
ROP16 of Toxoplasma gondii Inhibits Innate Immunity by Triggering cGAS-STING Pathway Inactivity through the Polyubiquitination of STING

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Parasite Culture
2.2. Antibodies and Reagents
2.3. Plasmid Construction
2.4. Transfection of RAW264.7 Cells
2.5. Quantitative Real-Time Reverse Transcription PCR (qRT-PCR)
2.6. Immunofluorescence Assay
2.7. Co-Immunoprecipitation
2.8. Western Blotting Assay
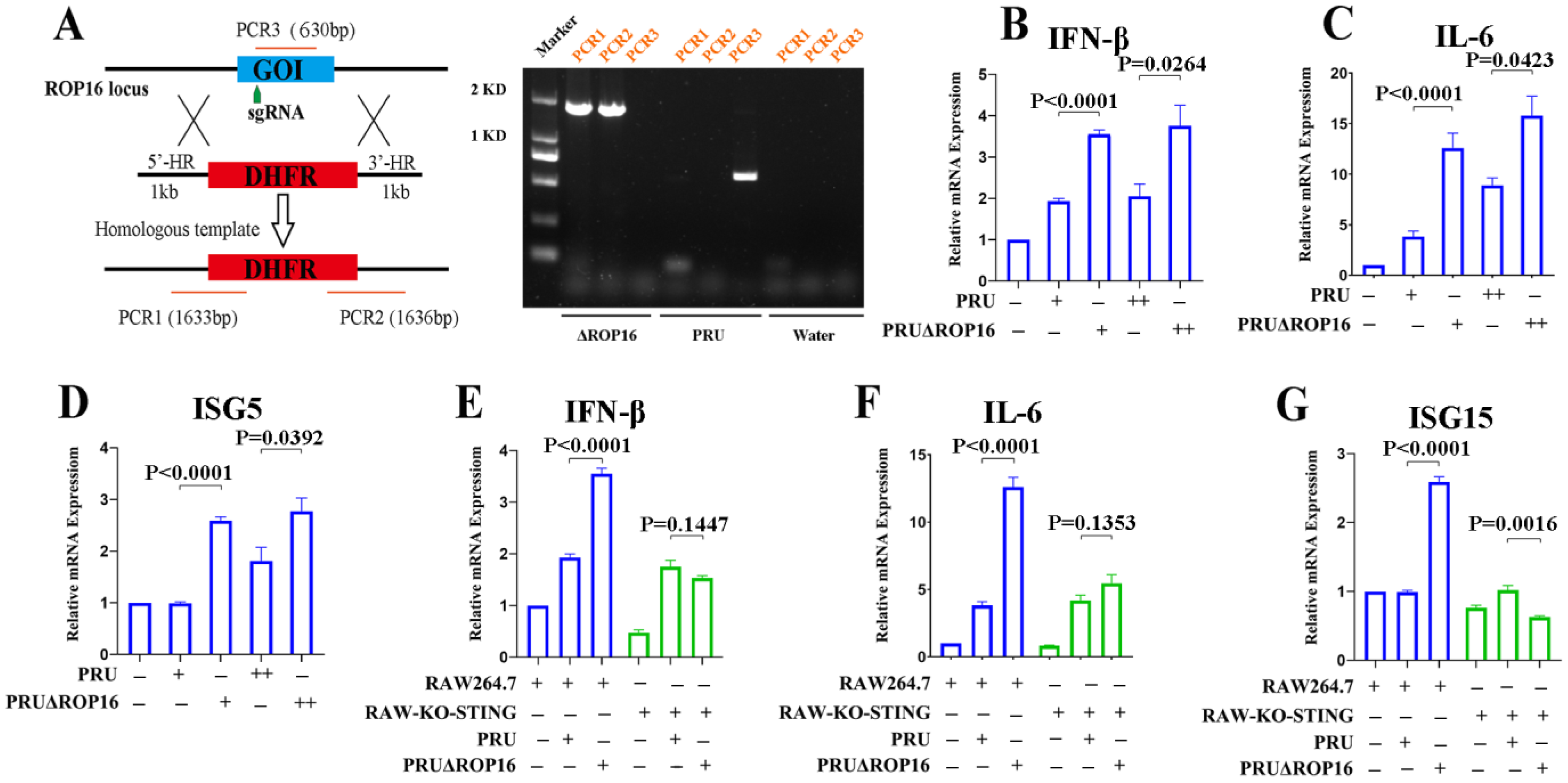
2.9. Generation of ROP16 Knockout PRU (PRUΔROP16)
2.10. T. gondii PRU Tachyzoites Infection In Vitro
2.11. Statistical Analysis
3. Results
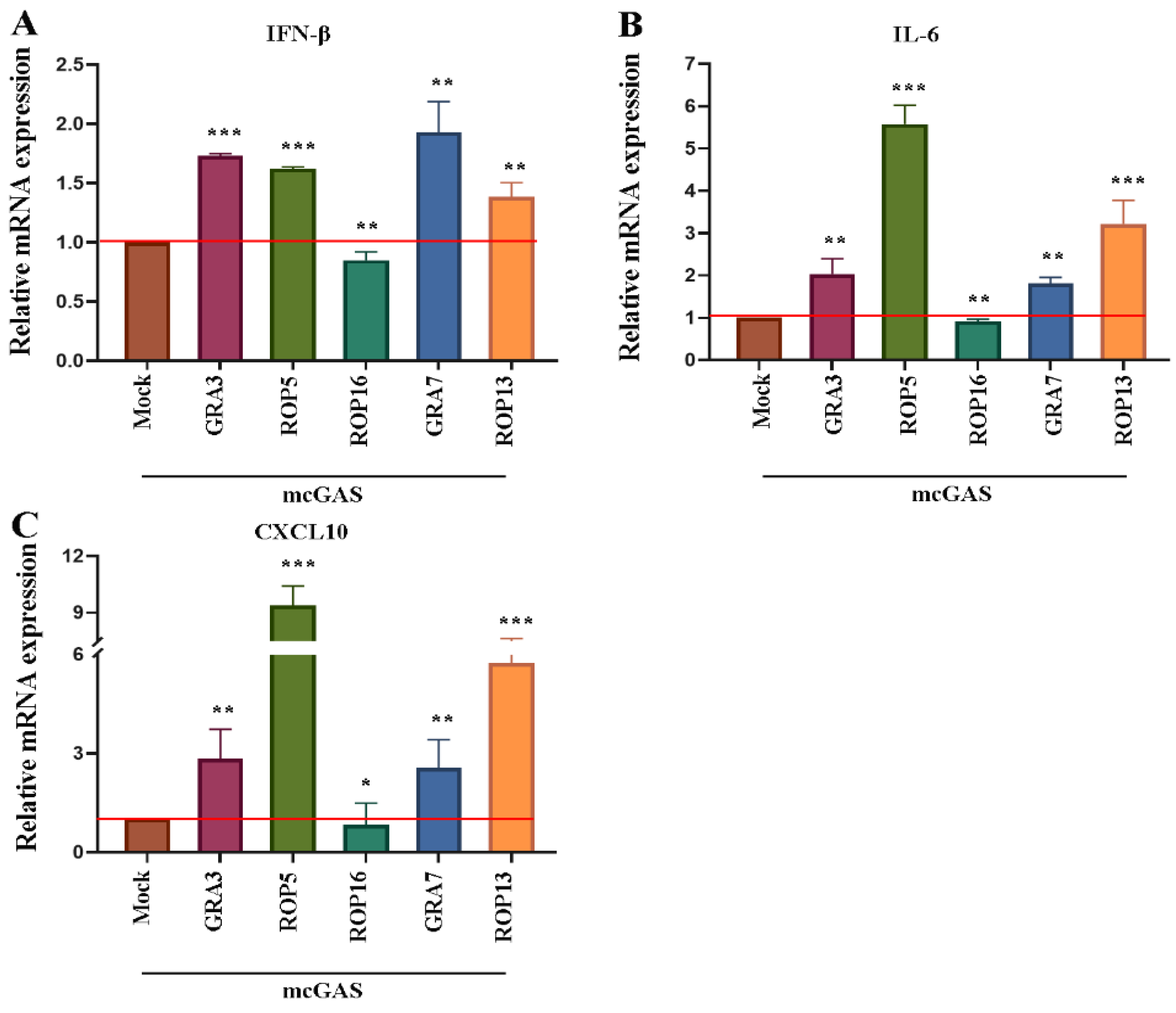
3.1. T. gondii ROP16 Is a Potential Inhibitor of the cGAS-STING Pathway
3.2. T. gondii ROP16 Inhibited cGAS-STING-Mediated Signaling to Regulate Immune Responses
3.3. T. gondii PRU ROP16-Deficiency Potentiates Innate Immune Responses
3.4. T. gondii ROP16 Interacted with STING
3.5. T. gondii ROP16 Inhibits cGAS-STING Signaling Based on the Predicted NLS Domain
3.6. T. gondii ROP16 Inhibits the K63-Linked Polyubiquitylation of STING
3.7. T. gondii ROP16 Plays an Important Role in T. gondii Immune Evasion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, Q.; Wang, Z.-D.; Huang, S.-Y.; Zhu, X.-Q. Diagnosis of toxoplasmosis and typing of Toxoplasma gondii. Parasites Vectors 2015, 8, 292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert-Gangneux, F.; Dardé, M.-L. Epidemiology of and Diagnostic Strategies for Toxoplasmosis. Clin. Microbiol. Rev. 2012, 25, 264–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochanowsky, J.A.; Koshy, A.A. Toxoplasma gondii. Curr. Biol. 2018, 28, R770–R771. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.A.; Moretto, M. Immune responses to Toxoplasma gondii. Curr. Opin. Immunol. 2022, 77, 102226. [Google Scholar] [CrossRef] [PubMed]
- Lima, T.S.; Lodoen, M.B. Mechanisms of Human Innate Immune Evasion by Toxoplasma gondii. Front. Cell. Infect. Microbiol. 2019, 9, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, C.A.; Sibley, L.D. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat. Rev. Genet. 2012, 10, 766–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihara, F.; Fereig, R.M.; Himori, Y.; Kameyama, K.; Umeda, K.; Tanaka, S.; Ikeda, R.; Yamamoto, M.; Nishikawa, Y. Toxoplasma gondii Dense Granule Proteins 7, 14, and 15 Are Involved in Modification and Control of the Immune Response Mediated via NF-κB Pathway. Front. Immunol. 2020, 11, 1709. [Google Scholar] [CrossRef]
- Sasai, M.; Yamamoto, M. Innate, adaptive, and cell-autonomous immunity against Toxoplasma gondii infection. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yarovinsky, F. Innate immunity to Toxoplasma gondii infection. Nat. Rev. Immunol. 2014, 14, 109–121. [Google Scholar] [CrossRef]
- Pifer, R.; Yarovinsky, F. Innate responses to Toxoplasma gondii in mice and humans. Trends Parasitol. 2011, 27, 388–393. [Google Scholar] [CrossRef] [Green Version]
- Butterworth, S.; Torelli, F.; Lockyer, E.J.; Wagener, J.; Song, O.-R.; Broncel, M.; Russell, M.R.G.; Moreira-Souza, A.C.A.; Young, J.C.; Treeck, M. Toxoplasma gondii virulence factor ROP1 reduces parasite susceptibility to murine and human innate immune restriction. PLoS Pathog. 2022, 18, e1011021. [Google Scholar] [CrossRef] [PubMed]
- Sher, A.; Tosh, K.; Jankovic, D. Innate recognition of Toxoplasma gondii in humans involves a mechanism distinct from that utilized by rodents. Cell. Mol. Immunol. 2016, 14, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.; Ham, Y.S.; Gil, W.J.; Yang, C.-S. The strategies of NLRP3 inflammasome to combat Toxoplasma gondii. Front. Immunol. 2022, 13, 1002387. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ablasser, A.; Schmid-Burgk, J.L.; Hemmerling, I.; Horvath, G.L.; Schmidt, T.; Latz, E.; Hornung, V. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 2013, 503, 530–534. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Chiu, Y.-H.; Chen, Z.J. The cGAS-cGAMP-STING Pathway of Cytosolic DNA Sensing and Signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Hopfner, K.-P.; Hornung, V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef]
- Cheng, Z.; Dai, T.; He, X.; Zhang, Z.; Xie, F.; Wang, S.; Zhang, L.; Zhou, F. The interactions between cGAS-STING pathway and pathogens. Signal Transduct. Target. Ther. 2020, 5, 91. [Google Scholar] [CrossRef]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS–STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Wang, P.; Li, S.; Zhao, Y.; Zhang, B.; Li, Y.; Liu, S.; Du, H.; Cao, L.; Ou, M.; Ye, X.; et al. The GRA15 protein from Toxoplasma gondii enhances host defense responses by activating the interferon stimulator STING. J. Biol. Chem. 2019, 294, 16494–16508. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Yao, L.; Zhou, L.; Yang, P.; Zou, W.; Xu, L.; Li, S.; Peng, H. Toxoplasma gondii ROP18I inhibits host innate immunity through cGAS-STING signaling. FASEB J. 2022, 36, e22171. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.D.C.; Hu, K.; Whitmarsh, R.J.; Hassan, M.A.; Julien, L.; Lu, D.; Chen, L.; Hunter, C.A.; Saeij, J.P.J. Toxoplasma gondii Rhoptry 16 Kinase Promotes Host Resistance to Oral Infection and Intestinal Inflammation Only in the Context of the Dense Granule Protein GRA15. Infect. Immun. 2013, 81, 2156–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-De-Los-Ríos, A.; Murillo-Leon, M.; Mantilla-Muriel, L.E.; Arenas, A.F.; Vargas-Montes, M.; Cardona, N.; De-La-Torre, A.; Sepulveda-Arias, J.C.; Gómez-Marín, J.E. Influence of Two Major Toxoplasma Gondii Virulence Factors (ROP16 and ROP18) on the Immune Response of Peripheral Blood Mononuclear Cells to Human Toxoplasmosis Infection. Front. Cell. Infect. Microbiol. 2019, 9, 413. [Google Scholar] [CrossRef]
- Chen, L.; Christian, D.A.; Kochanowsky, J.A.; Phan, A.T.; Clark, J.T.; Wang, S.; Berry, C.; Oh, J.; Chen, X.; Roos, D.S.; et al. The Toxoplasma gondii virulence factor ROP16 acts in cis and trans, and suppresses T cell responses. J. Exp. Med. 2020, 217, e20181757. [Google Scholar] [CrossRef]
- Kochanowsky, J.A.; Thomas, K.K.; Koshy, A.A. ROP16-Mediated Activation of STAT6 Suppresses Host Cell Reactive Oxygen Species Production, Facilitating Type III Toxoplasma gondii Growth and Survival. mBio 2021, 12, e03305-20. [Google Scholar] [CrossRef] [PubMed]
- Pierog, P.L.; Zhao, Y.; Singh, S.; Dai, J.; Yap, G.S.; Fitzgerald-Bocarsly, P. Toxoplasma gondii Inactivates Human Plasmacytoid Dendritic Cells by Functional Mimicry of IL-10. J. Immunol. 2018, 200, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.; Zhou, Y.; Wang, Y.; Li, L.; Song, Y.; Hou, L.; Zhao, J. Screening and Identification of the Host Proteins Interacting with Toxoplasma gondii Rhoptry Protein ROP16. Front. Microbiol. 2017, 8, 2408. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Shen, B.; Brown, K.; Long, S.; Sibley, L.D. Development of CRISPR/Cas9 for Efficient Genome Editing in Toxoplasma gondii. Methods Mol. Biol. 2017, 1498, 79–103. [Google Scholar] [CrossRef] [PubMed]
- Shastri, A.J.; Marino, N.D.; Franco, M.; Lodoen, M.B.; Boothroyd, J.C. GRA25 Is a Novel Virulence Factor of Toxoplasma gondii and Influences the Host Immune Response. Infect. Immun. 2014, 82, 2595–2605. [Google Scholar] [CrossRef]
- Jensen, K.D.C.; Camejo, A.; Melo, M.B.; Cordeiro, C.; Julien, L.; Grotenbreg, G.M.; Frickel, E.-M.; Ploegh, H.L.; Young, L.; Saeij, J.P.J. Toxoplasma gondii Superinfection and Virulence during Secondary Infection Correlate with the Exact ROP5/ROP18 Allelic Combination. mBio 2015, 6, e02280-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bando, H.; Sakaguchi, N.; Lee, Y.; Pradipta, A.; Ma, J.S.; Tanaka, S.; Lai, D.-H.; Liu, J.; Lun, Z.-R.; Nishikawa, Y.; et al. Toxoplasma Effector TgIST Targets Host IDO1 to Antagonize the IFN-γ-Induced Anti-parasitic Response in Human Cells. Front. Immunol. 2018, 9, 2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeij, J.P.J.; Coller, S.; Boyle, J.P.; Jerome, M.E.; White, M.W.; Boothroyd, J.C. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 2007, 445, 324–327. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Du, F.; Xu, P.; Shu, C.; Sankaran, B.; Bell, S.L.; Liu, M.; Lei, Y.; Gao, X.; Fu, X.; et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature 2019, 569, 718–722. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.; Zhang, C.; Chen, Z.J.; Bai, X.-C.; Zhang, X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP–AMP. Nature 2019, 567, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, M.-X.; Zhang, Q.; Zhu, G.-F.; Yuan, L.; Zhang, D.-E.; Zhu, Q.; Yao, J.; Shu, H.-B.; Zhong, B. USP18 recruits USP20 to promote innate antiviral response through deubiquitinating STING/MITA. Cell Res. 2016, 26, 1302–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Hu, M.-M.; Wang, Y.-Y.; Shu, H.-B. TRIM32 Protein Modulates Type I Interferon Induction and Cellular Antiviral Response by Targeting MITA/STING Protein for K63-linked Ubiquitination. J. Biol. Chem. 2012, 287, 28646–28655. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Jiang, F.; Kong, L.; Wu, H.; Zhang, H.; Chen, X.; Zhao, J.; Cai, B.; Li, Y.; Ma, C.; et al. OTUD5 promotes innate antiviral and antitumor immunity through deubiquitinating and stabilizing STING. Cell. Mol. Immunol. 2020, 18, 1945–1955. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Q.-W.; Yu, T.; Pan, M.; Fan, Y.-M.; Huang, S.-Y. ROP16 of Toxoplasma gondii Inhibits Innate Immunity by Triggering cGAS-STING Pathway Inactivity through the Polyubiquitination of STING. Cells 2023, 12, 1862. https://doi.org/10.3390/cells12141862
Jin Q-W, Yu T, Pan M, Fan Y-M, Huang S-Y. ROP16 of Toxoplasma gondii Inhibits Innate Immunity by Triggering cGAS-STING Pathway Inactivity through the Polyubiquitination of STING. Cells. 2023; 12(14):1862. https://doi.org/10.3390/cells12141862
Chicago/Turabian StyleJin, Qi-Wang, Ting Yu, Ming Pan, Yi-Min Fan, and Si-Yang Huang. 2023. "ROP16 of Toxoplasma gondii Inhibits Innate Immunity by Triggering cGAS-STING Pathway Inactivity through the Polyubiquitination of STING" Cells 12, no. 14: 1862. https://doi.org/10.3390/cells12141862