691–694. [Google Scholar] [CrossRef]
- Kumar, A.; Girimaji, S.C.; Duvvari, M.R.; Blanton, S.H. Mutations in STIL, Encoding a Pericentriolar and Centrosomal Protein, Cause Primary Microcephaly. Am. J. Hum. Genet. 2009, 84, 286–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arquint, C.; Sonnen, K.F.; Stierhof, Y.-D.; Nigg, E.A. Cell-Cycle-Regulated Expression of STIL Controls Centriole Number in Human Cells. J. Cell Sci. 2012, 125, 1342–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arquint, C.; Nigg, E.A. STIL Microcephaly Mutations Interfere with APC/C-Mediated Degradation and Cause Centriole Amplification. Curr. Biol. 2014, 24, 351–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleylein-Sohn, J.; Westendorf, J.; Le Clech, M.; Habedanck, R.; Stierhof, Y.-D.; Nigg, E.A. Plk4-Induced Centriole Biogenesis in Human Cells. Dev. Cell 2007, 13, 190–202. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Lee, S.; Chang, J.; Rhee, K. A Novel Function of CEP135 as a Platform Protein of C-NAP1 for Its Centriolar Localization. Exp. Cell Res. 2008, 314, 3692–3700. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Lipinszki, Z.; Rangone, H.; Min, M.; Mykura, C.; Chao-Chu, J.; Schneider, S.; Dzhindzhev, N.S.; Gottardo, M.; Riparbelli, M.G.; et al. Conserved Molecular Interactions in Centriole-to-Centrosome Conversion. Nat. Cell Biol. 2016, 18, 87–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, M.S.; Baig, S.M.; Neumann, S.; Nürnberg, G.; Farooq, M.; Ahmad, I.; Alef, T.; Hennies, H.C.; Technau, M.; Altmüller, J.; et al. A Truncating Mutation of CEP135 Causes Primary Microcephaly and Disturbed Centrosomal Function. Am. J. Hum. Genet. 2012, 90, 871–878. [Google Scholar] [CrossRef] [Green Version]
- Guernsey, D.L.; Matsuoka, M.; Jiang, H.; Evans, S.; Macgillivray, C.; Nightingale, M.; Perry, S.; Ferguson, M.; LeBlanc, M.; Paquette, J.; et al. Mutations in Origin Recognition Complex Gene ORC4 Cause Meier-Gorlin Syndrome. Nat. Genet. 2011, 43, 360–364. [Google Scholar] [CrossRef]
- Yang, Y.J.; Baltus, A.E.; Mathew, R.S.; Murphy, E.A.; Evrony, G.D.; Gonzalez, D.M.; Wang, E.P.; Marshall-Walker, C.A.; Barry, B.J.; Murn, J.; et al. Microcephaly Gene Links Trithorax and REST/NRSF to Control Neural Stem Cell Proliferation and Differentiation. Cell 2012, 151, 1097–1112. [Google Scholar] [CrossRef] [Green Version]
- Garapaty, S.; Xu, C.-F.; Trojer, P.; Mahajan, M.A.; Neubert, T.A.; Samuels, H.H. Identification and Characterization of a Novel Nuclear Protein Complex Involved in Nuclear Hormone Receptor-Mediated Gene Regulation. J. Biol. Chem. 2009, 284, 7542–7552. [Google Scholar] [CrossRef] [Green Version]
- Awad, S.; Al-Dosari, M.S.; Al-Yacoub, N.; Colak, D.; Salih, M.A.; Alkuraya, F.S.; Poizat, C. Mutation in PHC1 Implicates Chromatin Remodeling in Primary Microcephaly Pathogenesis. Hum. Mol. Genet. 2013, 22, 2200–2213. [Google Scholar] [CrossRef] [Green Version]
- Meyerson, M.; Harlow, E. Identification of G1 Kinase Activity for Cdk6, a Novel Cyclin D Partner. Mol. Cell. Biol. 1994, 14, 2077–2086. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.S.; Baig, S.M.; Neumann, S.; Peche, V.S.; Szczepanski, S.; Nürnberg, G.; Tariq, M.; Jameel, M.; Khan, T.N.; Fatima, A.; et al. CDK6 Associates with the Centrosome during Mitosis and Is Mutated in a Large Pakistani Family with Primary Microcephaly. Hum. Mol. Genet. 2013, 22, 5199–5214. [Google Scholar] [CrossRef] [PubMed]
- Thrower, D.A.; Jordan, M.A.; Schaar, B.T.; Yen, T.J.; Wilson, L. Mitotic HeLa Cells Contain a CENP-E-Associated Minus End-Directed Microtubule Motor. EMBO J. 1995, 14, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Bancroft, J.; Auckland, P.; Samora, C.P.; McAinsh, A.D. Chromosome Congression Is Promoted by CENP-Q- and CENP-E-Dependent Pathways. J. Cell Sci. 2015, 128, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Chan, G.K.; Schaar, B.T.; Yen, T.J. Characterization of the Kinetochore Binding Domain of CENP-E Reveals Interactions with the Kinetochore Proteins CENP-F and HBUBR1. J. Cell Biol. 1998, 143, 49–63. [Google Scholar] [CrossRef] [Green Version]
- Mirzaa, G.M.; Vitre, B.; Carpenter, G.; Abramowicz, I.; Gleeson, J.G.; Paciorkowski, A.R.; Cleveland, D.W.; Dobyns, W.B.; O’Driscoll, M. Mutations in CENPE Define a Novel Kinetochore-Centromeric Mechanism for Microcephalic Primordial Dwarfism. Hum. Genet. 2014, 133, 1023–1039. [Google Scholar] [CrossRef] [Green Version]
- Strnad, P.; Leidel, S.; Vinogradova, T.; Euteneuer, U.; Khodjakov, A.; Gönczy, P. Regulated HsSAS-6 Levels Ensure Formation of a Single Procentriole per Centriole during the Centrosome Duplication Cycle. Dev. Cell 2007, 13, 203–213. [Google Scholar] [CrossRef] [Green Version]
- Van Breugel, M.; Hirono, M.; Andreeva, A.; Yanagisawa, H.; Yamaguchi, S.; Nakazawa, Y.; Morgner, N.; Petrovich, M.; Ebong, I.-O.; Robinson, C.V.; et al. Structures of SAS-6 Suggest Its Organization in Centrioles. Science 2011, 331, 1196–1199. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.A.; Rupp, V.M.; Orpinell, M.; Hussain, M.S.; Altmuller, J.; Steinmetz, M.O.; Enzinger, C.; Thiele, H.; Hohne, W.; Nurnberg, G.; et al. A Missense Mutation in the PISA Domain of HsSAS-6 Causes Autosomal Recessive Primary Microcephaly in a Large Consanguineous Pakistani Family. Hum. Mol. Genet. 2017, 23, 5940–5949. [Google Scholar] [CrossRef]
- Nguyen, L.N.; Ma, D.; Shui, G.; Wong, P.; Cazenave-Gassiot, A.; Zhang, X.; Wenk, M.R.; Goh, E.L.K.; Silver, D.L. Mfsd2a Is a Transporter for the Essential Omega-3 Fatty Acid Docosahexaenoic Acid. Nature 2014, 509, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zvi, A.; Lacoste, B.; Kur, E.; Andreone, B.J.; Mayshar, Y.; Yan, H.; Gu, C. Mfsd2a Is Critical for the Formation and Function of the Blood-Brain Barrier. Nature 2014, 509, 507–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guemez-Gamboa, A.; Nguyen, L.N.; Yang, H.; Zaki, M.S.; Kara, M.; Ben-Omran, T.; Akizu, N.; Rosti, R.O.; Rosti, B.; Scott, E.; et al. Inactivating Mutations in MFSD2A, Required for Omega-3 Fatty Acid Transport in Brain, Cause a Lethal Microcephaly Syndrome. Nat. Genet. 2015, 47, 809–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asencio, C.; Davidson, I.F.; Santarella-Mellwig, R.; Ly-Hartig, T.B.N.; Mall, M.; Wallenfang, M.R.; Mattaj, I.W.; Gorjánácz, M. Coordination of Kinase and Phosphatase Activities by Lem4 Enables Nuclear Envelope Reassembly during Mitosis. Cell 2012, 150, 122–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, N.; Chung, H.; Jolly, A.; Withers, M.; Tepe, B.; Arenkiel, B.R.; Shah, P.S.; Krogan, N.J.; Aydin, H.; Geckinli, B.B.; et al. Mutations in ANKLE2, a ZIKA Virus Target, Disrupt an Asymmetric Cell Division Pathway in Drosophila Neuroblasts to Cause Microcephaly. Dev. Cell 2019, 51, 713–729.e6. [Google Scholar] [CrossRef]
- Di Cunto, F.; Calautti, E.; Hsiao, J.; Ong, L.; Topley, G.; Turco, E.; Dotto, G.P. Citron Rho-Interacting Kinase, a Novel Tissue-Specific Ser/Thr Kinase Encompassing the Rho-Rac-Binding Protein Citron. J. Biol. Chem. 1998, 273, 29706–29711. [Google Scholar] [CrossRef] [Green Version]
- Harding, B.N.; Moccia, A.; Drunat, S.; Soukarieh, O.; Tubeuf, H.; Chitty, L.S.; Verloes, A.; Gressens, P.; El Ghouzzi, V.; Joriot, S.; et al. Mutations in Citron Kinase Cause Recessive Microlissencephaly with Multinucleated Neurons. Am. J. Hum. Genet. 2016, 99, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Bielas, S.L.; Zaki, M.S.; Ismail, S.; Farfara, D.; Um, K.; Rosti, R.O.; Scott, E.C.; Tu, S.; Chi, N.C.; et al. Biallelic Mutations in Citron Kinase Link Mitotic Cytokinesis to Human Primary Microcephaly. Am. J. Hum. Genet. 2016, 99, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Gruneberg, U.; Neef, R.; Li, X.; Chan, E.H.Y.; Chalamalasetty, R.B.; Nigg, E.A.; Barr, F.A. KIF14 and Citron Kinase Act Together to Promote Efficient Cytokinesis. J. Cell Biol. 2006, 172, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Gai, M.; Bianchi, F.T.; Vagnoni, C.; Verni, F.; Bonaccorsi, S.; Pasquero, S.; Berto, G.E.; Sgro, F.; Chiotto, A.M.; Annaratone, L.; et al. ASPM and CITK Regulate Spindle Orientation by Affecting the Dynamics of Astral Microtubules. EMBO Rep. 2016, 17, 1396–1409. [Google Scholar] [CrossRef] [Green Version]
- Kadir, R.; Harel, T.; Markus, B.; Perez, Y.; Bakhrat, A.; Cohen, I.; Volodarsky, M.; Feintsein-Linial, M.; Chervinski, E.; Zlotogora, J.; et al. ALFY-Controlled DVL3 Autophagy Regulates Wnt Signaling, Determining Human Brain Size. PLoS Genet. 2016, 12, e1005919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, T.H.; Lamark, T.; Isakson, P.; Finley, K.; Larsen, K.B.; Brech, A.; Øvervatn, A.; Stenmark, H.; Bjørkøy, G.; Simonsen, A.; et al. P62/SQSTM1 and ALFY Interact to Facilitate the Formation of P62 Bodies/ALIS and Their Degradation by Autophagy. Autophagy 2010, 6, 330–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiStasio, A.; Driver, A.; Sund, K.; Donlin, M.; Muraleedharan, R.M.; Pooya, S.; Kline-Fath, B.; Kaufman, K.M.; Prows, C.A.; Schorry, E.; et al. Copb2 Is Essential for Embryogenesis and Hypomorphic Mutations Cause Human Microcephaly. Hum. Mol. Genet. 2017, 26, 4836–4848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carleton, M.; Mao, M.; Biery, M.; Warrener, P.; Kim, S.; Buser, C.; Marshall, C.G.; Fernandes, C.; Annis, J.; Linsley, P.S. RNA Interference-Mediated Silencing of Mitotic Kinesin KIF14 Disrupts Cell Cycle Progression and Induces Cytokinesis Failure. Mol. Cell. Biol. 2006, 26, 3853–3863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Zhao, J.; Bibikova, M.; Leverson, J.D.; Bossy-Wetzel, E.; Fan, J.-B.; Abraham, R.T.; Jiang, W. Functional Analysis of Human Microtubule-Based Motor Proteins, the Kinesins and Dyneins, in Mitosis/Cytokinesis Using RNA Interference. Mol. Biol. Cell 2005, 16, 3187–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moawia, A.; Shaheen, R.; Rasool, S.; Waseem, S.S.; Ewida, N.; Budde, B.; Kawalia, A.; Motameny, S.; Khan, K.; Fatima, A.; et al. Mutations of KIF14 Cause Primary Microcephaly by Impairing Cytokinesis. Ann. Neurol. 2017, 82, 562–577. [Google Scholar] [CrossRef]
- Martin, C.-A.; Murray, J.E.; Carroll, P.; Leitch, A.; Mackenzie, K.J.; Halachev, M.; Fetit, A.E.; Keith, C.; Bicknell, L.S.; Fluteau, A.; et al. Mutations in Genes Encoding Condensin Complex Proteins Cause Microcephaly through Decatenation Failure at Mitosis. Genes Dev. 2016, 30, 2158–2172. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.; Cuvier, O.; Hirano, T. Chromosome Condensation by a Human Condensin Complex in Xenopus Egg Extracts. J. Biol. Chem. 2001, 276, 5417–5420. [Google Scholar] [CrossRef] [Green Version]
- Zuccolo, M.; Alves, A.; Galy, V.; Bolhy, S.; Formstecher, E.; Racine, V.; Sibarita, J.-B.; Fukagawa, T.; Shiekhattar, R.; Yen, T.; et al. The Human Nup107-160 Nuclear Pore Subcomplex Contributes to Proper Kinetochore Functions. EMBO J. 2007, 26, 1853–1864. [Google Scholar] [CrossRef] [Green Version]
- Braun, D.A.; Lovric, S.; Schapiro, D.; Schneider, R.; Marquez, J.; Asif, M.; Hussain, M.S.; Daga, A.; Widmeier, E.; Rao, J.; et al. Mutations in Multiple Components of the Nuclear Pore Complex Cause Nephrotic Syndrome. J. Clin. Investig. 2018, 128, 4313–4328. [Google Scholar] [CrossRef] [Green Version]
- Perez, Y.; Bar-Yaacov, R.; Kadir, R.; Wormser, O.; Shelef, I.; Birk, O.S.; Flusser, H.; Birnbaum, R.Y. Mutations in the Microtubule-Associated Protein MAP11 (C7orf43) Cause Microcephaly in Humans and Zebrafish. Brain 2019, 142, 574–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristofoli, F.; Moss, T.; Moore, H.W.; Devriendt, K.; Flanagan-Steet, H.; May, M.; Jones, J.; Roelens, F.; Fons, C.; Fernandez, A.; et al. De Novo Variants in LMNB1 Cause Pronounced Syndromic Microcephaly and Disruption of Nuclear Envelope Integrity. Am. J. Hum. Genet. 2020, 107, 753–762. [Google Scholar] [CrossRef]
- Parry, D.A.; Martin, C.-A.; Greene, P.; Marsh, J.A.; Genomics England Research Consortium; Blyth, M.; Cox, H.; Donnelly, D.; Greenhalgh, L.; Greville-Heygate, S.; et al. Heterozygous Lamin B1 and Lamin B2 Variants Cause Primary Microcephaly and Define a Novel Laminopathy. Genet. Med. 2021, 23, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R.D.; Gruenbaum, Y.; Moir, R.D.; Shumaker, D.K.; Spann, T.P. Nuclear Lamins: Building Blocks of Nuclear Architecture. Genes Dev. 2002, 16, 533–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, M.-Y.; Wang, S.; Heidinger, J.M.; Shumaker, D.K.; Adam, S.A.; Goldman, R.D.; Zheng, Y. A Mitotic Lamin B Matrix Induced by RanGTP Required for Spindle Assembly. Science 2006, 311, 1887–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farooq, M.; Lindbæk, L.; Krogh, N.; Doganli, C.; Keller, C.; Mönnich, M.; Gonçalves, A.B.; Sakthivel, S.; Mang, Y.; Fatima, A.; et al. RRP7A Links Primary Microcephaly to Dysfunction of Ribosome Biogenesis, Resorption of Primary Cilia, and Neurogenesis. Nat. Commun. 2020, 11, 5816. [Google Scholar] [CrossRef]
- Singh, S.; Vanden Broeck, A.; Miller, L.; Chaker-Margot, M.; Klinge, S. Nucleolar Maturation of the Human Small Subunit Processome. Science 2021, 373, eabj5338. [Google Scholar] [CrossRef]
- Khan, A.; Alaamery, M.; Massadeh, S.; Obaid, A.; Kashgari, A.A.; Walsh, C.A.; Eyaid, W. PDCD6IP, Encoding a Regulator of the ESCRT Complex, Is Mutated in Microcephaly. Clin. Genet. 2020, 98, 80–85. [Google Scholar] [CrossRef]
- Klebig, C.; Korinth, D.; Meraldi, P. Bub1 Regulates Chromosome Segregation in a Kinetochore-Independent Manner. J. Cell Biol. 2009, 185, 841–858. [Google Scholar] [CrossRef]
- Carvalhal, S.; Bader, I.; Rooimans, M.A.; Oostra, A.B.; Balk, J.A.; Feichtinger, R.G.; Beichler, C.; Speicher, M.R.; van Hagen, J.M.; Waisfisz, Q.; et al. Biallelic BUB1 Mutations Cause Microcephaly, Developmental Delay, and Variable Effects on Cohesion and Chromosome Segregation. Sci. Adv. 2022, 8, eabk0114. [Google Scholar] [CrossRef]
- Seeley, T.W.; Wang, L.; Zhen, J.Y. Phosphorylation of Human MAD1 by the BUB1 Kinase in Vitro. Biochem. Biophys. Res. Commun. 1999, 257, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.L.; Scott, M.I.F.; Holt, S.V.; Hussein, D.; Taylor, S.S. Bub1 Is Required for Kinetochore Localization of BubR1, Cenp-E, Cenp-F and Mad2, and Chromosome Congression. J. Cell Sci. 2004, 117, 1577–1589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flory, M.R.; Moser, M.J.; Monnat, R.J.; Davis, T.N. Identification of a Human Centrosomal Calmodulin-Binding Protein That Shares Homology with Pericentrin. Proc. Natl. Acad. Sci. USA 2000, 97, 5919–5923. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hansen, D.; Killilea, A.; Joshi, H.C.; Palazzo, R.E.; Balczon, R. Kendrin/Pericentrin-B, a Centrosome Protein with Homology to Pericentrin That Complexes with PCM-1. J. Cell Sci. 2001, 114, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, K.; Nishimura, T.; Hayakawa, A.; Ono, Y.; Takahashi, M. Involvement of a Centrosomal Protein Kendrin in the Maintenance of Centrosome Cohesion by Modulating Nek2A Kinase Activity. Biochem. Biophys. Res. Commun. 2010, 398, 217–223. [Google Scholar] [CrossRef]
- Buchman, J.J.; Tseng, H.-C.; Zhou, Y.; Frank, C.L.; Xie, Z.; Tsai, L.-H. Cdk5rap2 Interacts with Pericentrin to Maintain the Neural Progenitor Pool in the Developing Neocortex. Neuron 2010, 66, 386–402. [Google Scholar] [CrossRef] [Green Version]
- Hall-Jackson, C.A.; Cross, D.A.; Morrice, N.; Smythe, C. ATR Is a Caffeine-Sensitive, DNA-Activated Protein Kinase with a Substrate Specificity Distinct from DNA-PK. Oncogene 1999, 18, 6707–6713. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 Is an Essential Kinase That Is Regulated by Atr and Required for the G(2)/M DNA Damage Checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar] [CrossRef]
- Casper, A.M.; Nghiem, P.; Arlt, M.F.; Glover, T.W. ATR Regulates Fragile Site Stability. Cell 2002, 111, 779–789. [Google Scholar] [CrossRef] [Green Version]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP Promotes DNA End Resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Qvist, P.; Huertas, P.; Jimeno, S.; Nyegaard, M.; Hassan, M.J.; Jackson, S.P.; Børglum, A.D. CtIP Mutations Cause Seckel and Jawad Syndromes. PLoS Genet. 2011, 7, e1002310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Chen, J. N Terminus of CtIP Is Critical for Homologous Recombination-Mediated Double-Strand Break Repair. J. Biol. Chem. 2009, 284, 31746–31752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Chen, J. DNA Damage-Induced Cell Cycle Checkpoint Control Requires CtIP, a Phosphorylation-Dependent Binding Partner of BRCA1 C-Terminal Domains. Mol. Cell. Biol. 2004, 24, 9478–9486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loffler, H.; Rebacz, B.; Ho, A.D.; Lukas, J.; Bartek, J.; Kramer, A. Chk1-Dependent Regulation of Cdc25B Functions to Coordinate Mitotic Events. Cell Cycle 2006, 5, 2543–2547. [Google Scholar] [CrossRef] [Green Version]
- Sir, J.-H.; Barr, A.R.; Nicholas, A.K.; Carvalho, O.P.; Khurshid, M.; Sossick, A.; Reichelt, S.; D’Santos, C.; Woods, C.G.; Gergely, F. A Primary Microcephaly Protein Complex Forms a Ring around Parental Centrioles. Nat. Genet. 2011, 43, 1147–1153. [Google Scholar] [CrossRef] [Green Version]
- Dauber, A.; Lafranchi, S.H.; Maliga, Z.; Lui, J.C.; Moon, J.E.; McDeed, C.; Henke, K.; Zonana, J.; Kingman, G.A.; Pers, T.H.; et al. Novel Microcephalic Primordial Dwarfism Disorder Associated with Variants in the Centrosomal Protein Ninein. J. Clin. Endocrinol. Metab. 2012, 97, E2140–E2151. [Google Scholar] [CrossRef]
- Ou, Y.Y.; Mack, G.J.; Zhang, M.; Rattner, J.B. CEP110 and Ninein Are Located in a Specific Domain of the Centrosome Associated with Centrosome Maturation. J. Cell Sci. 2002, 115, 1825–1835. [Google Scholar] [CrossRef]
- Huang, N.; Xia, Y.; Zhang, D.; Wang, S.; Bao, Y.; He, R.; Teng, J.; Chen, J. Hierarchical Assembly of Centriole Subdistal Appendages via Centrosome Binding Proteins CCDC120 and CCDC68. Nat. Commun. 2017, 8, 15057. [Google Scholar] [CrossRef] [Green Version]
- Duxin, J.P.; Dao, B.; Martinsson, P.; Rajala, N.; Guittat, L.; Campbell, J.L.; Spelbrink, J.N.; Stewart, S.A. Human Dna2 Is a Nuclear and Mitochondrial DNA Maintenance Protein. Mol. Cell. Biol. 2009, 29, 4274–4282. [Google Scholar] [CrossRef] [Green Version]
- Shaheen, R.; Faqeih, E.; Ansari, S.; Abdel-Salam, G.; Al-Hassnan, Z.N.; Al-Shidi, T.; Alomar, R.; Sogaty, S.; Alkuraya, F.S. Genomic Analysis of Primordial Dwarfism Reveals Novel Disease Genes. Genome Res. 2014, 24, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Zhou, M.; Guo, Z.; Lu, H.; Qian, L.; Dai, H.; Qiu, J.; Yakubovskaya, E.; Bogenhagen, D.F.; Demple, B.; et al. Human DNA2 Is a Mitochondrial Nuclease/Helicase for Efficient Processing of DNA Replication and Repair Intermediates. Mol. Cell 2008, 32, 325–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN Constitute Two DNA End Resection Machineries for Human DNA Break Repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harley, M.E.; Murina, O.; Leitch, A.; Higgs, M.R.; Bicknell, L.S.; Yigit, G.; Blackford, A.N.; Zlatanou, A.; Mackenzie, K.J.; Reddy, K.; et al. TRAIP Promotes DNA Damage Response during Genome Replication and Is Mutated in Primordial Dwarfism. Nat. Genet. 2016, 48, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, S.; Smedegaard, S.; Nakamura, K.; Mortuza, G.B.; Räschle, M.; Ibañez de Opakua, A.; Oka, Y.; Feng, Y.; Blanco, F.J.; Mann, M.; et al. TRAIP Is a PCNA-Binding Ubiquitin Ligase That Protects Genome Stability after Replication Stress. J. Cell Biol. 2016, 212, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Sonneville, R.; Bhowmick, R.; Hoffmann, S.; Mailand, N.; Hickson, I.D.; Labib, K. TRAIP Drives Replisome Disassembly and Mitotic DNA Repair Synthesis at Sites of Incomplete DNA Replication. eLife 2019, 8, e48686. [Google Scholar] [CrossRef]
- Chapard, C.; Meraldi, P.; Gleich, T.; Bachmann, D.; Hohl, D.; Huber, M. TRAIP Is a Regulator of the Spindle Assembly Checkpoint. J. Cell Sci. 2014, 127, 5149–5156. [Google Scholar] [CrossRef] [Green Version]
- Potts, P.R.; Yu, H. Human MMS21/NSE2 Is a SUMO Ligase Required for DNA Repair. Mol. Cell. Biol. 2005, 25, 7021–7032. [Google Scholar] [CrossRef] [Green Version]
- Potts, P.R.; Porteus, M.H.; Yu, H. Human SMC5/6 Complex Promotes Sister Chromatid Homologous Recombination by Recruiting the SMC1/3 Cohesin Complex to Double-Strand Breaks. EMBO J. 2006, 25, 3377–3388. [Google Scholar] [CrossRef] [Green Version]
- Payne, F.; Colnaghi, R.; Rocha, N.; Seth, A.; Harris, J.; Carpenter, G.; Bottomley, W.E.; Wheeler, E.; Wong, S.; Saudek, V.; et al. Hypomorphism in Human NSMCE2 Linked to Primordial Dwarfism and Insulin Resistance. J. Clin. Investig. 2014, 124, 4028–4038. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.M.; Preble, A.M.; Patel, U.K.; O’Connell, K.L.; Dias, D.P.; Moritz, M.; Agard, D.; Stults, J.T.; Stearns, T. GCP5 and GCP6: Two New Members of the Human Gamma-Tubulin Complex. Mol. Biol. Cell 2001, 12, 3340–3352. [Google Scholar] [CrossRef]
- Bettencourt-Dias, M.; Rodrigues-Martins, A.; Carpenter, L.; Riparbelli, M.; Lehmann, L.; Gatt, M.K.; Carmo, N.; Balloux, F.; Callaini, G.; Glover, D.M. SAK/PLK4 Is Required for Centriole Duplication and Flagella Development. Curr. Biol. 2005, 15, 2199–2207. [Google Scholar] [CrossRef] [Green Version]
- Habedanck, R.; Stierhof, Y.-D.; Wilkinson, C.J.; Nigg, E.A. The Polo Kinase Plk4 Functions in Centriole Duplication. Nat. Cell Biol. 2005, 7, 1140–1146. [Google Scholar] [CrossRef]
- Fava, F.; Raynaud-Messina, B.; Leung-Tack, J.; Mazzolini, L.; Li, M.; Guillemot, J.C.; Cachot, D.; Tollon, Y.; Ferrara, P.; Wright, M. Human 76p: A New Member of the Gamma-Tubulin-Associated Protein Family. J. Cell Biol. 1999, 147, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Rapley, J.; Nicolàs, M.; Groen, A.; Regué, L.; Bertran, M.T.; Caelles, C.; Avruch, J.; Roig, J. The NIMA-Family Kinase Nek6 Phosphorylates the Kinesin Eg5 at a Novel Site Necessary for Mitotic Spindle Formation. J. Cell Sci. 2008, 121, 3912–3921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostergaard, P.; Simpson, M.A.; Mendola, A.; Vasudevan, P.; Connell, F.C.; van Impel, A.; Moore, A.T.; Loeys, B.L.; Ghalamkarpour, A.; Onoufriadis, A.; et al. Mutations in KIF11 Cause Autosomal-Dominant Microcephaly Variably Associated with Congenital Lymphedema and Chorioretinopathy. Am. J. Hum. Genet. 2012, 90, 356–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Splinter, D.; Razafsky, D.S.; Schlager, M.A.; Serra-Marques, A.; Grigoriev, I.; Demmers, J.; Keijzer, N.; Jiang, K.; Poser, I.; Hyman, A.A.; et al. BICD2, Dynactin, and LIS1 Cooperate in Regulating Dynein Recruitment to Cellular Structures. Mol. Biol. Cell 2012, 23, 4226–4241. [Google Scholar] [CrossRef] [PubMed]
- McKenney, R.J.; Huynh, W.; Tanenbaum, M.E.; Bhabha, G.; Vale, R.D. Activation of Cytoplasmic Dynein Motility by Dynactin-Cargo Adapter Complexes. Science 2014, 345, 337–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaaban, S.; Carter, A.P. Structure of Dynein-Dynactin on Microtubules Shows Tandem Adaptor Binding. Nature 2022, 610, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Ellis, N.A.; Lennon, D.J.; Proytcheva, M.; Alhadeff, B.; Henderson, E.E.; German, J. Somatic Intragenic Recombination within the Mutated Locus BLM Can Correct the High Sister-Chromatid Exchange Phenotype of Bloom Syndrome Cells. Am. J. Hum. Genet. 1995, 57, 1019–1027. [Google Scholar]
- Ellis, N.A.; Groden, J.; Ye, T.Z.; Straughen, J.; Lennon, D.J.; Ciocci, S.; Proytcheva, M.; German, J. The Bloom’s Syndrome Gene Product Is Homologous to RecQ Helicases. Cell 1995, 83, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Karow, J.K.; Chakraverty, R.K.; Hickson, I.D. The Bloom’s Syndrome Gene Product Is a 3′-5′ DNA Helicase. J. Biol. Chem. 1997, 272, 30611–30614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langland, G.; Elliott, J.; Li, Y.; Creaney, J.; Dixon, K.; Groden, J. The BLM Helicase Is Necessary for Normal DNA Double-Strand Break Repair. Cancer Res. 2002, 62, 2766–2770. [Google Scholar] [PubMed]
- Grawunder, U.; Wilm, M.; Wu, X.; Kulesza, P.; Wilson, T.E.; Mann, M.; Lieber, M.R. Activity of DNA Ligase IV Stimulated by Complex Formation with XRCC4 Protein in Mammalian Cells. Nature 1997, 388, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Grawunder, U.; Zimmer, D.; Fugmann, S.; Schwarz, K.; Lieber, M.R. DNA Ligase IV Is Essential for V(D)J Recombination and DNA Double-Strand Break Repair in Human Precursor Lymphocytes. Mol. Cell 1998, 2, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Lu, H.; Tippin, B.; Shimazaki, N.; Goodman, M.F.; Lieber, M.R. XRCC4:DNA Ligase IV Can Ligate Incompatible DNA Ends and Can Ligate across Gaps. EMBO J. 2007, 26, 1010–1023. [Google Scholar] [CrossRef] [Green Version]
- Rosin, N.; Elcioglu, N.H.; Beleggia, F.; Isguven, P.; Altmuller, J.; Thiele, H.; Steindl, K.; Joset, P.; Rauch, A.; Nurnberg, P.; et al. Mutations in XRCC4 Cause Primary Microcephaly, Short Stature and Increased Genomic Instability. Hum. Mol. Genet. 2015, 24, 3708–3717. [Google Scholar] [CrossRef] [Green Version]
- Bicknell, L.S.; Walker, S.; Klingseisen, A.; Stiff, T.; Leitch, A.; Kerzendorfer, C.; Martin, C.-A.; Yeyati, P.; Al Sanna, N.; Bober, M.; et al. Mutations in ORC1, Encoding the Largest Subunit of the Origin Recognition Complex, Cause Microcephalic Primordial Dwarfism Resembling Meier-Gorlin Syndrome. Nat. Genet. 2011, 43, 350–355. [Google Scholar] [CrossRef]
- Ohta, S.; Tatsumi, Y.; Fujita, M.; Tsurimoto, T.; Obuse, C. The ORC1 Cycle in Human Cells: II. Dynamic Changes in the Human ORC Complex during the Cell Cycle. J. Biol. Chem. 2003, 278, 41535–41540. [Google Scholar] [CrossRef] [Green Version]
- Wohlschlegel, J.A.; Dwyer, B.T.; Dhar, S.K.; Cvetic, C.; Walter, J.C.; Dutta, A. Inhibition of Eukaryotic DNA Replication by Geminin Binding to Cdt1. Science 2000, 290, 2309–2312. [Google Scholar] [CrossRef]
- Varma, D.; Chandrasekaran, S.; Sundin, L.J.R.; Reidy, K.T.; Wan, X.; Chasse, D.A.D.; Nevis, K.R.; DeLuca, J.G.; Salmon, E.D.; Cook, J.G. Recruitment of the Human Cdt1 Replication Licensing Protein by the Loop Domain of Hec1 Is Required for Stable Kinetochore-Microtubule Attachment. Nat. Cell Biol. 2012, 14, 593–603. [Google Scholar] [CrossRef] [Green Version]
- Walter, D.; Hoffmann, S.; Komseli, E.-S.; Rappsilber, J.; Gorgoulis, V.; Sørensen, C.S. SCF(Cyclin F)-Dependent Degradation of CDC6 Suppresses DNA Re-Replication. Nat. Commun. 2016, 7, 10530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, P.; Chen, J.; Thome, K.C.; Lawlis, S.J.; Hou, Z.H.; Hendricks, M.; Parvin, J.D.; Dutta, A. Human CDC6/Cdc18 Associates with Orc1 and Cyclin-Cdk and Is Selectively Eliminated from the Nucleus at the Onset of S Phase. Mol. Cell. Biol. 1998, 18, 2758–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrage, L.C.; Charng, W.-L.; Eldomery, M.K.; Willer, J.R.; Davis, E.E.; Lugtenberg, D.; Zhu, W.; Leduc, M.S.; Akdemir, Z.C.; Azamian, M.; et al. De Novo GMNN Mutations Cause Autosomal-Dominant Primordial Dwarfism Associated with Meier-Gorlin Syndrome. Am. J. Hum. Genet. 2015, 97, 904–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pefani, D.-E.; Dimaki, M.; Spella, M.; Karantzelis, N.; Mitsiki, E.; Kyrousi, C.; Symeonidou, I.-E.; Perrakis, A.; Taraviras, S.; Lygerou, Z. Idas, a Novel Phylogenetically Conserved Geminin-Related Protein, Binds to Geminin and Is Required for Cell Cycle Progression. J. Biol. Chem. 2011, 286, 23234–23246. [Google Scholar] [CrossRef] [Green Version]

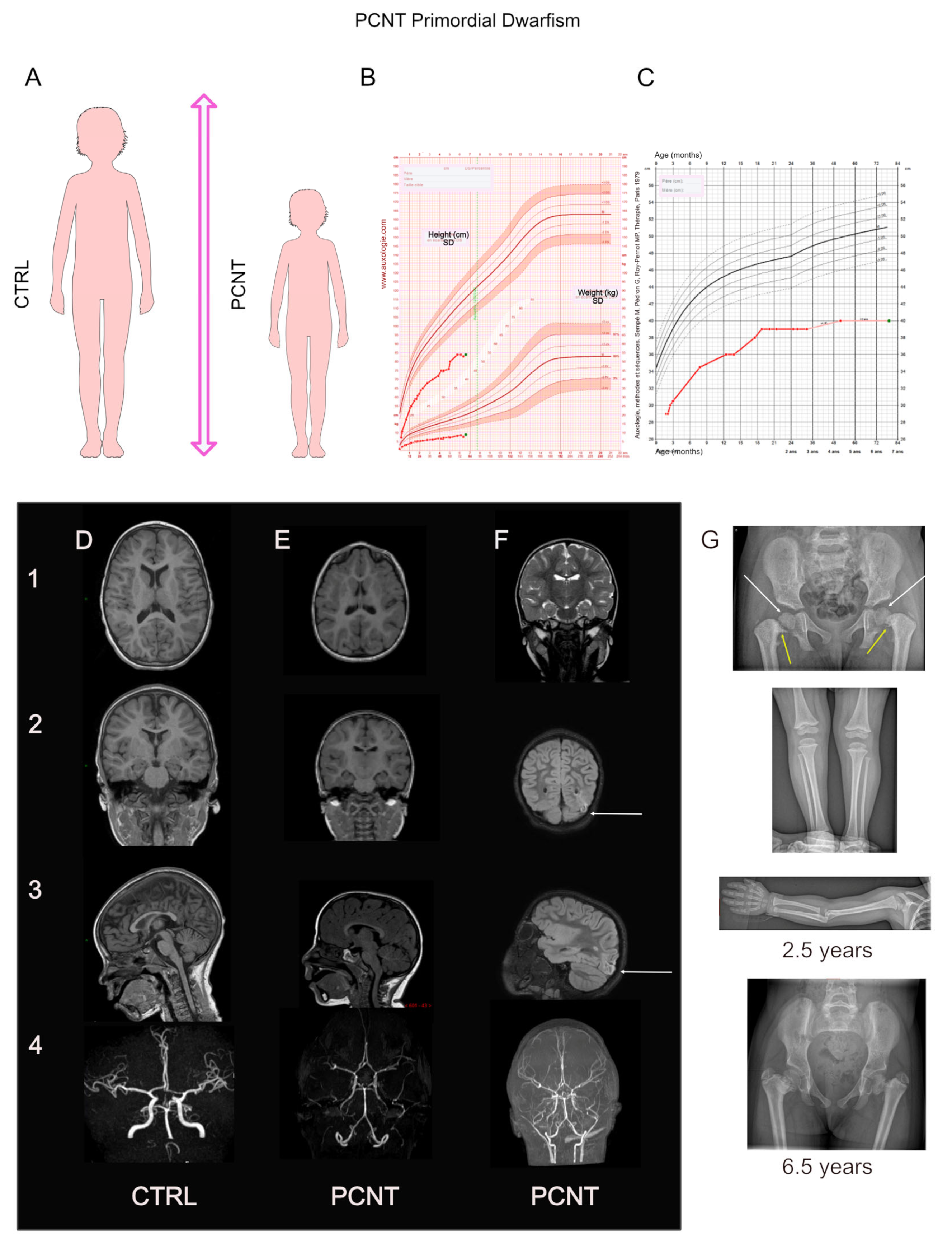

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farcy, S.; Hachour, H.; Bahi-Buisson, N.; Passemard, S. Genetic Primary Microcephalies: When Centrosome Dysfunction Dictates Brain and Body Size. Cells 2023, 12, 1807. https://doi.org/10.3390/cells12131807
Farcy S, Hachour H, Bahi-Buisson N, Passemard S. Genetic Primary Microcephalies: When Centrosome Dysfunction Dictates Brain and Body Size. Cells. 2023; 12(13):1807. https://doi.org/10.3390/cells12131807
Chicago/Turabian StyleFarcy, Sarah, Hassina Hachour, Nadia Bahi-Buisson, and Sandrine Passemard. 2023. "Genetic Primary Microcephalies: When Centrosome Dysfunction Dictates Brain and Body Size" Cells 12, no. 13: 1807. https://doi.org/10.3390/cells12131807
APA StyleFarcy, S., Hachour, H., Bahi-Buisson, N., & Passemard, S. (2023). Genetic Primary Microcephalies: When Centrosome Dysfunction Dictates Brain and Body Size. Cells, 12(13), 1807. https://doi.org/10.3390/cells12131807