



Insight into Cancer Immunity: MHCs, Immune Cells and Commensal Microbiota
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. MHC-I Reduction and Immune Evasion
3. TsMHC-II Function and Tumor Immune Surveillance
4. NK Cells and Anti-Cancer Immunity
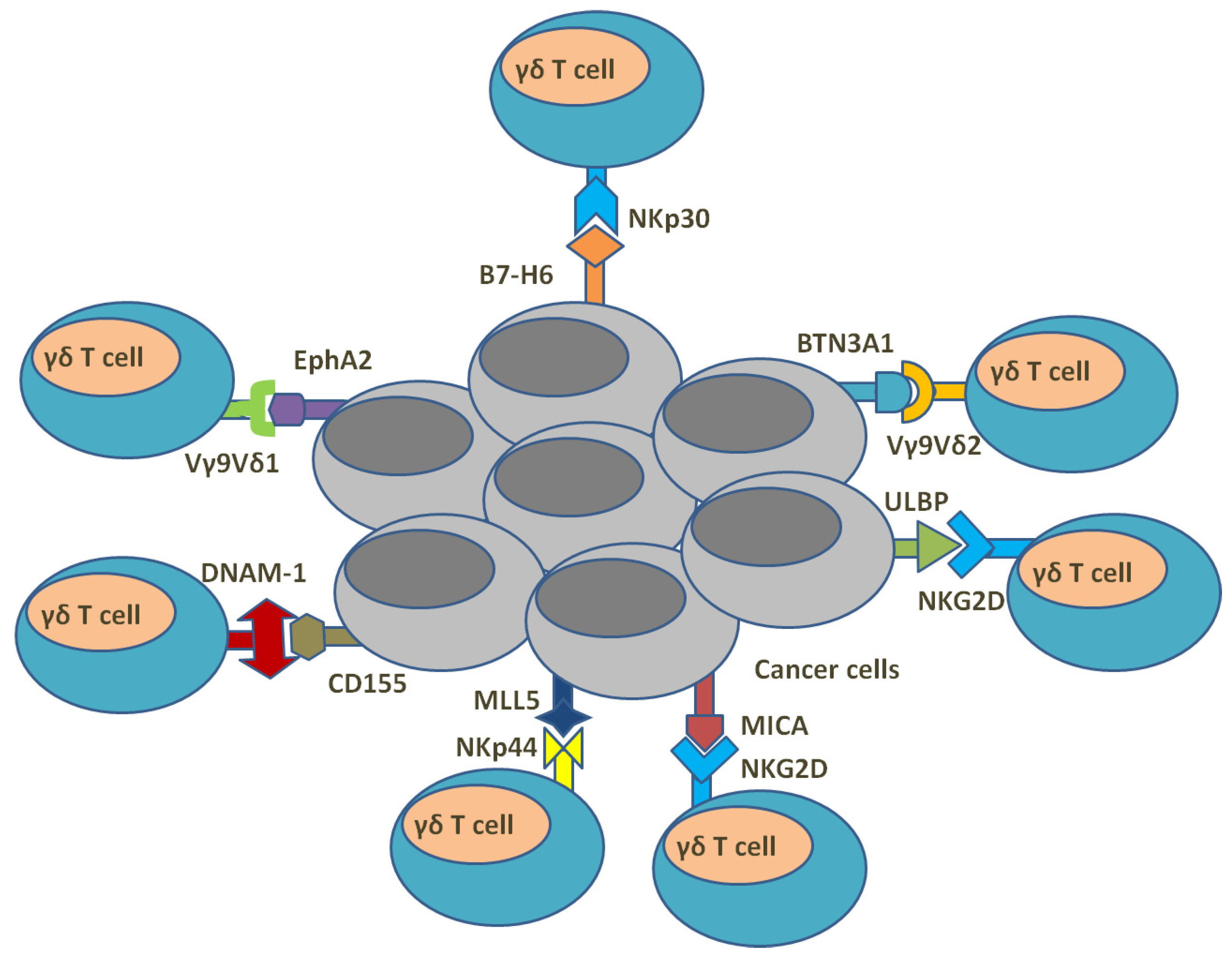
5. γδ. T Cells and Anti-Cancer Immunity
6. Eosinophils and Tumor Microenvironment
7. Microbiota and Anti-Cancer Immunity
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef]
- Parham, P.; Lomen, C.E.; Lawlor, D.A.; Ways, J.P.; Holmes, N.; Coppin, H.L.; Salter, R.D.; Wan, A.M.; Ennis, P.D. Nature of polymorphism in HLA-A, -B, and -C molecules. Proc. Natl. Acad. Sci. USA 1988, 85, 4005–4009. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, P.J.; Parham, P. Structure, function, and diversity of class I major histocompatibility complex molecules. Annu. Rev. Biochem. 1990, 59, 253–288. [Google Scholar] [CrossRef] [PubMed]
- Shawar, S.M.; Vyas, J.M.; Rodgers, J.R.; Rich, R.R. Antigen presentation by major histocompatibility complex class I-B molecules. Annu. Rev. Immunol. 1994, 12, 839–880. [Google Scholar] [CrossRef]
- D’Souza, M.P.; Adams, E.; Altman, J.D.; Birnbaum, M.E.; Boggiano, C.; Casorati, G.; Chien, Y.H.; Conley, A.; Eckle, S.B.G.; Fruh, K.; et al. Casting a wider net: Immunosurveillance by nonclassical MHC molecules. PLoS Pathog. 2019, 15, e1007567. [Google Scholar] [CrossRef]
- Hicklin, D.J.; Marincola, F.M.; Ferrone, S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol. Med. Today 1999, 5, 178–186. [Google Scholar] [CrossRef]
- Koopman, L.A.; Corver, W.E.; van der Slik, A.R.; Giphart, M.J.; Fleuren, G.J. Multiple genetic alterations cause frequent and heterogeneous human histocompatibility leukocyte antigen class I loss in cervical cancer. J. Exp. Med. 2000, 191, 961–976. [Google Scholar] [CrossRef]
- Paschen, A.; Mendez, R.M.; Jimenez, P.; Sucker, A.; Ruiz-Cabello, F.; Song, M.; Garrido, F.; Schadendorf, D. Complete loss of HLA class I antigen expression on melanoma cells: A result of successive mutational events. Int. J. Cancer 2003, 103, 759–767. [Google Scholar] [CrossRef]
- Bukur, J.; Jasinski, S.; Seliger, B. The role of classical and non-classical HLA class I antigens in human tumors. Semin. Cancer Biol. 2012, 22, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Sokol, L.; Koelzer, V.H.; Rau, T.T.; Karamitopoulou, E.; Zlobec, I.; Lugli, A. Loss of tapasin correlates with diminished CD8(+) T-cell immunity and prognosis in colorectal cancer. J. Transl. Med. 2015, 13, 279. [Google Scholar] [CrossRef] [PubMed]
- Campoli, M.; Chang, C.C.; Ferrone, S. HLA class I antigen loss, tumor immune escape and immune selection. Vaccine 2002, 20 (Suppl. S4), A40–A45. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, T.; Lara, E.; Romero, J.M.; Maleno, I.; Real, L.M.; Ruiz-Cabello, F.; Valero, P.; Camacho, F.M.; Garrido, F. HLA class I expression in metastatic melanoma correlates with tumor development during autologous vaccination. Cancer Immunol. Immunother. CII 2007, 56, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B.; Ruiz-Cabello, F.; Garrido, F. IFN inducibility of major histocompatibility antigens in tumors. Adv. Cancer Res. 2008, 101, 249–276. [Google Scholar]
- Campoli, M.; Ferrone, S. HLA antigen changes in malignant cells: Epigenetic mechanisms and biologic significance. Oncogene 2008, 27, 5869–5885. [Google Scholar] [CrossRef]
- Meissner, T.B.; Li, A.; Biswas, A.; Lee, K.H.; Liu, Y.J.; Bayir, E.; Iliopoulos, D.; van den Elsen, P.J.; Kobayashi, K.S. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc. Natl. Acad. Sci. USA 2010, 107, 13794–13799. [Google Scholar] [CrossRef]
- Yoshihama, S.; Roszik, J.; Downs, I.; Meissner, T.B.; Vijayan, S.; Chapuy, B.; Sidiq, T.; Shipp, M.A.; Lizee, G.A.; Kobayashi, K.S. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 5999–6004. [Google Scholar] [CrossRef]
- Cui, J.; Zhu, L.; Xia, X.; Wang, H.Y.; Legras, X.; Hong, J.; Ji, J.; Shen, P.; Zheng, S.; Chen, Z.J.; et al. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell 2010, 141, 483–496. [Google Scholar] [CrossRef]
- Benko, S.; Magalhaes, J.G.; Philpott, D.J.; Girardin, S.E. NLRC5 limits the activation of inflammatory pathways. J. Immunol. 2010, 185, 1681–1691. [Google Scholar] [CrossRef]
- Staehli, F.; Ludigs, K.; Heinz, L.X.; Seguin-Estevez, Q.; Ferrero, I.; Braun, M.; Schroder, K.; Rebsamen, M.; Tardivel, A.; Mattmann, C.; et al. NLRC5 deficiency selectively impairs MHC class I- dependent lymphocyte killing by cytotoxic T cells. J. Immunol. 2012, 188, 3820–3828. [Google Scholar] [CrossRef]
- Kuenzel, S.; Till, A.; Winkler, M.; Hasler, R.; Lipinski, S.; Jung, S.; Grotzinger, J.; Fickenscher, H.; Schreiber, S.; Rosenstiel, P. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J. Immunol. 2010, 184, 1990–2000. [Google Scholar] [CrossRef]
- Neerincx, A.; Rodriguez, G.M.; Steimle, V.; Kufer, T.A. NLRC5 controls basal MHC class I gene expression in an MHC enhanceosome-dependent manner. J. Immunol. 2012, 188, 4940–4950. [Google Scholar] [CrossRef] [PubMed]
- Robbins, G.R.; Truax, A.D.; Davis, B.K.; Zhang, L.; Brickey, W.J.; Ting, J.P. Regulation of class I major histocompatibility complex (MHC) by nucleotide-binding domain, leucine-rich repeat-containing (NLR) proteins. J. Biol. Chem. 2012, 287, 24294–24303. [Google Scholar] [CrossRef]
- Lupfer, C.R.; Stokes, K.L.; Kuriakose, T.; Kanneganti, T.D. Deficiency of the NOD-Like Receptor NLRC5 Results in Decreased CD8(+) T Cell Function and Impaired Viral Clearance. J. Virol. 2017, 91, e00377-17. [Google Scholar] [CrossRef]
- Sun, T.; Ferrero, R.L.; Girardin, S.E.; Gommerman, J.L.; Philpott, D.J. NLRC5 deficiency has a moderate impact on immunodominant CD8(+) T cell responses during rotavirus infection of adult mice. Immunol. Cell Biol. 2019, 97, 552–562. [Google Scholar] [CrossRef]
- Kobayashi, K.S. NLRC5/CITA: A novel regulator of class I major histocompatibility complex genes. J. Immunodefic. Disord. 2012, 1, 1000e102. [Google Scholar] [CrossRef] [PubMed]
- Biswas, A.; Meissner, T.B.; Kawai, T.; Kobayashi, K.S. Cutting edge: Impaired MHC class I expression in mice deficient for Nlrc5/class I transactivator. J. Immunol. 2012, 189, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Wang, Y.; Chen, F.; Huang, Y.; Zhu, S.; Leng, Q.; Wang, H.; Shi, Y.; Qian, Y. NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res. 2012, 22, 836–847. [Google Scholar] [CrossRef]
- Dang, A.T.; Strietz, J.; Zenobi, A.; Khameneh, H.J.; Brandl, S.M.; Lozza, L.; Conradt, G.; Kaufmann, S.H.E.; Reith, W.; Kwee, I.; et al. NLRC5 promotes transcription of BTN3A1-3 genes and Vgamma9Vdelta2 T cell-mediated killing. iScience 2021, 24, 101900. [Google Scholar] [CrossRef]
- Zhang, P.; Yu, C.; Yu, J.; Li, Z.; Lan, H.Y.; Zhou, Q. Arid2-IR promotes NF-kappaB-mediated renal inflammation by targeting NLRC5 transcription. Cell Mol. Life Sci. 2020, 78, 2387–2404. [Google Scholar] [CrossRef]
- Zhou, Q.; Huang, X.R.; Yu, J.; Yu, X.; Lan, H.Y. Long Noncoding RNA Arid2-IR Is a Novel Therapeutic Target for Renal Inflammation. Mol. Ther. J. Am. Soc. Gene Ther. 2015, 23, 1034–1043. [Google Scholar] [CrossRef]
- Zong, Z.; Song, Y.; Xue, Y.; Ruan, X.; Liu, X.; Yang, C.; Zheng, J.; Cao, S.; Li, Z.; Liu, Y. Knockdown of LncRNA SCAMP1 suppressed malignant biological behaviours of glioma cells via modulating miR-499a-5p/LMX1A/NLRC5 pathway. J. Cell Mol. Med. 2019, 23, 5048–5062. [Google Scholar] [CrossRef]
- Periyasamy, P.; Thangaraj, A.; Bendi, V.S.; Buch, S. HIV-1 Tat-mediated microglial inflammation involves a novel miRNA-34a-NLRC5-NFkappaB signaling axis. Brain Behav. Immun. 2019, 80, 227–237. [Google Scholar] [CrossRef]
- Li, J.; Yu, L.; Shen, Z.; Li, Y.; Chen, B.; Wei, W.; Chen, X.; Wang, Q.; Tong, F.; Lou, H.; et al. miR-34a and its novel target, NLRC5, are associated with HPV16 persistence. Infect. Genet. Evol. 2016, 44, 293–299. [Google Scholar] [CrossRef]
- Zong, Y.; Zhang, Y.; Hou, D.; Xu, J.; Cui, F.; Qin, Y.; Sun, X. The lncRNA XIST promotes the progression of breast cancer by sponging miR-125b-5p to modulate NLRC5. Am. J. Transl. Res. 2020, 12, 3501–3511. [Google Scholar]
- Agudo, J.; Park, E.S.; Rose, S.A.; Alibo, E.; Sweeney, R.; Dhainaut, M.; Kobayashi, K.S.; Sachidanandam, R.; Baccarini, A.; Merad, M.; et al. Quiescent Tissue Stem Cells Evade Immune Surveillance. Immunity 2018, 48, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Schatton, T.; Schutte, U.; Frank, N.Y.; Zhan, Q.; Hoerning, A.; Robles, S.C.; Zhou, J.; Hodi, F.S.; Spagnoli, G.C.; Murphy, G.F.; et al. Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res. 2010, 70, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Di Tomaso, T.; Mazzoleni, S.; Wang, E.; Sovena, G.; Clavenna, D.; Franzin, A.; Mortini, P.; Ferrone, S.; Doglioni, C.; Marincola, F.M.; et al. Immunobiological characterization of cancer stem cells isolated from glioblastoma patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 800–813. [Google Scholar] [CrossRef] [PubMed]
- Maccalli, C.; Volonte, A.; Cimminiello, C.; Parmiani, G. Immunology of cancer stem cells in solid tumours. A review. Eur. J. Cancer 2014, 50, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Bruttel, V.S.; Wischhusen, J. Cancer stem cell immunology: Key to understanding tumorigenesis and tumor immune escape? Front. Immunol. 2014, 5, 360. [Google Scholar] [CrossRef] [PubMed]
- Sultan, M.; Coyle, K.M.; Vidovic, D.; Thomas, M.L.; Gujar, S.; Marcato, P. Hide-and-seek: The interplay between cancer stem cells and the immune system. Carcinogenesis 2017, 38, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.J.; Steel, J.C.; Morris, J.C. Reduction of MHC-I expression limits T-lymphocyte-mediated killing of Cancer-initiating cells. BMC Cancer 2018, 18, 469. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, G.M.; Bobbala, D.; Serrano, D.; Mayhue, M.; Champagne, A.; Saucier, C.; Steimle, V.; Kufer, T.A.; Menendez, A.; Ramanathan, S.; et al. NLRC5 elicits antitumor immunity by enhancing processing and presentation of tumor antigens to CD8(+) T lymphocytes. Oncoimmunology 2016, 5, e1151593. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B.; Wollscheid, U.; Momburg, F.; Blankenstein, T.; Huber, C. Characterization of the major histocompatibility complex class I deficiencies in B16 melanoma cells. Cancer Res. 2001, 61, 1095–1099. [Google Scholar]
- Kim, H.; Kim, H.; Feng, Y.; Li, Y.; Tamiya, H.; Tocci, S.; Ronai, Z.A. PRMT5 control of cGAS/STING and NLRC5 pathways defines melanoma response to antitumor immunity. Sci. Transl. Med. 2020, 12, eaaz5683. [Google Scholar] [CrossRef]
- Zebertavage, L.K.; Alice, A.; Crittenden, M.R.; Gough, M.J. Transcriptional Upregulation of NLRC5 by Radiation Drives STINGand Interferon-Independent MHC-I Expression on Cancer Cells and T Cell Cytotoxicity. Sci. Rep. 2020, 10, 7376. [Google Scholar] [CrossRef]
- Kalbasi, A.; Tariveranmoshabad, M.; Hakimi, K.; Kremer, S.; Campbell, K.M.; Funes, J.M.; Vega-Crespo, A.; Parisi, G.; Champekar, A.; Nguyen, C.; et al. Uncoupling interferon signaling and antigen presentation to overcome immunotherapy resistance due to JAK1 loss in melanoma. Sci. Transl. Med. 2020, 12, eabb0152. [Google Scholar] [CrossRef]
- Peng, Y.Y.; He, Y.H.; Chen, C.; Xu, T.; Li, L.; Ni, M.M.; Meng, X.M.; Huang, C.; Li, J. NLRC5 regulates cell proliferation, migration and invasion in hepatocellular carcinoma by targeting the Wnt/beta-catenin signaling pathway. Cancer Lett. 2016, 376, 10–21. [Google Scholar] [CrossRef]
- Wang, Q.; Ding, H.; He, Y.; Li, X.; Cheng, Y.; Xu, Q.; Yang, Y.; Liao, G.; Meng, X.; Huang, C.; et al. NLRC5 mediates cell proliferation, migration, and invasion by regulating the Wnt/beta-catenin signalling pathway in clear cell renal cell carcinoma. Cancer Lett. 2019, 444, 9–19. [Google Scholar] [CrossRef]
- He, Y.H.; Li, M.F.; Zhang, X.Y.; Meng, X.M.; Huang, C.; Li, J. NLRC5 promotes cell proliferation via regulating the AKT/VEGF-A signaling pathway in hepatocellular carcinoma. Toxicology 2016, 359–360, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Dong, Z.; Shi, Y.; Sun, S.; Wei, B.; Zhan, L. NLRC5 promotes cell migration and invasion by activating the PI3K/AKT signaling pathway in endometrial cancer. J. Int. Med. Res. 2020, 48, 300060520925352. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, M.; Cao, L.; Cong, L.; Gao, Y.; Lu, J.; Feng, J.; Shen, B.; Liu, D. miR-4319 Suppresses the Growth of Esophageal Squamous Cell Carcinoma Via Targeting NLRC5. Curr. Mol. Pharm. 2020, 13, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Arnett, H.A.; Viney, J.L. Immune modulation by butyrophilins. Nat. Rev. Immunol. 2014, 14, 559–569. [Google Scholar] [CrossRef]
- Mayor, A.; Martinon, F.; De Smedt, T.; Petrilli, V.; Tschopp, J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat. Immunol. 2007, 8, 497–503. [Google Scholar] [CrossRef]
- Budhwani, M.; Mazzieri, R.; Dolcetti, R. Plasticity of Type I Interferon-Mediated Responses in Cancer Therapy: From Anti-tumor Immunity to Resistance. Front. Oncol. 2018, 8, 322. [Google Scholar] [CrossRef]
- Xi, S.; Dyer, K.F.; Kimak, M.; Zhang, Q.; Gooding, W.E.; Chaillet, J.R.; Chai, R.L.; Ferrell, R.E.; Zamboni, B.; Hunt, J.; et al. Decreased STAT1 expression by promoter methylation in squamous cell carcinogenesis. J. Natl. Cancer Inst. 2006, 98, 181–189. [Google Scholar] [CrossRef]
- Komyod, W.; Bohm, M.; Metze, D.; Heinrich, P.C.; Behrmann, I. Constitutive suppressor of cytokine signaling 3 expression confers a growth advantage to a human melanoma cell line. Mol. Cancer Res. 2007, 5, 271–281. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal. Transduct Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef]
- Kungulovski, G.; Jeltsch, A. Epigenome Editing: State of the Art, Concepts, and Perspectives. Trends Genet. 2016, 32, 101–113. [Google Scholar] [CrossRef]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef] [PubMed]
- Janssen, E.M.; Lemmens, E.E.; Wolfe, T.; Christen, U.; von Herrath, M.G.; Schoenberger, S.P. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature 2003, 421, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.C.; Bevan, M.J. Defective CD8 T Cell Memory Following Acute Infection Without CD4 T Cell Help. Science 2003, 300, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, B.J.; Craft, J.E.; Kaech, S.M. The multifaceted role of CD4+ T cells in CD8+ T cell memory. Nat. Rev. Immunol. 2016, 16, 102–111. [Google Scholar] [CrossRef]
- Ossendorp, F.; Mengedé, E.; Camps, M.; Filius, R.; Melief, C.J. Specific T helper cell requirement for optimal induction of cytotoxic T lymphocytes against major histocompatibility complex class II negative tumors. J. Exp. Med. 1998, 187, 693–702. [Google Scholar] [CrossRef]
- Spitzer, M.H.; Carmi, Y.; Reticker-Flynn, N.E.; Kwek, S.S.; Madhireddy, D.; Martins, M.M.; Gherardini, P.F.; Prestwood, T.R.; Chabon, J.; Bendall, S.C.; et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell 2017, 168, 487–502. [Google Scholar] [CrossRef]
- Ahrends, T.; Babala, N.; Xiao, Y.; Yagita, H.; van Eenennaam, H.; Borst, J. CD27 Agonism Plus PD-1 Blockade Recapitulates CD4+ T-cell Help in Therapeutic Anticancer Vaccination. Cancer Res. 2016, 76, 2921–2931. [Google Scholar] [CrossRef]
- Quezada, S.A.; Simpson, T.R.; Peggs, K.S.; Merghoub, T.; Vider, J.; Fan, X.; Blasberg, R.; Yagita, H.; Muranski, P.; Antony, P.A.; et al. Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J. Exp. Med. 2010, 207, 637–650. [Google Scholar] [CrossRef]
- Bos, R.; Sherman, L.A. CD4+ T-Cell Help in the Tumor Milieu Is Required for Recruitment and Cytolytic Function of CD8+ T Lymphocytes. Cancer Res. 2010, 70, 8368–8377. [Google Scholar] [CrossRef]
- Homet Moreno, B.; Zaretsky, J.M.; Garcia-Diaz, A.; Tsoi, J.; Parisi, G.; Robert, L.; Meeth, K.; Ndoye, A.; Bosenberg, M.; Weeraratna, A.T.; et al. Response to Programmed Cell Death-1 Blockade in a Murine Melanoma Syngeneic Model Requires Costimulation, CD4, and CD8 T Cells. Cancer Immunol. Res. 2016, 4, 845–857. [Google Scholar] [CrossRef]
- Kambayashi, T.; Laufer, T.M. Atypical MHC class II-expressing antigen-presenting cells: Can anything replace a dendritic cell? Nat. Rev. Immunol. 2014, 14, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Estrada, M.V.; Salgado, R.; Sanchez, V.; Doxie, D.B.; Opalenik, S.R.; Vilgelm, A.E.; Feld, E.; Johnson, A.S.; Greenplate, A.R.; et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat. Commun. 2016, 7, 10582. [Google Scholar] [CrossRef] [PubMed]
- Forero, A.; Li, Y.; Chen, D.; Grizzle, W.E.; Updike, K.L.; Merz, N.D.; Downs-Kelly, E.; Burwell, T.C.; Vaklavas, C.; Buchsbaum, D.J.; et al. Expression of the MHC Class II Pathway in Triple-Negative Breast Cancer Tumor Cells Is Associated with a Good Prognosis and Infiltrating Lymphocytes. Cancer Immunol. Res. 2016, 4, 390–399. [Google Scholar] [CrossRef]
- Park, I.A.; Hwang, S.H.; Song, I.H.; Heo, S.H.; Kim, Y.A.; Bang, W.S.; Park, H.S.; Lee, M.; Gong, G.; Lee, H.J. Expression of the MHC class II in triple-negative breast cancer is associated with tumor-infiltrating lymphocytes and interferon signaling. PLoS ONE 2017, 12, e0182786. [Google Scholar] [CrossRef]
- Armstrong, T.D.; Clements, V.K.; Martin, B.K.; Ting, J.P.; Ostrand-Rosenberg, S. Major histocompatibility complex class II-transfected tumor cells present endogenous antigen and are potent inducers of tumor-specific immunity. Proc. Natl. Acad. Sci. USA 1997, 94, 6886–6891. [Google Scholar] [CrossRef] [PubMed]
- Roemer, M.G.M.; Redd, R.A.; Cader, F.Z.; Pak, C.J.; Abdelrahman, S.; Ouyang, J.; Sasse, S.; Younes, A.; Fanale, M.; Santoro, A.; et al. Major Histocompatibility Complex Class II and Programmed Death Ligand 1 Expression Predict Outcome After Programmed Death 1 Blockade in Classic Hodgkin Lymphoma. J. Clin. Oncol. 2018, 36, 942–950. [Google Scholar] [CrossRef]
- Rodig, S.J.; Gusenleitner, D.; Jackson, D.G.; Gjini, E.; Giobbie-Hurder, A.; Jin, C.; Chang, H.; Lovitch, S.B.; Horak, C.; Weber, J.S.; et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med. 2018, 10, eaar3342. [Google Scholar] [CrossRef]
- Trowsdale, J. Genomic structure and function in the MHC. Trends Genet. 1993, 9, 117–122. [Google Scholar] [CrossRef]
- Unanue, E.R.; Turk, V.; Neefjes, J. Variations in MHC Class II Antigen Processing and Presentation in Health and Disease. Annu. Rev. Immunol. 2016, 34, 265–297. [Google Scholar] [CrossRef]
- Arnold, P.Y.; La Gruta, N.L.; Miller, T.; Vignali, K.M.; Adams, P.S.; Woodland, D.L.; Vignali, D.A. The majority of immunogenic epitopes generate CD4+ T cells that are dependent on MHC class II-bound peptide-flanking residues. J. Immunol. 2002, 169, 739–749. [Google Scholar] [CrossRef]
- Mortara, L.; Castellani, P.; Meazza, R.; Tosi, G.; De Lerma Barbaro, A.; Procopio, F.A.; Comes, A.; Zardi, L.; Ferrini, S.; Accolla, R.S. CIITA-induced MHC class II expression in mammary adenocarcinoma leads to a Th1 polarization of the tumor microenvironment, tumor rejection, and specific antitumor memory. Clin. Cancer. Res. 2006, 12, 3435–3443. [Google Scholar] [CrossRef]
- Baskar, S.; Clements, V.K.; Glimcher, L.H.; Nabavi, N.; Ostrand-Rosenberg, S. Rejection of MHC class II-transfected tumor cells requires induction of tumor-encoded B7-1 and/or B7-2 costimulatory molecules. J. Immunol. 1996, 156, 3821–3827. [Google Scholar] [CrossRef]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin. Cancer. Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Concha, A.; Ruiz-Cabello, F.; Cabrera, T.; Nogales, F.; Collado, A.; Garrido, F. Different patterns of HLA-DR antigen expression in normal epithelium, hyperplastic and neoplastic malignant lesions of the breast. Eur. J. Immunogenet. 1995, 22, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Oldford, S.A.; Robb, J.D.; Codner, D.; Gadag, V.; Watson, P.H.; Drover, S. Tumor cell expression of HLA-DM associates with a Th1 profile and predicts improved survival in breast carcinoma patients. Int. Immunol. 2006, 18, 1591–1602. [Google Scholar] [CrossRef]
- Oldford, S.A.; Robb, J.D.; Watson, P.H.; Drover, S. HLA-DRB alleles are differentially expressed by tumor cells in breast carcinoma. Int. J. Cancer 2004, 112, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Jabrane-Ferrat, N.; Faille, A.; Loiseau, P.; Poirier, O.; Charron, D.; Calvo, F. Effect of gamma interferon on HLA class-I and -II transcription and protein expression in human breast adenocarcinoma cell lines. Int. J. Cancer 1990, 45, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- da Silva, G.B.; Silva, T.G.; Duarte, R.A.; Neto, N.L.; Carrara, H.H.; Donadi, E.A.; Gonçalves, M.A.; Soares, E.G.; Soares, C.P. Expression of the Classical and Nonclassical HLA Molecules in Breast Cancer. Int. J. Breast Cancer 2013, 2013, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Feinmesser, M.; Sulkes, A.; Morgenstern, S.; Sulkes, J.; Stern, S.; Okon, E. HLA-DR and beta 2 microglobulin expression in medullary and atypical medullary carcinoma of the breast: Histopathologically similar but biologically distinct entities. J. Clin. Pathol. 2000, 53, 286–291. [Google Scholar] [CrossRef]
- Bártek, J.; Petrek, M.; Vojtĕsek, B.; Bártková, J.; Kovarík, J.; Rejthar, A. HLA-DR antigens on differentiating human mammary gland epithelium and breast tumours. Br. J. Cancer 1987, 56, 727–733. [Google Scholar] [CrossRef]
- Michel, S.; Linnebacher, M.; Alcaniz, J.; Voss, M.; Wagner, R.; Dippold, W.; Becker, C.; von Knebel Doeberitz, M.; Ferrone, S.; Kloor, M. Lack of HLA class II antigen expression in microsatellite unstable colorectal carcinomas is caused by mutations in HLA class II regulatory genes. Int. J. Cancer 2010, 127, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Li, S.-R.; Phillips, S.; Dorudi, S. Expression of HLA Class II in Colorectal Cancer: Evidence for Enhanced Immunogenicity of Microsatellite-Instability-Positive Tumours. Tumor Biol. 2001, 22, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Soos, J.M.; Krieger, J.I.; Stüve, O.; King, C.L.; Patarroyo, J.C.; Aldape, K.; Wosik, K.; Slavin, A.J.; Nelson, P.A.; Antel, J.P.; et al. Malignant glioma cells use MHC class II transactivator (CIITA) promoters III and IV to direct IFN-gamma-inducible CIITA expression and can function as nonprofessional antigen presenting cells in endocytic processing and CD4(+) T-cell activation. Glia 2001, 36, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Bordeaux, J.; Kim, J.Y.; Vaupel, C.; Rimm, D.L.; Ho, T.H.; Joseph, R.W.; Daud, A.I.; Conry, R.M.; Gaughan, E.M.; et al. Quantitative Spatial Profiling of PD-1/PD-L1 Interaction and HLA-DR/IDO-1 Predicts Improved Outcomes of anti-PD-1 Therapies in Metastatic Melanoma. Clin. Cancer Res. 2018, 24, 5250–5260. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, T.; Kamma, H.; Fujiwara, M.; Matsui, M.; Horiguchi, H.; Satoh, H.; Fujimoto, M.; Yokoyama, K.; Ogata, T. Lack of class II transactivator causes severe deficiency of HLA-DR expression in small cell lung cancer. J. Pathol. 1999, 187, 191–199. [Google Scholar] [CrossRef]
- Callahan, M.J.; Nagymanyoki, Z.; Bonome, T.; Johnson, M.E.; Litkouhi, B.; Sullivan, E.H.; Hirsch, M.S.; Matulonis, U.A.; Liu, J.; Birrer, M.J.; et al. Increased HLA-DMB Expression in the Tumor Epithelium Is Associated with Increased CTL Infiltration and Improved Prognosis in Advanced-Stage Serous Ovarian Cancer. Clin. Cancer Res. 2008, 14, 7667–7673. [Google Scholar] [CrossRef]
- Turner, T.B.; Meza-Perez, S.; Londoño, A.; Katre, A.; Peabody, J.E.; Smith, H.J.; Forero, A.; Norian, L.A.; Straughn, J.M., Jr.; Buchsbaum, D.J.; et al. Epigenetic modifiers upregulate MHC II and impede ovarian cancer tumor growth. Oncotarget 2017, 8, 44159–44170. [Google Scholar] [CrossRef]
- Younger, A.R.; Amria, S.; Jeffrey, W.A.; Mahdy, A.E.; Goldstein, O.G.; Norris, J.S.; Haque, A. HLA class II antigen presentation by prostate cancer cells. Prostate Cancer Prostatic Dis. 2008, 11, 334–341. [Google Scholar] [CrossRef]
- Kim, H.-J.; Cantor, H. CD4 T-cell Subsets and Tumor Immunity: The Helpful and the Not-so-Helpful. Cancer Immunol. Res. 2014, 2, 91–98. [Google Scholar] [CrossRef]
- Linnemann, C.; van Buuren, M.M.; Bies, L.; Verdegaal, E.M.; Schotte, R.; Calis, J.J.; Behjati, S.; Velds, A.; Hilkmann, H.; Atmioui, D.E.; et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med. 2015, 21, 81–85. [Google Scholar] [CrossRef]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Löwer, M.; Diekmann, J.; Boegel, S.; Schrörs, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Dadmarz, R.; Sgagias, M.K.; Rosenberg, S.A.; Schwartzentruber, D.J. CD4+ T lymphocytes infiltrating human breast cancer recognise autologous tumor in an MHC-class-II restricted fashion. Cancer Immunol. Immunother. 1995, 40, 1–9. [Google Scholar] [PubMed]
- Levine, A.G.; Arvey, A.; Jin, W.; Rudensky, A.Y. Continuous requirement for the TCR in regulatory T cell function. Nat. Immunol. 2014, 15, 1070–1078. [Google Scholar] [CrossRef]
- Plitas, G.; Konopacki, C.; Wu, K.; Bos, P.D.; Morrow, M.; Putintseva, E.V.; Chudakov, D.M.; Rudensky, A.Y. Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity 2016, 45, 1122–1134. [Google Scholar] [CrossRef]
- Jang, J.E.; Hajdu, C.H.; Liot, C.; Miller, G.; Dustin, M.L.; Bar-Sagi, D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep. 2017, 20, 558–571. [Google Scholar] [CrossRef]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Driessens, G.; Kline, J.; Gajewski, T.F. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol. Rev. 2009, 229, 126–144. [Google Scholar] [CrossRef]
- Haabeth, O.A.; Tveita, A.A.; Fauskanger, M.; Schjesvold, F.; Lorvik, K.B.; Hofgaard, P.O.; Omholt, H.; Munthe, L.A.; Dembic, Z.; Corthay, A.; et al. How Do CD4(+) T Cells Detect and Eliminate Tumor Cells That Either Lack or Express MHC Class II Molecules? Front. Immunol. 2014, 5, 174. [Google Scholar] [CrossRef] [PubMed]
- Jilaveanu, L.B.; Sznol, J.; Aziz, S.A.; Duchen, D.; Kluger, H.M.; Camp, R.L. CD70 expression patterns in renal cell carcinoma. Hum. Pathol. 2012, 43, 1394–1399. [Google Scholar] [CrossRef]
- Shibahara, I.; Saito, R.; Zhang, R.; Chonan, M.; Shoji, T.; Kanamori, M.; Sonoda, Y.; Kumabe, T.; Kanehira, M.; Kikuchi, T.; et al. OX40 ligand expressed in glioblastoma modulates adaptive immunity depending on the microenvironment: A clue for successful immunotherapy. Mol. Cancer 2015, 14, 41. [Google Scholar] [CrossRef] [PubMed]
- Sartoris, S.; Valle, M.T.; Barbaro, A.L.; Tosi, G.; Cestari, T.; D’Agostino, A.; Megiovanni, A.M.; Manca, F.; Accolla, R.S. HLA class II expression in uninducible hepatocarcinoma cells after transfection of AIR-1 gene product CIITA: Acquisition of antigen processing and presentation capacity. J. Immunol. 1998, 161, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Steimle, V.; Siegrist, C.A.; Mottet, A.; Lisowska-Grospierre, B.; Mach, B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science 1994, 265, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Fontes, J.D.; Peterlin, M.; Flavell, R.A. Class II transactivator (CIITA) is sufficient for the inducible expression of major histocompatibility complex class II genes. J. Exp. Med. 1994, 180, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Meraz, M.A.; White, J.M.; Sheehan, K.C.; Bach, E.A.; Rodig, S.J.; Dighe, A.S.; Kaplan, D.H.; Riley, J.K.; Greenlund, A.C.; Campbell, D.; et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell 1996, 84, 431–442. [Google Scholar] [CrossRef]
- Martins, I.; Deshayes, F.; Baton, F.; Forget, A.; Ciechomska, I.; Sylla, K.; Aoudjit, F.; Charron, D.; Al-Daccak, R.; Alcaide-Loridan, C. Pathologic expression of MHC class II is driven by mitogen-activated protein kinases. Eur. J. Immunol. 2007, 37, 788–797. [Google Scholar] [CrossRef]
- Mottok, A.; Woolcock, B.; Chan, F.C.; Tong, K.M.; Chong, L.; Farinha, P.; Telenius, A.; Chavez, E.; Ramchandani, S.; Drake, M.; et al. Genomic Alterations in CIITA Are Frequent in Primary Mediastinal Large B Cell Lymphoma and Are Associated with Diminished MHC Class II Expression. Cell Rep. 2015, 13, 1418–1431. [Google Scholar] [CrossRef]
- Steidl, C.; Shah, S.P.; Woolcock, B.W.; Rui, L.; Kawahara, M.; Farinha, P.; Johnson, N.A.; Zhao, Y.; Telenius, A.; Neriah, S.B.; et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 2011, 471, 377–381. [Google Scholar] [CrossRef]
- Steidl, C.; Woolcock, B.; Rogic, S.; Ben-Neriah, S.; Telenius, A.; Drake, M.; Siebert, R.; Gascoyne, R.D. Inactivating Gene Alterations of MHC Class II Transactivator CIITA Are Recurrent in Primary Mediastinal B Cell Lymphoma and Hodgkin Lymphoma. Blood 2011, 118, 437. [Google Scholar] [CrossRef]
- Yazawa, T.; Ito, T.; Kamma, H.; Suzuki, T.; Okudela, K.; Hayashi, H.; Horiguchi, H.; Ogata, T.; Mitsui, H.; Ikeda, M.; et al. Complicated mechanisms of class II transactivator transcription deficiency in small cell lung cancer and neuroblastoma. Am. J. Pathol. 2002, 161, 291–300. [Google Scholar] [CrossRef]
- Holling, T.M.; Bergevoet, M.W.; Wilson, L.; Van Eggermond, M.C.; Schooten, E.; Steenbergen, R.D.; Snijders, P.J.; Jager, M.J.; Van den Elsen, P.J. A role for EZH2 in silencing of IFN-gamma inducible MHC2TA transcription in uveal melanoma. J. Immunol. 2007, 179, 5317–5325. [Google Scholar] [CrossRef]
- Martin, B.K.; Frelinger, J.G.; Ting, J.P. Combination gene therapy with CD86 and the MHC class II transactivator in the control of lung tumor growth. J. Immunol. 1999, 162, 6663–6670. [Google Scholar] [CrossRef] [PubMed]
- Bou Nasser Eddine, F.; Forlani, G.; Lombardo, L.; Tedeschi, A.; Tosi, G.; Accolla, R.S. CIITA-driven MHC class II expressing tumor cells can efficiently prime naive CD4 + TH cells in vivo and vaccinate the host against parental MHC-II-negative tumor cells. Oncoimmunology 2017, 6, e1261777. [Google Scholar] [CrossRef] [PubMed]
- Frangione, V.; Mortara, L.; Castellani, P.; De Lerma Barbaro, A.; Accolla, R.S. CIITA-driven MHC-II positive tumor cells: Preventive vaccines and superior generators of antitumor CD4+ T lymphocytes for immunotherapy. Int. J. Cancer 2010, 127, 1614–1624. [Google Scholar] [CrossRef] [PubMed]
- Pulaski, B.A.; Ostrand-Rosenberg, S. Reduction of established spontaneous mammary carcinoma metastases following immunotherapy with major histocompatibility complex class II and B7.1 cell-based tumor vaccines. Cancer Res. 1998, 58, 1486–1493. [Google Scholar]
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting Natural Killer Cells in Cancer Immunotherapy. Nat. Immunol. 2016, 17, 1025–1036. [Google Scholar] [CrossRef]
- Tseng, H.C.; Bui, V.; Man, Y.G.; Cacalano, N.; Jewett, A. Induction of Split Anergy Conditions Natural Killer Cells to Promote Differentiation of Stem Cells Through Cell-Cell Contact and Secreted Factors. Front. Immunol. 2014, 5, 269. [Google Scholar] [CrossRef]
- Kaur, K.; Topchyan, P.; Kozlowska, A.K.; Ohanian, N.; Chiang, J.; Maung, P.O.; Park, S.H.; Ko, M.W.; Fang, C.; Nishimura, I.; et al. Super-Charged NK Cells Inhibit Growth and Progression of Stem-Like/Poorly Differentiated Oral Tumors In Vivo in Humanized BLT Mice; Effect on Tumor Differentiation and Response to Chemotherapeutic Drugs. Oncoimmunology 2018, 7, e1426518. [Google Scholar] [CrossRef]
- Ruscetti, M.; Leibold, J.; Bott, M.J.; Fennell, M.; Kulick, A.; Salgado, N.R.; Chen, C.C.; Ho, Y.J.; Sanchez-Rivera, F.J.; Feucht, J.; et al. NK Cell-Mediated Cytotoxicity Contributes to Tumor Control by a Cytostatic Drug Combination. Science 2018, 362, 1416–1422. [Google Scholar] [CrossRef]
- Ferlazzo, G.; Morandi, B. Cross-Talks Between Natural Killer Cells and Distinct Subsets of Dendritic Cells. Front. Immunol. 2014, 5, 159. [Google Scholar] [CrossRef]
- Walzer, T.; Dalod, M.; Robbins, S.H.; Zitvogel, L.; Vivier, E. Natural-Killer Cells and Dendritic Cells: “L’union Fait La Force”. Blood 2005, 106, 2252–2258. [Google Scholar] [CrossRef]
- Moretta, L.; Ferlazzo, G.; Bottino, C.; Vitale, M.; Pende, D.; Mingari, M.C.; Moretta, A. Effector and Regulatory Events During Natural Killer-Dendritic Cell Interactions. Immunol. Rev. 2006, 214, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Kalinski, P.; Mailliard, R.B.; Giermasz, A.; Zeh, H.J.; Basse, P.; Bartlett, D.L.; Kirkwood, J.M.; Lotze, M.T.; Herberman, R.B. Natural Killer-Dendritic Cell Cross-Talk in Cancer Immunotherapy. Expert Opin. Biol. Ther. 2005, 5, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Srivastava, R.M.; Lopez-Albaitero, A.; Ferrone, S.; Ferris, R.L. Natural Killer (NK): Dendritic Cell (DC) Cross Talk Induced by Therapeutic Monoclonal Antibody Triggers Tumor Antigen-Specific T Cell Immunity. Immunol. Res. 2011, 50, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors At the Tumor Site Enhances Tumor Responses to Therapeutic Pd-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723:e4. [Google Scholar] [CrossRef] [PubMed]
- Oba, T.; Long, M.D.; Keler, T.; Marsh, H.C.; Minderman, H.; Abrams, S.I.; Liu, S.; Ito, F. Overcoming Primary and Acquired Resistance to anti-PD-L1 Therapy by Induction and Activation of Tumor-Residing Cdc1s. Nat. Commun. 2020, 11, 5415. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 Into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A Natural Killer-Dendritic Cell Axis Defines Checkpoint Therapy-Responsive Tumor Microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef]
- Fujihara, A.; Kurooka, M.; Miki, T.; Kaneda, Y. Intratumoral Injection of Inactivated Sendai Virus Particles Elicits Strong Antitumor Activity by Enhancing Local CXCL10 Expression and Systemic NK Cell Activation. Cancer Immunol. Immunother. 2008, 57, 73–84. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.S. Targeting Checkpoint Receptors and Molecules for Therapeutic Modulation of Natural Killer Cells. Front. Immunol. 2018, 9, 2041. [Google Scholar] [CrossRef]
- Klose, C.; Berchtold, S.; Schmidt, M.; Beil, J.; Smirnow, I.; Venturelli, S.; Burkard, M.; Handgretinger, R.; Lauer, U.M. Biological Treatment of Pediatric Sarcomas by Combined Virotherapy and NK Cell Therapy. BMC Cancer 2019, 19, 1172. [Google Scholar] [CrossRef]
- Imai, K.; Matsuyama, S.; Miyake, S.; Suga, K.; Nakachi, K. Natural Cytotoxic Activity of Peripheral-Blood Lymphocytes and Cancer Incidence: An 11-Year Follow-Up Study of a General Population. Lancet 2000, 356, 1795–1799. [Google Scholar] [CrossRef] [PubMed]
- Balch, C.M.; Tilden, A.B.; Dougherty, P.A.; Cloud, G.A. Depressed Levels of Granular Lymphocytes With Natural Killer (NK) Cell Function in 247 Cancer Patients. Ann. Surg. 1983, 198, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Angka, L.; Khan, S.T.; Kilgour, M.K.; Xu, R.; Kennedy, M.A.; Auer, R.C. Dysfunctional Natural Killer Cells in the Aftermath of Cancer Surgery. Int. J. Mol. Sci. 2017, 18, 1787. [Google Scholar] [CrossRef] [PubMed]
- Cong, J.; Wang, X.; Zheng, X.; Wang, D.; Fu, B.; Sun, R.; Tian, Z.; Wei, H. Dysfunction of Natural Killer Cells by FBP1-Induced Inhibition of Glycolysis During Lung Cancer Progression. Cell Metab. 2018, 28, 243–255.e5. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.J.; Xu, L.J.; Yang, L.; Tsai, Y.; Keng, P.C.; Chen, Y.; Lee, S.O.; Chen, Y. Radiation Alters PDL1/NKG2D Ligand Levels in Lung Cancer Cells and Leads to Immune Escape From NK Cell Cytotoxicity Via IL-6-MEK/Erk Signaling Pathway. Oncotarget 2017, 8, 80506–80520. [Google Scholar] [CrossRef]
- Mantovani, S.; Oliviero, B.; Lombardi, A.; Varchetta, S.; Mele, D.; Sangiovanni, A.; Rossi, G.; Donadon, M.; Torzilli, G.; Soldani, C.; et al. Deficient Natural Killer Cell Nkp30-Mediated Function and Altered Ncr3 Splice Variants in Hepatocellular Carcinoma. Hepatology 2019, 69, 1165–1179. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, Y.; Lian, J.; Yang, H.; Li, F.; Zhao, S.; Qi, Y.; Zhang, Y.; Huang, L. TNF-α-Induced Tim-3 Expression Marks the Dysfunction of Infiltrating Natural Killer Cells in Human Esophageal Cancer. J. Trans. Med. 2019, 17, 165. [Google Scholar] [CrossRef]
- Ciurea, S.O.; Schafer, J.R.; Bassett, R.; Denman, C.J.; Cao, K.; Willis, D.; Rondon, G.; Chen, J.; Soebbing, D.; Kaur, I.; et al. Phase 1 Clinical Trial Using mbIL21 Ex Vivo-Expanded Donor-Derived NK Cells After Haploidentical Transplantation. Blood 2017, 130, 1857–1868. [Google Scholar] [CrossRef]
- Baggio, L.; Laureano, Á.M.; Silla, L.; Lee, D.A. Natural Killer Cell Adoptive Immunotherapy: Coming of Age. Clin. Immunol. 2017, 177, 3–11. [Google Scholar] [CrossRef]
- Pistoia, V.; Tumino, N.; Vacca, P.; Veneziani, I.; Moretta, A.; Locatelli, F.; Moretta, L. Human gammadelta T-Cells: From Surface Receptors to the Therapy of High-Risk Leukemias. Front. Immunol. 2018, 9, 984. [Google Scholar] [CrossRef]
- Minculescu, L.; Sengelov, H. The role of gamma delta T cells in haematopoietic stem cell transplantation. Scand. J. Immunol. 2015, 81, 459–468. [Google Scholar] [CrossRef]
- Handgretinger, R.; Schilbach, K. The potential role of gammadelta T cells after allogeneic HCT for leukemia. Blood 2018, 131, 1063–1072. [Google Scholar] [CrossRef]
- Parker, C.M.; Groh, V.; Band, H.; Porcelli, S.A.; Morita, C.; Fabbi, M.; Glass, D.; Strominger, J.L.; Brenner, M.B. Evidence for extrathymic changes in the T cell receptor gamma/delta repertoire. J. Exp. Med. 1990, 171, 1597–1612. [Google Scholar] [CrossRef] [PubMed]
- Kunzmann, V.; Wilhelm, M. Anti-lymphoma effect of gammadelta T cells. Leuk. Lymphoma 2005, 46, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, B.; Simoes, A.E.; Caiado, F.; Tieppo, P.; Correia, D.V.; Carvalho, T.; da Silva, M.G.; Dechanet-Merville, J.; Schumacher, T.N.; Prinz, I.; et al. Broad Cytotoxic Targeting of Acute Myeloid Leukemia by Polyclonal Delta One T Cells. Cancer Immunol. Res. 2019, 7, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Kabelitz, D.; He, W. The multifunctionality of human Vgamma9Vdelta2 gammadelta T cells: Clonal plasticity or distinct subsets? Scand. J. Immunol. 2012, 76, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Silva-Santos, B.; Mensurado, S.; Coffelt, S.B. gammadelta T cells: Pleiotropic immune effectors with therapeutic potential in cancer. Nat. Rev. Cancer 2019, 19, 392–404. [Google Scholar] [CrossRef]
- Sandberg, Y.; Almeida, J.; Gonzalez, M.; Lima, M.; Barcena, P.; Szczepanski, T.; van Gastel-Mol, E.J.; Wind, H.; Balanzategui, A.; van Dongen, J.J.; et al. TCRgammadelta+ large granular lymphocyte leukemias reflect the spectrum of normal antigen-selected TCRgammadelta+ T-cells. Leukemia 2006, 20, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Wang, Y.; Li, P.; Zhao, J. Gamma Delta T Cells and Their Pathogenic Role in Psoriasis. Front. Immunol. 2021, 12, 627139. [Google Scholar] [CrossRef] [PubMed]
- Kenna, T.; Golden-Mason, L.; Norris, S.; Hegarty, J.E.; O’Farrelly, C.; Doherty, D.G. Distinct subpopulations of gamma delta T cells are present in normal and tumor-bearing human liver. Clin. Immunol. 2004, 113, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Davey, M.S.; Willcox, C.R.; Hunter, S.; Kasatskaya, S.A.; Remmerswaal, E.B.M.; Salim, M.; Mohammed, F.; Bemelman, F.J.; Chudakov, D.M.; Oo, Y.H.; et al. The human Vdelta2+ T-cell compartment comprises distinct innate-like Vgamma9+ and adaptive Vgamma9− subsets. Nat. Commun. 2018, 9, 1760. [Google Scholar] [CrossRef] [PubMed]
- Rigau, M.; Ostrouska, S.; Fulford, T.S.; Johnson, D.N.; Woods, K.; Ruan, Z.; McWilliam, H.E.G.; Hudson, C.; Tutuka, C.; Wheatley, A.K.; et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by gammadelta T cells. Science 2020, 367, eaay5516. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Recio, C.; Aranda-Tavio, H.; Guerra-Rodriguez, M.; Garcia-Castellano, J.M.; Fernandez-Perez, L. The Mevalonate Pathway, a Metabolic Target in Cancer Therapy. Front. Oncol. 2021, 11, 626971. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef]
- Luoma, A.M.; Castro, C.D.; Adams, E.J. gammadelta T cell surveillance via CD1 molecules. Trends Immunol. 2014, 35, 613–621. [Google Scholar] [CrossRef]
- Bai, L.; Picard, D.; Anderson, B.; Chaudhary, V.; Luoma, A.; Jabri, B.; Adams, E.J.; Savage, P.B.; Bendelac, A. The majority of CD1d-sulfatide-specific T cells in human blood use a semiinvariant Vdelta1 TCR. Eur. J. Immunol. 2012, 42, 2505–2510. [Google Scholar] [CrossRef]
- Russano, A.M.; Agea, E.; Corazzi, L.; Postle, A.D.; De Libero, G.; Porcelli, S.; de Benedictis, F.M.; Spinozzi, F. Recognition of pollen-derived phosphatidyl-ethanolamine by human CD1d-restricted gamma delta T cells. J. Allergy Clin. Immunol. 2006, 117, 1178–1184. [Google Scholar] [CrossRef]
- Agea, E.; Russano, A.; Bistoni, O.; Mannucci, R.; Nicoletti, I.; Corazzi, L.; Postle, A.D.; De Libero, G.; Porcelli, S.A.; Spinozzi, F. Human CD1-restricted T cell recognition of lipids from pollens. J. Exp. Med. 2005, 202, 295–308. [Google Scholar] [CrossRef]
- Mangan, B.A.; Dunne, M.R.; O’Reilly, V.P.; Dunne, P.J.; Exley, M.A.; O’Shea, D.; Scotet, E.; Hogan, A.E.; Doherty, D.G. Cutting edge: CD1d restriction and Th1/Th2/Th17 cytokine secretion by human Vdelta3 T cells. J. Immunol. 2013, 191, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Zhang, C.; Guo, H.; Gao, S.H.; Yang, F.Y.; Zhou, G.B.; Wang, G.Z. Comprehensive analysis of BTN3A1 in cancers: Mining of omics data and validation in patient samples and cellular models. FEBS Open Bio 2021, 11, 2586–2599. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Dhodapkar, M.V. Natural Killer T Cells in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1178. [Google Scholar] [CrossRef]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Simoes, A.E.; Di Lorenzo, B.; Silva-Santos, B. Molecular Determinants of Target Cell Recognition by Human gammadelta T Cells. Front. Immunol. 2018, 9, 929. [Google Scholar] [CrossRef]
- Groh, V.; Rhinehart, R.; Secrist, H.; Bauer, S.; Grabstein, K.H.; Spies, T. Broad tumor-associated expression and recognition by tumor-derived gamma delta T cells of MICA and MICB. Proc. Natl. Acad. Sci. USA 1999, 96, 6879–6884. [Google Scholar] [CrossRef]
- Kong, Y.; Cao, W.; Xi, X.; Ma, C.; Cui, L.; He, W. The NKG2D ligand ULBP4 binds to TCRgamma9/delta2 and induces cytotoxicity to tumor cells through both TCRgammadelta and NKG2D. Blood 2009, 114, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Lanca, T.; Correia, D.V.; Moita, C.F.; Raquel, H.; Neves-Costa, A.; Ferreira, C.; Ramalho, J.S.; Barata, J.T.; Moita, L.F.; Gomes, A.Q.; et al. The MHC class Ib protein ULBP1 is a nonredundant determinant of leukemia/lymphoma susceptibility to gammadelta T-cell cytotoxicity. Blood 2010, 115, 2407–2411. [Google Scholar] [CrossRef] [PubMed]
- Kabelitz, D.; Serrano, R.; Kouakanou, L.; Peters, C.; Kalyan, S. Cancer immunotherapy with gammadelta T cells: Many paths ahead of us. Cell Mol. Immunol. 2020, 17, 925–939. [Google Scholar] [CrossRef]
- Zhao, Y.; Niu, C.; Cui, J. Gamma-delta (gammadelta) T cells: Friend or foe in cancer development? J. Transl. Med. 2018, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Todaro, M.; D’Asaro, M.; Caccamo, N.; Iovino, F.; Francipane, M.G.; Meraviglia, S.; Orlando, V.; La Mendola, C.; Gulotta, G.; Salerno, A.; et al. Efficient killing of human colon cancer stem cells by gammadelta T lymphocytes. J. Immunol. 2009, 182, 7287–7296. [Google Scholar] [CrossRef] [PubMed]
- Dieli, F.; Vermijlen, D.; Fulfaro, F.; Caccamo, N.; Meraviglia, S.; Cicero, G.; Roberts, A.; Buccheri, S.; D’Asaro, M.; Gebbia, N.; et al. Targeting human γδ T cells with zoledronate and interleukin-2 for immunotherapy of hormone-refractory prostate cancer. Cancer Res. 2007, 67, 7450–7457. [Google Scholar] [CrossRef] [PubMed]
- Couzi, L.; Pitard, V.; Sicard, X.; Garrigue, I.; Hawchar, O.; Merville, P.; Moreau, J.F.; Dechanet-Merville, J. Antibody-dependent anti-cytomegalovirus activity of human gammadelta T cells expressing CD16 (FcgammaRIIIa). Blood 2012, 119, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Tokuyama, H.; Hagi, T.; Mattarollo, S.R.; Morley, J.; Wang, Q.; So, H.F.; Moriyasu, F.; Nieda, M.; Nicol, A.J. V gamma 9 V delta 2 T cell cytotoxicity against tumor cells is enhanced by monoclonal antibody drugs–rituximab and trastuzumab. Int. J. Cancer 2008, 122, 2526–2534. [Google Scholar] [CrossRef]
- Dar, A.A.; Patil, R.S.; Chiplunkar, S.V. Insights into the Relationship between Toll Like Receptors and Gamma Delta T Cell Responses. Front. Immunol. 2014, 5, 366. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Golenbock, D.; Bowie, A.G. The History of Toll-like Receptors—Redefining Innate Immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef]
- Corsale, A.M.; Di Simone, M.; Lo Presti, E.; Picone, C.; Dieli, F.; Meraviglia, S. Metabolic Changes in Tumor Microenvironment: How Could They Affect gammadelta T Cells Functions? Cells 2021, 10, 2896. [Google Scholar] [CrossRef]
- Fowler, D.W.; Copier, J.; Dalgleish, A.G.; Bodman-Smith, M.D. Zoledronic acid renders human M1 and M2 macrophages susceptible to Vdelta2+ gammadelta T cell cytotoxicity in a perforin-dependent manner. Cancer Immunol. Immunother. 2017, 66, 1205–1215. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Brandes, M.; Willimann, K.; Moser, B. Professional antigen-presentation function by human gammadelta T Cells. Science 2005, 309, 264–268. [Google Scholar] [CrossRef]
- Brandes, M.; Willimann, K.; Bioley, G.; Levy, N.; Eberl, M.; Luo, M.; Tampe, R.; Levy, F.; Romero, P.; Moser, B. Cross-presenting human gammadelta T cells induce robust CD8+ alphabeta T cell responses. Proc. Natl. Acad. Sci. USA 2009, 106, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Muto, M.; Baghdadi, M.; Maekawa, R.; Wada, H.; Seino, K. Myeloid molecular characteristics of human gammadelta T cells support their acquisition of tumor antigen-presenting capacity. Cancer Immunol. Immunother. 2015, 64, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Scotet, E.; Nedellec, S.; Devilder, M.C.; Allain, S.; Bonneville, M. Bridging innate and adaptive immunity through gammadelta T-dendritic cell crosstalk. Front. Biosci. 2008, 13, 6872–6885. [Google Scholar] [CrossRef]
- van Beek, J.J.; Wimmers, F.; Hato, S.V.; de Vries, I.J.; Skold, A.E. Dendritic cell cross talk with innate and innate-like effector cells in antitumor immunity: Implications for DC vaccination. Crit. Rev. Immunol. 2014, 34, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Maniar, A.; Zhang, X.; Lin, W.; Gastman, B.R.; Pauza, C.D.; Strome, S.E.; Chapoval, A.I. Human gammadelta T lymphocytes induce robust NK cell-mediated antitumor cytotoxicity through CD137 engagement. Blood 2010, 116, 1726–1733. [Google Scholar] [CrossRef]
- Ladel, C.H.; Blum, C.; Kaufmann, S.H. Control of natural killer cell-mediated innate resistance against the intracellular pathogen Listeria monocytogenes by gamma/delta T lymphocytes. Infect. Immun. 1996, 64, 1744–1749. [Google Scholar] [CrossRef]
- Jacobsen, E.A.; Jackson, D.J.; Heffler, E.; Mathur, S.K.; Bredenoord, A.J.; Pavord, I.D.; Akuthota, P.; Roufosse, F.; Rothenberg, M.E. Eosinophil knockout humans: Uncovering the role of eosinophils through eosinophil-directed biological therapies. Annu. Rev. Immunol. 2021, 39, 719–757. [Google Scholar] [CrossRef]
- Reinbach, G. Ueber das Verhalten der Leukocyten bei malignen Tumoren. Arch. Klin. Chir. 1893, 46, 486–562. [Google Scholar]
- Grisaru-Tal, S.; Itan, M.; Grass, D.G.; Torres-Roca, J.; Eschrich, S.A.; Gordon, Y.; Dolitzky, A.; Hazut, I.; Avlas, S.; Jacobsen, E.A.; et al. Primary tumors from mucosal barrier organs drive unique eosinophil infiltration patterns and clinical associations. Oncoimmunology 2020, 10, 1859732. [Google Scholar] [CrossRef]
- Grisaru-Tal, S.; Itan, M.; Klion, A.D.; Munitz, A. A new dawn for eosinophils in the tumour microenvironment. Nat. Rev. Cancer 2020, 20, 594–607. [Google Scholar] [CrossRef]
- Rodriguez-Rodriguez, N.; Gogoi, M.; McKenzie, A.N.J. Group 2 innate lymphoid cells: Team players in regulating asthma. Annu. Rev. Immunol. 2021, 39, 167–198. [Google Scholar] [CrossRef]
- Nussbaum, J.C.; Van Dyken, S.J.; von Moltke, J.; Cheng, L.E.; Mohapatra, A.; Molofsky, A.B.; Thornton, E.E.; Krummel, M.F.; Chawla, A.; Liang, H.E.; et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 2013, 502, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Maggi, E.; Veneziani, I.; Moretta, L.; Cosmi, L.; Annunziato, F. Group 2 innate lymphoid cells: A double-edged sword in cancer? Cancers 2020, 12, 3452. [Google Scholar] [CrossRef] [PubMed]
- Jacquelot, N.; Seillet, C.; Wang, M.; Pizzolla, A.; Liao, Y.; Hediyeh-Zadeh, S.; Grisaru-Tal, S.; Louis, C.; Huang, Q.; Schreuder, J.; et al. Blockade of the co-inhibitory molecule PD-1 unleashes ILC2-dependent antitumor immunity in melanoma. Nat. Immunol. 2021, 22, 851–864. [Google Scholar] [CrossRef]
- Ikutani, M.; Yanagibashi, T.; Ogasawara, M.; Tsuneyama, K.; Yamamoto, S.; Hattori, Y.; Kouro, T.; Itakura, A.; Nagai, Y.; Takaki, S.; et al. Identification of innate IL-5-producing cells and their role in lung eosinophil regulation and antitumor immunity. J. Immunol. 2012, 188, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.T.; Martin, M.U. Interleukin 33 is a guardian of barriers and a local alarmin. Nat. Immunol. 2016, 17, 122–131. [Google Scholar] [CrossRef]
- Lucarini, V.; Ziccheddu, G.; Macchia, I.; La Sorsa, V.; Peschiaroli, F.; Buccione, C.; Sistigu, A.; Sanchez, M.; Andreone, S.; D’Urso, M.T.; et al. IL-33 restricts tumor growth and inhibits pulmonary metastasis in melanoma-bearing mice through eosinophils. Oncoimmunology 2017, 6, e1317420. [Google Scholar] [CrossRef]
- Gao, K.; Li, X.; Zhang, L.; Bai, L.; Dong, W.; Gao, K.; Shi, G.; Xia, X.; Wu, L.; Zhang, L. Transgenic expression of IL-33 activates CD8+ T cells and NK cells and inhibits tumor growth and metastasis in mice. Cancer Lett. 2013, 335, 463–471. [Google Scholar] [CrossRef]
- Munitz, A.; Bachelet, I.; Fraenkel, S.; Katz, G.; Mandelboim, O.; Simon, H.U.; Moretta, L.; Colonna, M.; Levi-Schaffer, F. 2B4 (CD244) is expressed and functional on human eosinophils. J. Immunol. 2005, 174, 110–118. [Google Scholar] [CrossRef]
- Munitz, A.; Bachelet, I.; Eliashar, R.; Moretta, A.; Moretta, L.; Levi-Schaffer, F. The inhibitory receptor IRp60 (CD300a) suppresses the effects of IL-5, GM-CSF and eotaxin on human peripheral blood eosinophils. Blood 2006, 107, 1996–2003. [Google Scholar] [CrossRef]
- Pesce, S.; Thoren, F.B.; Cantoni, C.; Prato, C.; Moretta, L.; Moretta, A.; Marcenaro, E. The innate immune cross-talk between NK cells and eosinophils is regulated by the interaction of natural cytotoxicity receptors with eosinophil surface ligands. Front. Immunol. 2017, 8, 510. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Zhang, Q.; Miao, Y.; Kang, W.; Tian, Z.; Xu, D.; Xiao, W.; Fang, F. Interleukin-33 activates and recruits natural killer cells to inhibit pulmonary metastatic cancer development. Int. J. Cancer 2020, 146, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Schuijs, M.J.; Png, S.; Richard, A.C.; Tsyben, A.; Hamm, G.; Stockis, J.; Garcia, C.; Pinaud, S.; Nicholls, A.; Ros, X.R.; et al. ILC2-driven innate immune checkpoint mechanism antagonizes NK cell antimetastatic function in the lung. Nat. Immunol. 2020, 21, 998–1009. [Google Scholar] [CrossRef]
- Grisaru-Tal, S.; Dulberg, S.; Beck, L.; Zhang, C.; Itan, M.; Hediyeh-Zadeh, S.; Caldwell, J.; Rozenberg, P.; Dolitzky, A.; Avlas, S.; et al. Metastasis-entrained eosinophils enhance lymphocytemediated antitumor immunity. Cancer Res. 2021, 81, 5555–5571. [Google Scholar] [CrossRef]
- Reichman, H.; Itan, M.; Rozenberg, P.; Yarmolovski, T.; Brazowski, E.; Varol, C.; Gluck, N.; Shapira, S.; Arber, N.; Qimron, U.H.; et al. Activated eosinophils exert antitumorigenic activities in colorectal cancer. Cancer Immunol. Res. 2019, 7, 388–400. [Google Scholar] [CrossRef]
- Dolitzky, A.; Shapira, G.; Grisaru-Tal, S.; Hazut, I.; Avlas, S.; Gordon, Y.; Itan, M.; Shomron, N.; Munitz, A. Transcriptional profiling of mouse eosinophils identifies distinct gene signatures following cellular activation. Front. Immunol. 2021, 12, 802839. [Google Scholar] [CrossRef]
- Carretero, R.; Sektioglu, I.M.; Garbi, N.; Salgado, O.C.; Beckhove, P.; Hämmerling, G.J. Eosinophils orchestrate cancer rejection by normalizing tumor vessels and enhancing infiltration of CD8+ T cells. Nat. Immunol. 2015, 16, 609–617. [Google Scholar] [CrossRef]
- Akuthota, P.; Wang, H.B.; Spencer, L.A.; Weller, P.F. Immunoregulatory roles of eosinophils: A new look at a familiar cell. Clin. Exp. Allergy 2008, 38, 1254–1263. [Google Scholar] [CrossRef]
- Simon, H.U.; Plötz, S.; Simon, D.; Seitzer, U.; Braathen, L.R.; Menz, G.; Straumann, A.; Dummer, R.; Levi-Schaffer, F. Interleukin-2 primes eosinophil degranulation in hypereosinophilia and Wells’ syndrome. Eur. J. Immunol. 2003, 33, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Sosman, J.A.; Bartemes, K.; Offord, K.P.; Kita, H.; Fisher, S.G.; Kefer, C.; Ellis, T.A.; Fisher, R.I.; Higgins, T.J.; Gleich, G.J. Evidence for eosinophil activation in cancer patients receiving recombinant interleukin-4: Effects of interleukin-4 alone and following interleukin-2 administration. Clin. Cancer Res. 1995, 1, 805–812. [Google Scholar] [PubMed]
- Ellem, K.A.; O’Rourke, M.G.; Johnson, G.R.; Parry, G.; Misko, I.S.; Schmidt, C.W.; Parsons, P.G.; Burrows, S.R.; Cross, S.; Fell, A.; et al. A case report: Immune responses and clinical course of the first human use of granulocyte/macrophage-colony- stimulating-factor-transduced autologous melanoma cells for immunotherapy. Cancer Immunol. Immunother. 1997, 44, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, C.; Sevko, A.; Jiang, H.; Lichtenberger, R.; Reith, M.; Tarnanidis, K.; Holland-Letz, T.; Umansky, L.; Beckhove, P.; Sucker, A.; et al. Myeloid cells and related chronic inflammatory factors as novel predictive markers in melanoma treatment with ipilimumab. Clin. Cancer Res. 2015, 21, 5453–5459. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.; Leisgang, W.; Schuler, G.; Heinzerling, L. Eosinophilic count as a biomarker for prognosis of melanoma patients and its importance in the response to immunotherapy. Immunotherapy 2017, 9, 115–121. [Google Scholar] [CrossRef]
- Weide, B.; Martens, A.; Hassel, J.C.; Berking, C.; Postow, M.A.; Bisschop, K.; Simeone, E.; Mangana, J.; Schilling, B.; Di Giacomo, A.M.; et al. Baseline biomarkers for outcome of melanoma patients treated with pembrolizumab. Clin. Cancer Res. 2016, 22, 5487–5496. [Google Scholar] [CrossRef]
- Alves, A.; Dias, M.; Campainha, S.; Barroso, A. Peripheral blood eosinophilia may be a prognostic biomarker in non-small cell lung cancer patients treated with immunotherapy. J. Thorac. Dis. 2021, 13, 2716–2727. [Google Scholar] [CrossRef]
- Herrmann, T.; Ginzac, A.; Molnar, I.; Bailly, S.; Durando, X.; Mahammedi, H. Eosinophil counts as a relevant prognostic marker for response to nivolumab in the management of renal cell carcinoma: A retrospective study. Cancer Med. 2021, 10, 6705–6713. [Google Scholar] [CrossRef] [PubMed]
- Ghebeh, H.; Elshenawy, M.A.; AlSayed, A.D.; Al-Tweigeri, T. Peripheral blood eosinophil count is associated with response to chemoimmunotherapy in metastatic triple-negative breast cancer. Immunotherapy 2022, 14, 189–199. [Google Scholar] [CrossRef]
- Nishikawa, D.; Suzuki, H.; Beppu, S.; Terada, H.; Sawabe, M.; Kadowaki, S.; Sone, M.; Hanai, N. Eosinophil prognostic scores for patients with head and neck squamous cell carcinoma treated with nivolumab. Cancer Sci. 2021, 112, 339–346. [Google Scholar] [CrossRef]
- Lai, W.; Xie, H.; Liu, Y.; Zheng, F.; Zhang, Y.; Lei, Q.; Lv, L.; Dong, J.; Song, J.; Gao, X.; et al. Human pluripotent stem cell-derived eosinophils reveal potent cytotoxicity against solid tumors. Stem Cell Rep. 2021, 16, 1697–1704. [Google Scholar] [CrossRef]
- Lynch, J.B.; Hsiao, E.Y. Microbiomes as sources of emergent host phenotypes. Science 2019, 365, 1405–1409. [Google Scholar] [CrossRef]
- Amieva, M.; Peek, R.M. Pathobiology of Helicobacter pylori–induced gastric cancer. Gastroenterology 2016, 150, 64–78. [Google Scholar] [CrossRef]
- Moss, S.F. The clinical evidence linking Helicobacter pylori to gastric cancer. Cell Mol. Gastroenterol. Hepatol. 2017, 3, 183–191. [Google Scholar] [CrossRef]
- Flanagan, L.; Schmid, J.; Ebert, M.; Soucek, P.; Kunicka, T.; Liska, V.; Bruha, J.; Neary, P.; Dezeeuw, N.; Tommasino, M.; et al. Fusobacterium nucleatum associates with stages of colorectal neoplasia development, colorectal cancer and disease outcome. Eur. J. Clin. Microbiol. Infect Dis. 2014, 33, 1381–1390. [Google Scholar] [CrossRef]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef]
- Pushalkar, S.; Hundeyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef]
- Riquelme, E.; Zhang, Y.; Zhang, L.; Montiel, M.; Zoltan, M.; Dong, W.; Quesada, P.; Sahin, I.; Chandra, V.; San Lucas, A.; et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell 2019, 178, 795–806.e12. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut microbiota promotes obesity-associated liver cancer through PGE2-mediated suppression of antitumor immunity. Cancer Discov. 2017, 7, 522–538. [Google Scholar] [CrossRef]
- Baker, J.M.; Al-Nakkash, L.; Herbst-Kralovetz, M.M. Estrogen–gut microbiome axis: Physiological and clinical implications. Maturitas 2017, 103, 45–53. [Google Scholar] [CrossRef]
- Flores, R.; Shi, J.; Fuhrman, B.; Xu, X.; Veenstra, T.D.; Gail, M.H.; Gajer, P.; Ravel, J.; Goedert, J.J. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: A cross-sectional study. J. Transl. Med. 2012, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Kwa, M.; Plottel, C.S.; Blaser, M.J.; Adams, S. The intestinal microbiome and estrogen receptor-positive female breast cancer. J. Natl. Cancer Inst. 2016, 108, djw029. [Google Scholar]
- Rao, V.P.; Poutahidis, T.; Ge, Z.; Nambiar, P.R.; Boussahmain, C.; Wang, Y.Y.; Horwitz, B.H.; Fox, J.G.; Erdman, S.E. Innate immune inflammatory response against enteric bacteria Helicobacter hepaticus induces mammary adenocarcinoma in mice. Cancer Res. 2006, 66, 7395–7400. [Google Scholar] [CrossRef] [PubMed]
- Eslami, S.Z.; Majidzadeh, A.K.; Halvaei, S.; Babapirali, F.; Esmaeili, R. Microbiome and breast cancer: New role for an ancient population. Front. Oncol. 2020, 10, 120. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Xing, C.; Long, W.; Wang, H.Y.; Liu, Q.; Wang, R.F. Impact of microbiota on central nervous system and neurological diseases: The gut-brain axis. J. Neuroinflammation 2019, 16, 53. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Aziz, M.A.; Cooper, A.; Weyrich, L.S. Exploring relationships between host genome and microbiome: New insights from genome-wide association studies. Front. Microbiol. 2016, 7, 1611. [Google Scholar] [CrossRef]
- Ben-Shaanan, T.L.; Schiller, M.; Azulay-Debby, H.; Korin, B.; Boshnak, N.; Koren, T.; Krot, M.; Shakya, J.; Rahat, M.A.; Hakim, F.; et al. Modulation of anti-tumor immunity by the brain’s reward system. Nat. Commun. 2018, 9, 2723. [Google Scholar] [CrossRef]
- Xavier, J.B.; Young, V.B.; Skufca, J.; Ginty, F.; Testerman, T.; Pearson, A.T.; Macklin, P.; Mitchell, A.; Shmulevich, I.; Xie, L.; et al. The cancer microbiome: Distinguishing direct and indirect effects requires a systemic view. Trends Cancer 2020, 6, 192–204. [Google Scholar] [CrossRef]
- Fluckiger, A.; Daillère, R.; Sassi, M.; Sixt, B.S.; Liu, P.; Loos, F.; Richard, C.; Rabu, C.; Alou, M.T.; Goubet, A.G.; et al. Cross-reactivity between tumor MHC class I-restricted antigens and an enterococcal bacteriophage. Science 2020, 369, 936–942. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Borgerding, J.N.; Shang, J.; Britton, G.J.; Salmon, H.; Bigenwald, C.; Maier, B.; Rose, S.R.; Mogno, I.; Kamphorst, A.O.; Merad, M.; et al. Human microbial transplant restores T cell cytotoxicity and anti-tumor response to PD-L1 blockade in gnotobiotic mice. bioRxiv 2020, 242040. [Google Scholar] [CrossRef]
- Mager, L.F.; Burkhard, R.; Pett, N.; Cooke, N.C.A.; Brown, K.; Ramay, H.; Paik, S.; Stagg, J.; Groves, R.A.; Gallo, M.; et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020, 369, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Wolf, N.; Raulet, D.H.; Akkari, L.; Pittet, M.J.; Rodriguez, P.C.; Kaplan, R.N.; Munitz, A.; Zhang, Z.; Cheng, S.; et al. Innate immune cells in the tumor microenvironment. Cancer Cell 2021, 39, 725–729. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, M.; Li, Y.; Qin, X.; Qin, B.; Wang, Q. Insight into Cancer Immunity: MHCs, Immune Cells and Commensal Microbiota. Cells 2023, 12, 1882. https://doi.org/10.3390/cells12141882
Wen M, Li Y, Qin X, Qin B, Wang Q. Insight into Cancer Immunity: MHCs, Immune Cells and Commensal Microbiota. Cells. 2023; 12(14):1882. https://doi.org/10.3390/cells12141882
Chicago/Turabian StyleWen, Minting, Yingjing Li, Xiaonan Qin, Bing Qin, and Qiong Wang. 2023. "Insight into Cancer Immunity: MHCs, Immune Cells and Commensal Microbiota" Cells 12, no. 14: 1882. https://doi.org/10.3390/cells12141882
APA StyleWen, M., Li, Y., Qin, X., Qin, B., & Wang, Q. (2023). Insight into Cancer Immunity: MHCs, Immune Cells and Commensal Microbiota. Cells, 12(14), 1882. https://doi.org/10.3390/cells12141882