
Soluble Collectin 11 (CL-11) Acts as an Immunosuppressive Molecule Potentially Used by Stem Cell-Derived Retinal Epithelial Cells to Modulate T Cell Response

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. RPE Cell Culture and Phenotype
2.2. Cell Isolation
2.3. Co-Culture Assay
2.4. Cytokine Intracellular Staining
2.5. T Cell Activation, Proliferation, and Phenotype
2.6. Confocal Microscopy
2.7. Determination of CL-11 Binding to RPE and T Cells
2.8. Statistical Evaluation
3. Results
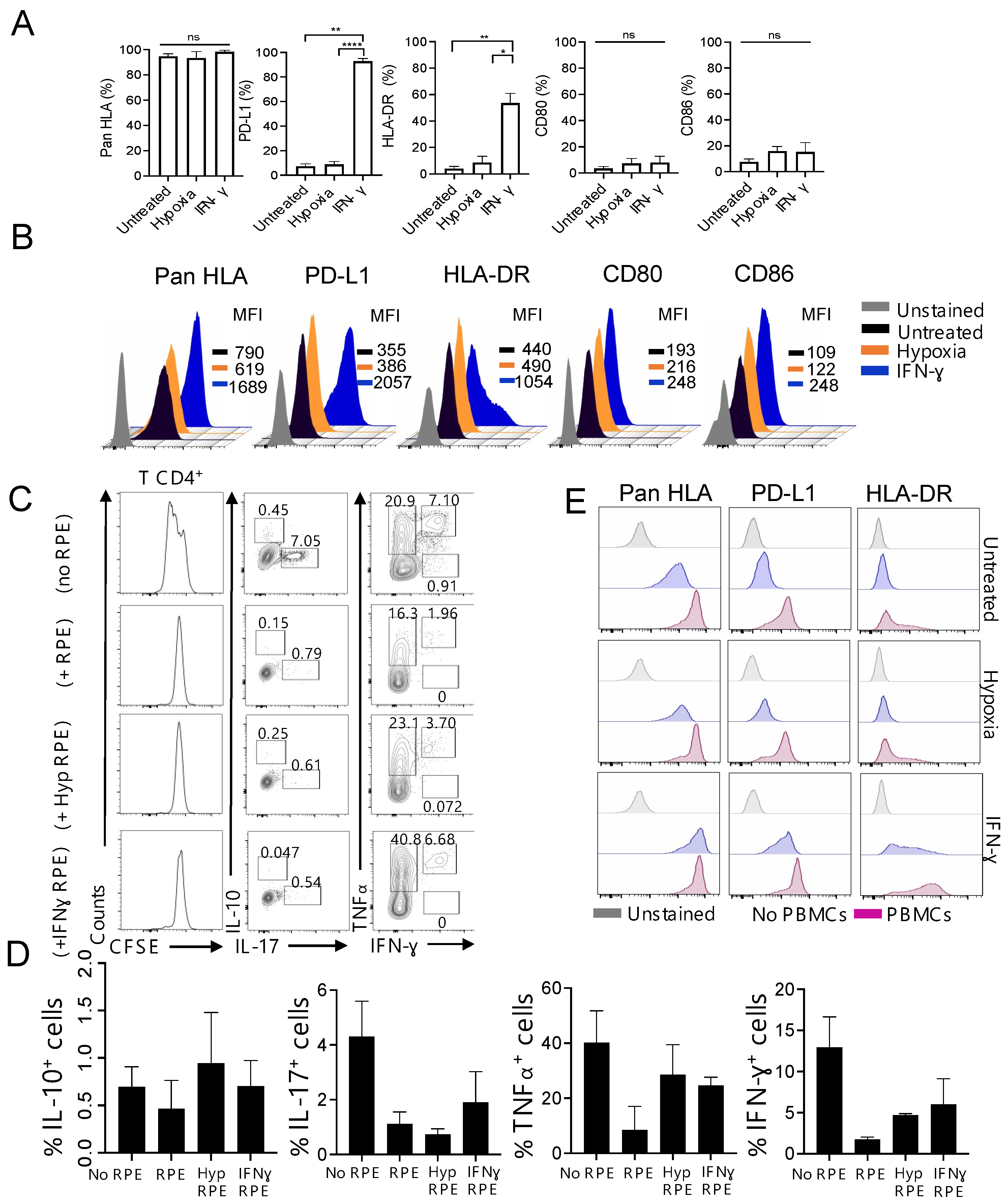
3.1. Foetal-Derived RPE Cells with a Modified Immunophenotype in Response to Inflammation Inhibit T Cell Proliferation In Vitro
3.2. CL-11 Produced by Inflammatory fRPE Cells Can Function as a Modulator of CD4+CD25− T Cell Activation In Vitro
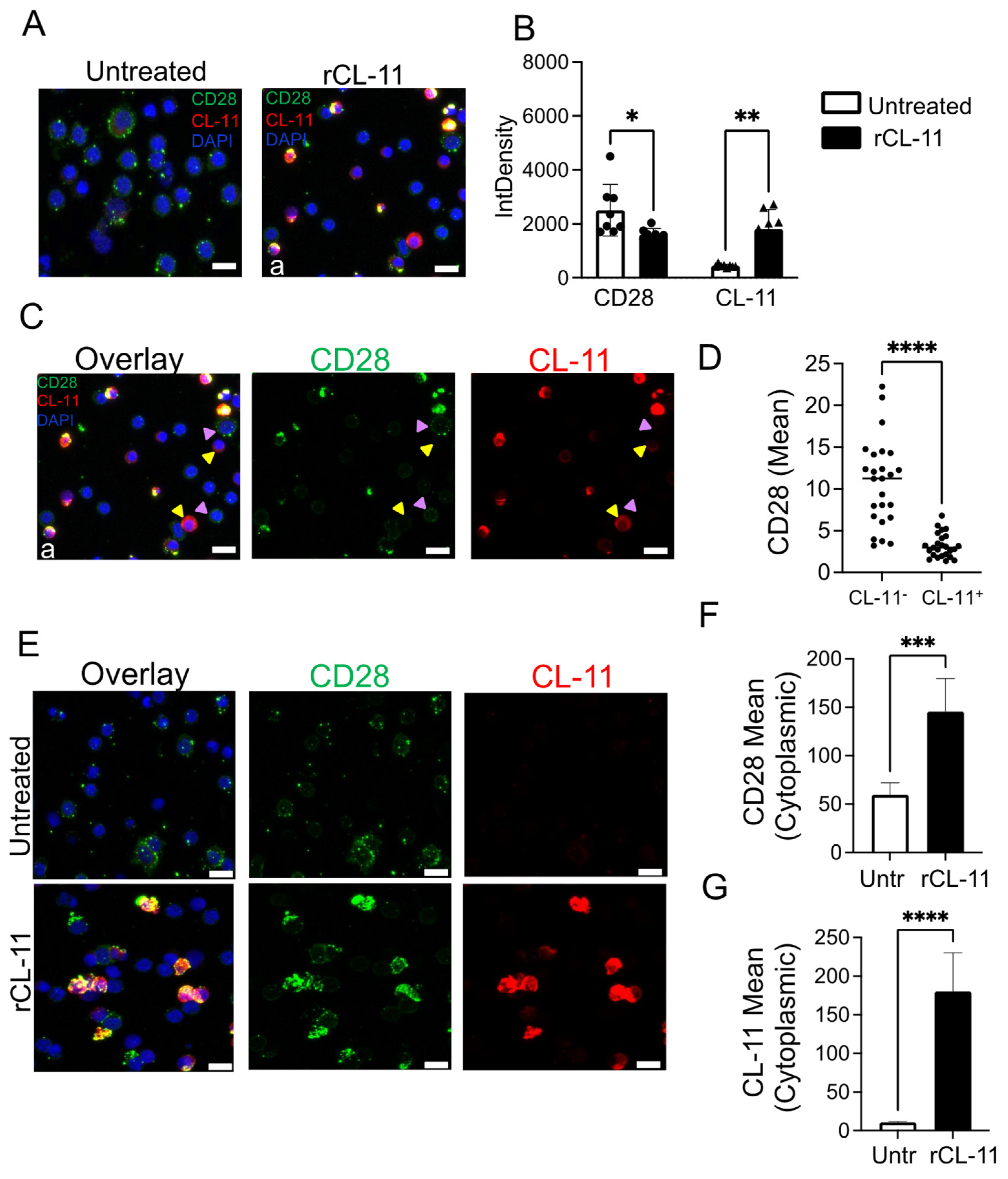
3.3. The Binding of CL-11 to T Cells Results in Increased Intracellular CD28 Detection
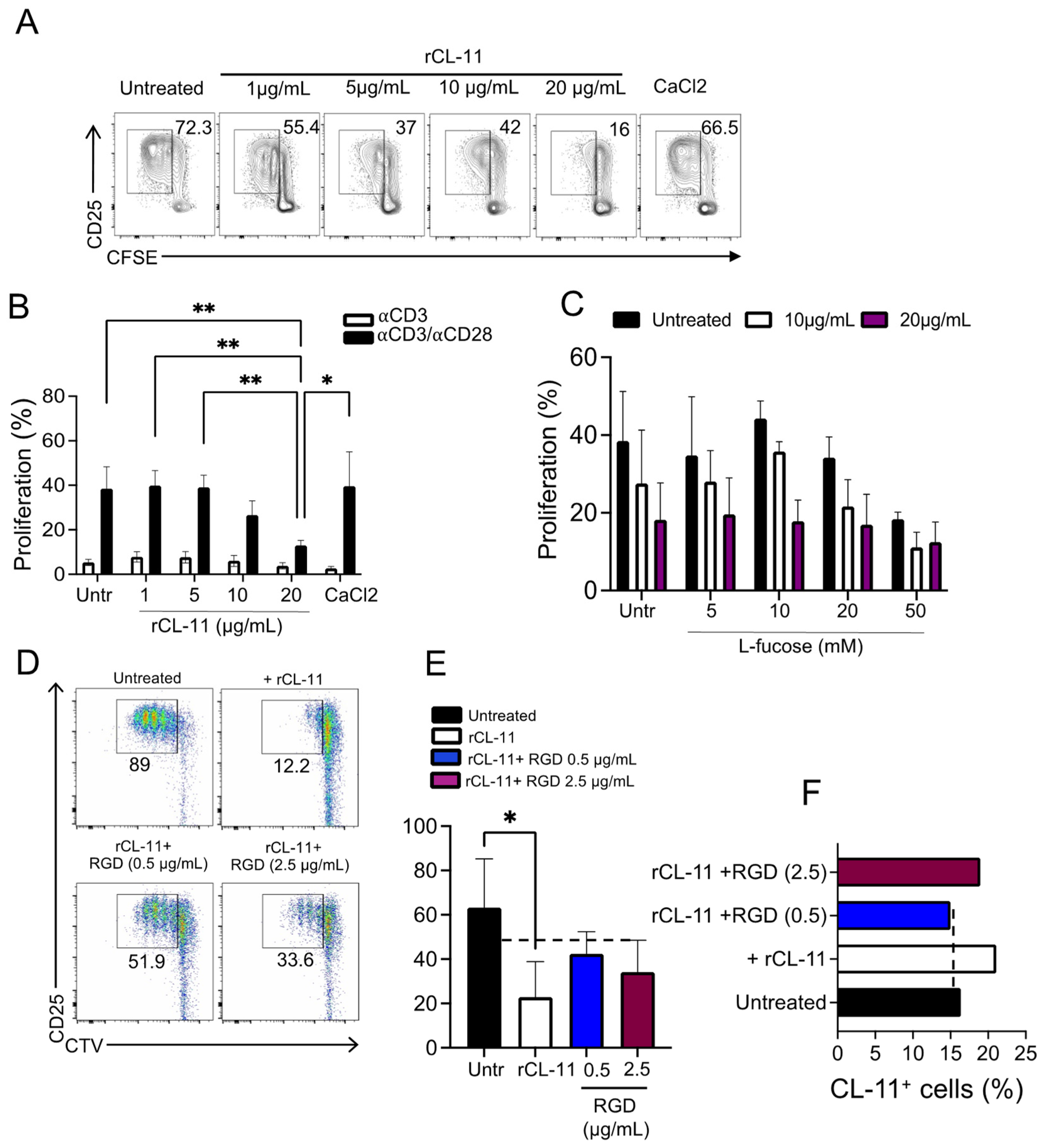
3.4. The Inhibition of T Cell Proliferation Was Mediated by the Binding of CL-11 through Its CLD Domain
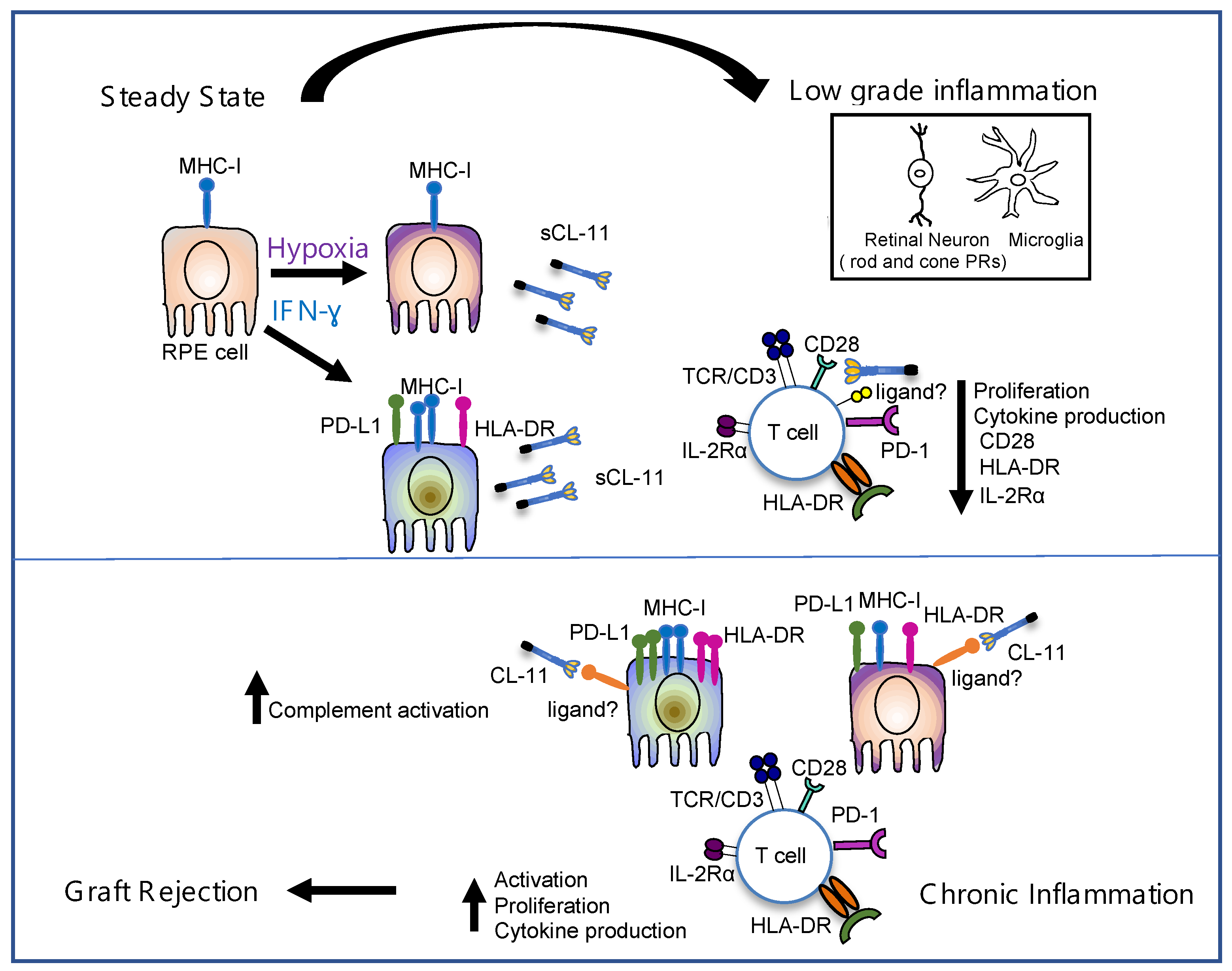
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bora, N.S.; Jha, P.; Bora, P.S. The role of complement in ocular pathology. Semin. Immunopathol. 2008, 30, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, V.L.; Caspi, R.R. Immune mechanisms in inflammatory and degenerative eye disease. Trends Immunol. 2015, 36, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Whitcup, S.M.; Sodhi, A.; Atkinson, J.P.; Holers, V.M.; Sinha, D.; Rohrer, B.; Dick, A.D. The role of the immune response in age-related macular degeneration. Int. J. Inflamm. 2013, 2013, 348092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benhar, I.; London, A.; Schwartz, M. The privileged immunity of immune privileged organs: The case of the eye. Front. Immunol. 2012, 3, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streilein, J.W. Ocular immune privilege: Therapeutic opportunities from an experiment of nature. Nat. Rev. Immunol. 2003, 3, 879–889. [Google Scholar] [CrossRef]
- Sheedlo, H.J.; Li, L.; Fan, W.; Turner, J.E. Retinal pigment epithelial cell support of photoreceptor survival in vitro. Vitr. Cell Dev. Biol. Anim. 1995, 31, 330–333. [Google Scholar] [CrossRef]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [Green Version]
- Sugita, S.; Usui, Y.; Horie, S.; Futagami, Y.; Aburatani, H.; Okazaki, T.; Honjo, T.; Takeuchi, M.; Mochizuki, M. T-cell suppression by programmed cell death 1 ligand 1 on retinal pigment epithelium during inflammatory conditions. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2862–2870. [Google Scholar] [CrossRef]
- Sugita, S.; Horie, S.; Nakamura, O.; Futagami, Y.; Takase, H.; Keino, H.; Aburatani, H.; Katunuma, N.; Ishidoh, K.; Yamamoto, Y.; et al. Retinal pigment epithelium-derived CTLA-2alpha induces TGFbeta-producing T regulatory cells. J. Immunol. 2008, 181, 7525–7536. [Google Scholar] [CrossRef] [Green Version]
- Kawazoe, Y.; Sugita, S.; Keino, H.; Yamada, Y.; Imai, A.; Horie, S.; Mochizuki, M. Retinoic acid from retinal pigment epithelium induces T regulatory cells. Exp. Eye Res. 2012, 94, 32–40. [Google Scholar] [CrossRef]
- Sugita, S.; Futagami, Y.; Smith, S.B.; Naggar, H.; Mochizuki, M. Retinal and ciliary body pigment epithelium suppress activation of T lymphocytes via transforming growth factor beta. Exp. Eye Res. 2006, 83, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Sugita, S.; Iwasaki, Y.; Makabe, K.; Kamao, H.; Mandai, M.; Shiina, T.; Ogasawara, K.; Hirami, Y.; Kurimoto, Y.; Takahashi, M. Successful Transplantation of Retinal Pigment Epithelial Cells from MHC Homozygote iPSCs in MHC-Matched Models. Stem. Cell Rep. 2016, 7, 635–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugita, S.; Iwasaki, Y.; Makabe, K.; Kimura, T.; Futagami, T.; Suegami, S.; Takahashi, M. Lack of T Cell Response to iPSC-Derived Retinal Pigment Epithelial Cells from HLA Homozygous Donors. Stem. Cell Rep. 2016, 7, 619–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugita, S.; Kamao, H.; Iwasaki, Y.; Okamoto, S.; Hashiguchi, T.; Iseki, K.; Hayashi, N.; Mandai, M.; Takahashi, M. Inhibition of T-cell activation by retinal pigment epithelial cells derived from induced pluripotent stem cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1051–1062. [Google Scholar] [CrossRef]
- Juel, H.B.; Faber, C.; Udsen, M.S.; Folkersen, L.; Nissen, M.H. Chemokine expression in retinal pigment epithelial ARPE-19 cells in response to coculture with activated T cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 8472–8480. [Google Scholar] [CrossRef] [Green Version]
- Zipfel, P.F.; Skerka, C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009, 9, 729–740. [Google Scholar] [CrossRef]
- Juel, H.B.; Kaestel, C.; Folkersen, L.; Faber, C.; Heegaard, N.H.; Borup, R.; Nissen, M.H. Retinal pigment epithelial cells upregulate expression of complement factors after co-culture with activated T cells. Exp. Eye Res. 2011, 92, 180–188. [Google Scholar] [CrossRef]
- Luo, C.; Zhao, J.; Madden, A.; Chen, M.; Xu, H. Complement expression in retinal pigment epithelial cells is modulated by activated macrophages. Exp. Eye Res. 2013, 112, 93–101. [Google Scholar] [CrossRef]
- Yang, P.; Tyrrell, J.; Han, I.; Jaffe, G.J. Expression and modulation of RPE cell membrane complement regulatory proteins. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3473–3481. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Forrester, J.V.; Xu, H. Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp. Eye Res. 2007, 84, 635–645. [Google Scholar] [CrossRef]
- Anderson, D.H.; Radeke, M.J.; Gallo, N.B.; Chapin, E.A.; Johnson, P.T.; Curletti, C.R.; Hancox, L.S.; Hu, J.; Ebright, J.N.; Malek, G.; et al. The pivotal role of the complement system in aging and age-related macular degeneration: Hypothesis re-visited. Prog. Retin. Eye Res. 2010, 29, 95–112. [Google Scholar] [CrossRef] [Green Version]
- Fritsche, L.G.; Chen, W.; Schu, M.; Yaspan, B.L.; Yu, Y.; Thorleifsson, G.; Zack, D.J.; Arakawa, S.; Cipriani, V.; Ripke, S.; et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 2013, 45, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Seddon, J.M.; Yu, Y.; Miller, E.C.; Reynolds, R.; Tan, P.L.; Gowrisankar, S.; Goldstein, J.I.; Triebwasser, M.; Anderson, H.E.; Zerbib, J.; et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat. Genet. 2013, 45, 1366–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fanelli, G.; Gonzalez-Cordero, A.; Gardner, P.J.; Peng, Q.; Fernando, M.; Kloc, M.; Farrar, C.A.; Naeem, A.; Garred, P.; Ali, R.R.; et al. Human stem cell-derived retinal epithelial cells activate complement via collectin 11 in response to stress. Sci. Rep. 2017, 7, 14625. [Google Scholar] [CrossRef] [Green Version]
- Hansen, S.; Selman, L.; Palaniyar, N.; Ziegler, K.; Brandt, J.; Kliem, A.; Jonasson, M.; Skjoedt, M.O.; Nielsen, O.; Hartshorn, K.; et al. Collectin 11 (CL-11, CL-K1) is a MASP-1/3-associated plasma collectin with microbial-binding activity. J. Immunol. 2010, 185, 6096–6104. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.J.; Skjoedt, M.O.; Garred, P. Collectin-11/MASP complex formation triggers activation of the lectin complement pathway—The fifth lectin pathway initiation complex. J. Innate Immun. 2013, 5, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.J. Acute kidney injury: Collectin-11: A signal for complement activation. Nat. Rev. Nephrol. 2016, 12, 378. [Google Scholar] [CrossRef] [PubMed]
- Nauser, C.L.; Sacks, S.H. Local complement synthesis-A process with near and far consequences for ischemia reperfusion injury and transplantation. Immunol. Rev. 2023, 313, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Farrar, C.A.; Tran, D.; Li, K.; Wu, W.; Peng, Q.; Schwaeble, W.; Zhou, W.; Sacks, S.H. Collectin-11 detects stress-induced L-fucose pattern to trigger renal epithelial injury. J. Clin. Investig. 2016, 126, 1911–1925. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Wu, W.; Ma, L.; Liu, C.; Bhuckory, M.B.; Wang, L.; Nandrot, E.F.; Xu, H.; Li, K.; Liu, Y.; et al. Collectin-11 Is an Important Modulator of Retinal Pigment Epithelial Cell Phagocytosis and Cytokine Production. J. Innate Immun. 2017, 9, 529–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, M.; Elgueta, R.; McCluskey, D.; Ortega-Prieto, A.M.; Stolarczyk, E.; Dazzi, F.; Lucendo-Villarin, B.; Meseguer-Ripolles, J.; Williams, J.; Fanelli, G.; et al. Pluripotent Stem Cell-Derived Hepatocytes Inhibit T Cell Proliferation. Cells 2021, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Sollazzo, D.; Trabanelli, S.; Barone, M.; Polverelli, N.; Perricone, M.; Forte, D.; Luatti, S.; Cavo, M.; Vianelli, N.; et al. Mutations in. Oncoimmunology 2017, 6, e1345402. [Google Scholar] [CrossRef] [Green Version]
- Howard, M.C.; Nauser, C.L.; Farrar, C.A.; Wallis, R.; Sacks, S.H. l-Fucose prevention of renal ischaemia/reperfusion injury in Mice. FASEB J. 2020, 34, 822–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idelson, M.; Alper, R.; Obolensky, A.; Yachimovich-Cohen, N.; Rachmilewitz, J.; Ejzenberg, A.; Beider, E.; Banin, E.; Reubinoff, B. Immunological Properties of Human Embryonic Stem Cell-Derived Retinal Pigment Epithelial Cells. Stem. Cell Rep. 2018, 11, 681–695. [Google Scholar] [CrossRef] [Green Version]
- Azuma, M. Co-signal Molecules in T-Cell Activation: Historical Overview and Perspective. Adv. Exp. Med. Biol. 2019, 1189, 3–23. [Google Scholar] [CrossRef]
- Detrick, B.; Rodrigues, M.; Chan, C.C.; Tso, M.O.; Hooks, J.J. Expression of HLA-DR antigen on retinal pigment epithelial cells in retinitis pigmentosa. Am. J. Ophthalmol. 1986, 101, 584–590. [Google Scholar] [CrossRef]
- Osusky, R.; Dorio, R.J.; Arora, Y.K.; Ryan, S.J.; Walker, S.M. MHC class II positive retinal pigment epithelial (RPE) cells can function as antigen-presenting cells for microbial superantigen. Ocul. Immunol. Inflamm. 1997, 5, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Plaza Reyes, A.; Petrus-Reurer, S.; Padrell Sánchez, S.; Kumar, P.; Douagi, I.; Bartuma, H.; Aronsson, M.; Westman, S.; Lardner, E.; André, H.; et al. Identification of cell surface markers and establishment of monolayer differentiation to retinal pigment epithelial cells. Nat. Commun. 2020, 11, 1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, T.; Nishida, T.; Takagi, E.; Miyao, K.; Koyama, D.; Sakemura, R.; Hanajiri, R.; Watanabe, K.; Imahashi, N.; Terakura, S.; et al. Programmed Death-Ligand 1 on Antigen-presenting Cells Facilitates the Induction of Antigen-specific Cytotoxic T Lymphocytes: Application to Adoptive T-Cell Immunotherapy. J. Immunother. 2016, 39, 306–315. [Google Scholar] [CrossRef]
- Couture, A.; Garnier, A.; Docagne, F.; Boyer, O.; Vivien, D.; Le-Mauff, B.; Latouche, J.B.; Toutirais, O. HLA-Class II Artificial Antigen Presenting Cells in CD4. Front. Immunol. 2019, 10, 1081. [Google Scholar] [CrossRef]
- Sugita, S.; Futatsugi, Y.; Ishida, M.; Edo, A.; Takahashi, M. Retinal Pigment Epithelial Cells Derived from Induced Pluripotent Stem (iPS) Cells Suppress or Activate T Cells via Costimulatory Signals. Int. J. Mol. Sci. 2020, 21, 6507. [Google Scholar] [CrossRef] [PubMed]
- Luckheeram, R.V.; Zhou, R.; Verma, A.D.; Xia, B. CD4⁺T cells: Differentiation and functions. Clin. Dev. Immunol. 2012, 2012, 925135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jankovic, D.; Kugler, D.G.; Sher, A. IL-10 production by CD4+ effector T cells: A mechanism for self-regulation. Mucosal Immunol. 2010, 3, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Sugita, S.; Kawazoe, Y.; Imai, A.; Usui, Y.; Iwakura, Y.; Isoda, K.; Ito, M.; Mochizuki, M. Mature dendritic cell suppression by IL-1 receptor antagonist on retinal pigment epithelium cells. Investig. Ophthalmol. Vis. Sci. 2013, 54, 3240–3249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregerson, D.S.; Sam, T.N.; McPherson, S.W. The antigen-presenting activity of fresh, adult parenchymal microglia and perivascular cells from retina. J. Immunol. 2004, 172, 6587–6597. [Google Scholar] [CrossRef] [Green Version]
- Ali, Y.M.; Lynch, N.J.; Haleem, K.S.; Fujita, T.; Endo, Y.; Hansen, S.; Holmskov, U.; Takahashi, K.; Stahl, G.L.; Dudler, T.; et al. The lectin pathway of complement activation is a critical component of the innate immune response to pneumococcal infection. PLoS Pathog. 2012, 8, e1002793. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Wu, J.; Xiong, S.; Zhang, L.; Lu, X.; Chen, S.; Wu, Q.; Wang, H.; Liu, Y.; Chen, Z.; et al. Mannan-binding lectin, a serum collectin, suppresses T-cell proliferation. FASEB J. 2017, 31, 2405–2417. [Google Scholar] [CrossRef] [Green Version]
- Fanelli, G.; Romano, M.; Nova-Lamperti, E.; Werner Sunderland, M.; Nerviani, A.; Scottà, C.; Bombardieri, M.; Quezada, S.A.; Sacks, S.H.; Noelle, R.J.; et al. PD-L1 signaling on human memory CD4+ T cells induces a regulatory phenotype. PLoS Biol. 2021, 19, e3001199. [Google Scholar] [CrossRef]
- Weng, N.P.; Akbar, A.N.; Goronzy, J. CD28(-) T cells: Their role in the age-associated decline of immune function. Trends Immunol. 2009, 30, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Selman, L.; Hansen, S. Structure and function of collectin liver 1 (CL-L1) and collectin 11 (CL-11, CL-K1). Immunobiology 2012, 217, 851–863. [Google Scholar] [CrossRef]
- Howard, M.C.; Nauser, C.L.; Vizitiu, D.A.; Sacks, S.H. Fucose as a new therapeutic target in renal transplantation. Pediatr. Nephrol. 2021, 36, 1065–1073. [Google Scholar] [CrossRef]
- Bellis, S.L. Advantages of RGD peptides for directing cell association with biomaterials. Biomaterials 2011, 32, 4205–4210. [Google Scholar] [CrossRef] [Green Version]
- West, E.E.; Kolev, M.; Kemper, C. Complement and the Regulation of T Cell Responses. Annu. Rev. Immunol. 2018, 36, 309–338. [Google Scholar] [CrossRef] [PubMed]
- Tavano, R.; Contento, R.L.; Baranda, S.J.; Soligo, M.; Tuosto, L.; Manes, S.; Viola, A. CD28 interaction with filamin-A controls lipid raft accumulation at the T-cell immunological synapse. Nat. Cell Biol. 2006, 8, 1270–1276. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Altman, A. Filamin A is required for T cell activation mediated by protein kinase C-theta. J. Immunol. 2006, 177, 1721–1728. [Google Scholar] [CrossRef]
- Siokis, A.; Robert, P.A.; Demetriou, P.; Dustin, M.L.; Meyer-Hermann, M. F-Actin-Driven CD28-CD80 Localization in the Immune Synapse. Cell Rep. 2018, 24, 1151–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zumerle, S.; Molon, B.; Viola, A. Membrane Rafts in T Cell Activation: A Spotlight on CD28 Costimulation. Front. Immunol. 2017, 8, 1467. [Google Scholar] [CrossRef] [PubMed]
- Swigut, T.; Shohdy, N.; Skowronski, J. Mechanism for down-regulation of CD28 by Nef. EMBO J. 2001, 20, 1593–1604. [Google Scholar] [CrossRef] [Green Version]
- Gardai, S.J.; Xiao, Y.Q.; Dickinson, M.; Nick, J.A.; Voelker, D.R.; Greene, K.E.; Henson, P.M. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell 2003, 115, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Gao, Z.; Lu, X.; Hu, F. Role of collectin-11 in innate defence against uropathogenic. Innate Immun. 2021, 27, 50–60. [Google Scholar] [CrossRef]
- Wang, J.; Ohno-Matsui, K.; Yoshida, T.; Shimada, N.; Ichinose, S.; Sato, T.; Mochizuki, M.; Morita, I. Amyloid-beta up-regulates complement factor B in retinal pigment epithelial cells through cytokines released from recruited macrophages/microglia: Another mechanism of complement activation in age-related macular degeneration. J. Cell Physiol. 2009, 220, 119–128. [Google Scholar] [CrossRef]
- Chen, M.; Muckersie, E.; Robertson, M.; Forrester, J.V.; Xu, H. Up-regulation of complement factor B in retinal pigment epithelial cells is accompanied by complement activation in the aged retina. Exp. Eye Res. 2008, 87, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Kazmierkiewicz, K.L.; Bowman, A.S.; Li, M.; Curcio, C.A.; Stambolian, D.E. Transcriptome of the human retina, retinal pigmented epithelium and choroid. Genomics 2015, 105, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Rutar, M.; Valter, K.; Natoli, R.; Provis, J.M. Synthesis and propagation of complement C3 by microglia/monocytes in the aging retina. PLoS ONE 2014, 9, e93343. [Google Scholar] [CrossRef] [PubMed]
- Pauly, D.; Agarwal, D.; Dana, N.; Schäfer, N.; Biber, J.; Wunderlich, K.A.; Jabri, Y.; Straub, T.; Zhang, N.R.; Gautam, A.K.; et al. Cell-Type-Specific Complement Expression in the Healthy and Diseased Retina. Cell Rep. 2019, 29, 2835–2848.e2834. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fanelli, G.; Romano, M.; Lombardi, G.; Sacks, S.H. Soluble Collectin 11 (CL-11) Acts as an Immunosuppressive Molecule Potentially Used by Stem Cell-Derived Retinal Epithelial Cells to Modulate T Cell Response. Cells 2023, 12, 1805. https://doi.org/10.3390/cells12131805
Fanelli G, Romano M, Lombardi G, Sacks SH. Soluble Collectin 11 (CL-11) Acts as an Immunosuppressive Molecule Potentially Used by Stem Cell-Derived Retinal Epithelial Cells to Modulate T Cell Response. Cells. 2023; 12(13):1805. https://doi.org/10.3390/cells12131805
Chicago/Turabian StyleFanelli, Giorgia, Marco Romano, Giovanna Lombardi, and Steven H. Sacks. 2023. "Soluble Collectin 11 (CL-11) Acts as an Immunosuppressive Molecule Potentially Used by Stem Cell-Derived Retinal Epithelial Cells to Modulate T Cell Response" Cells 12, no. 13: 1805. https://doi.org/10.3390/cells12131805
APA StyleFanelli, G., Romano, M., Lombardi, G., & Sacks, S. H. (2023). Soluble Collectin 11 (CL-11) Acts as an Immunosuppressive Molecule Potentially Used by Stem Cell-Derived Retinal Epithelial Cells to Modulate T Cell Response. Cells, 12(13), 1805. https://doi.org/10.3390/cells12131805