
Pyrroline-5-Carboxylate Reductase-2 Promotes Colorectal Carcinogenesis by Modulating Microtubule-Associated Serine/Threonine Kinase-like/Wnt/β-Catenin Signaling

 , , , , , , ,
, , , , , , , 

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Bioinformatic Assessment of the Public Databases
2.2. Cell Culture
2.3. Loss and Gain of Expression and Function in PYCR2 and MASTL
2.4. Cell Viability Assay
2.5. Cell Invasion Assay
2.6. Wound-Healing Assay
2.7. Soft Agar Assay
2.8. Sphere Forming Assay
2.9. Immunohistochemical (IHC) Analysis
2.10. Immunofluorescence (IF) Analysis
2.11. Fluorescence-Activated Cell Sorting (FACS) Analysis for Determining Apoptosis and Cell Cycle Progression
2.12. Western Blot Analysis
2.13. RT-qPCR Analysis
2.14. Xenograft Tumor Growth Assay
2.15. Colonoscopy-Guided Intramucosal Transplantation of CRC Cells for Tumor Development
2.16. Proteomics (LC-MS/MS) Analysis
2.17. Intracellular Proline Measurement
2.18. Statistical Analysis
3. Results
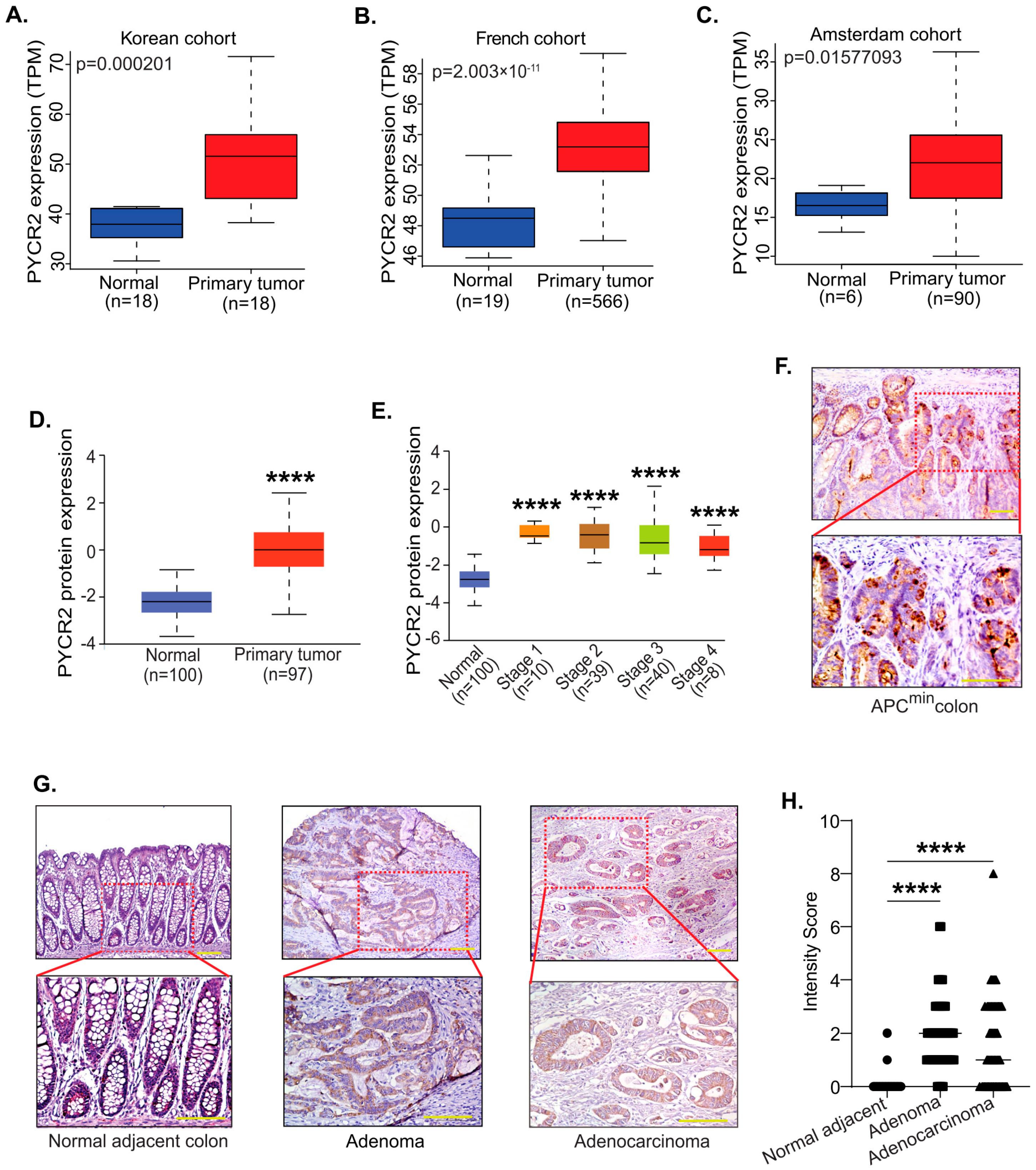
3.1. PYCR2 Expression Is Upregulated during Colon Carcinogenesis in Both Mice and Humans

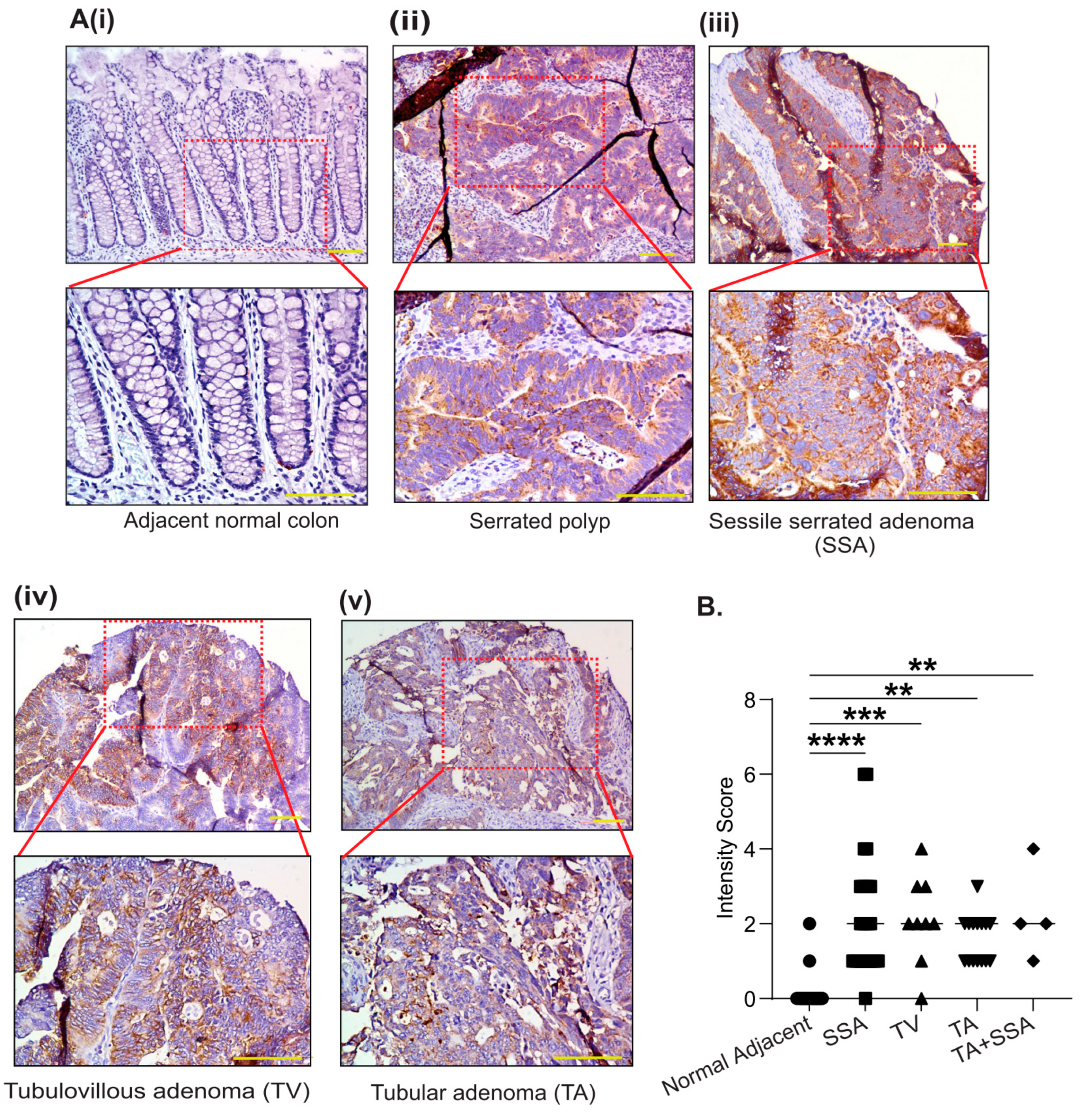
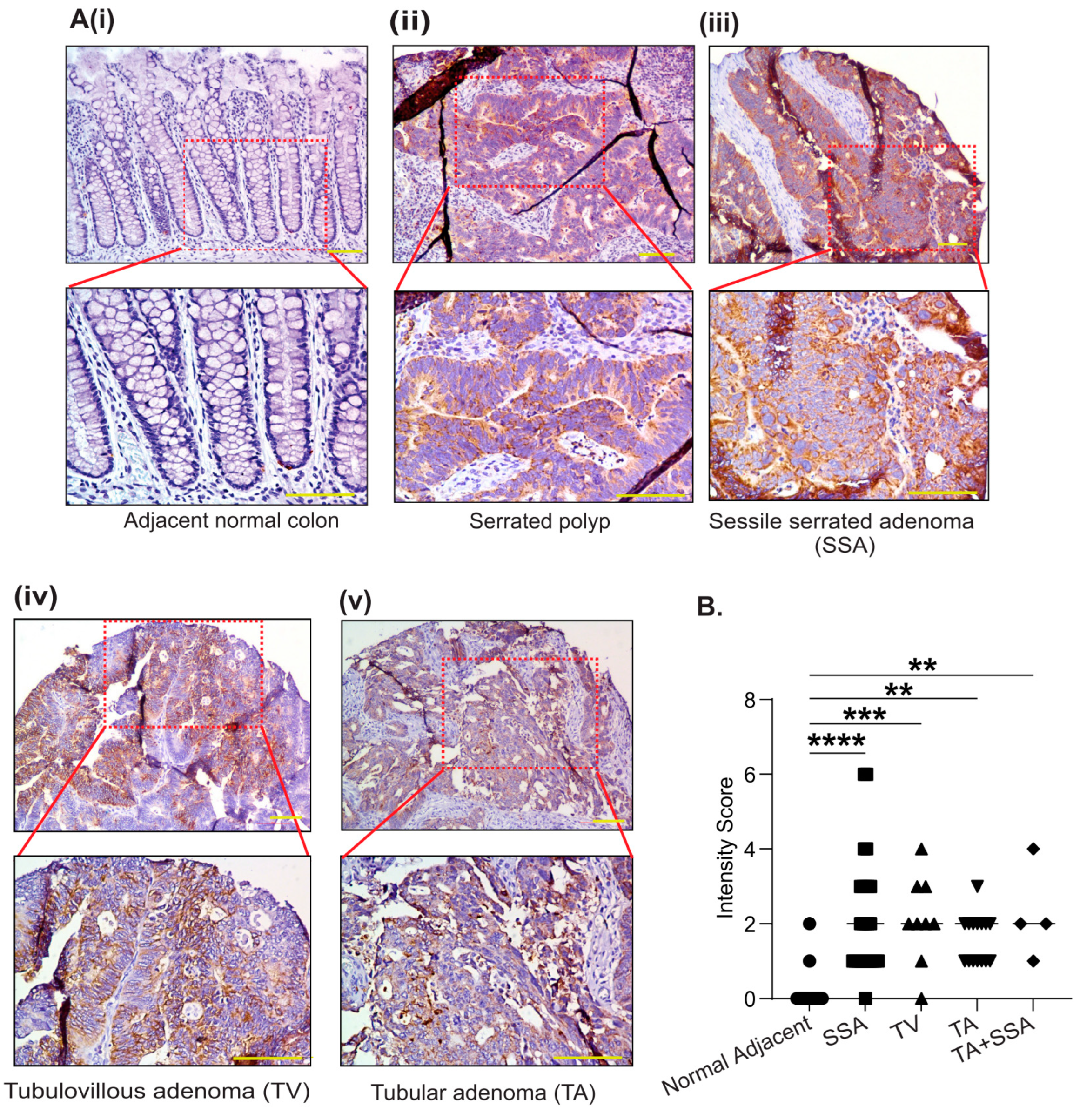
3.2. PYCR2 Upregulation in CRC Is an Early and Universal Event across CRC Phenotypes
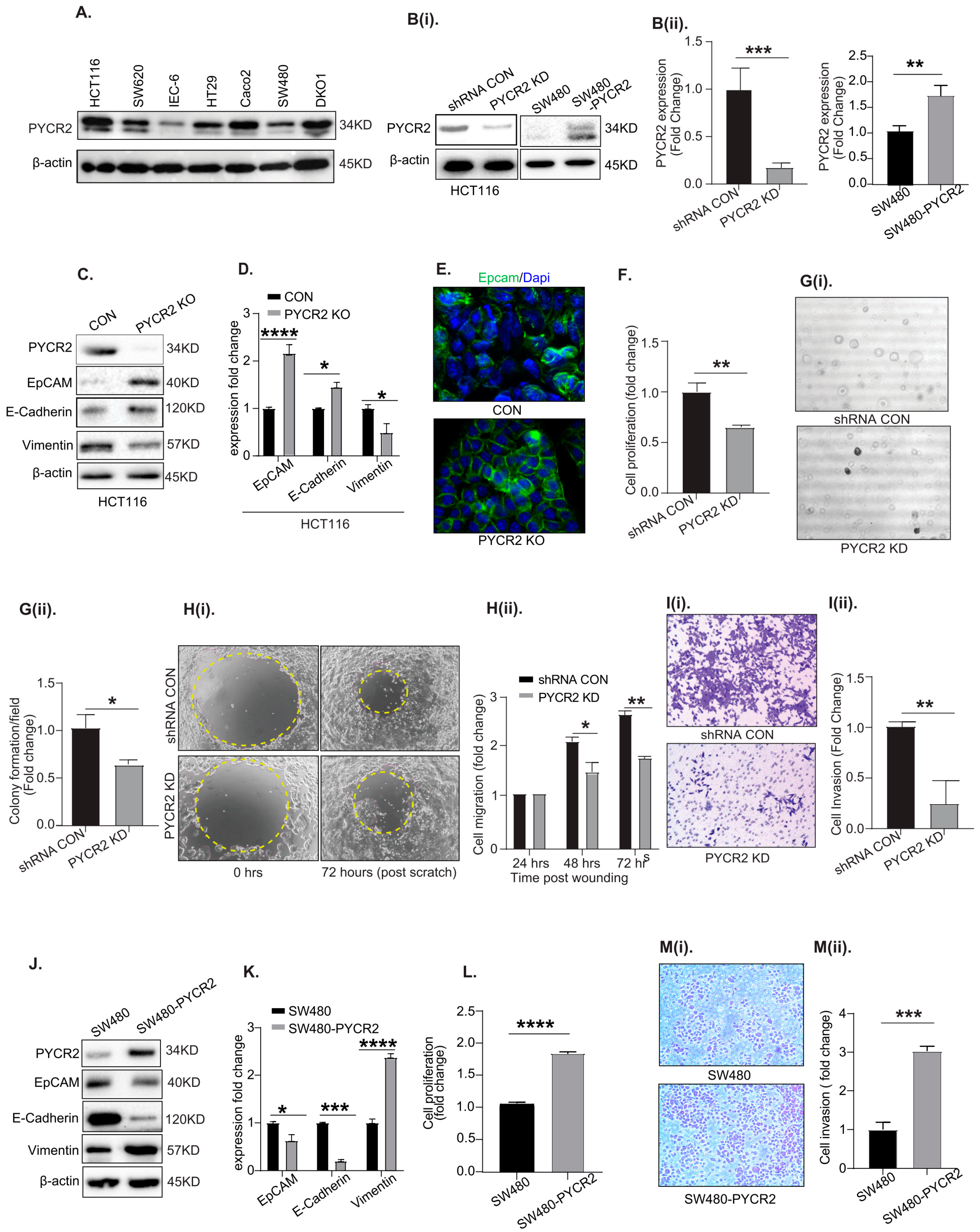
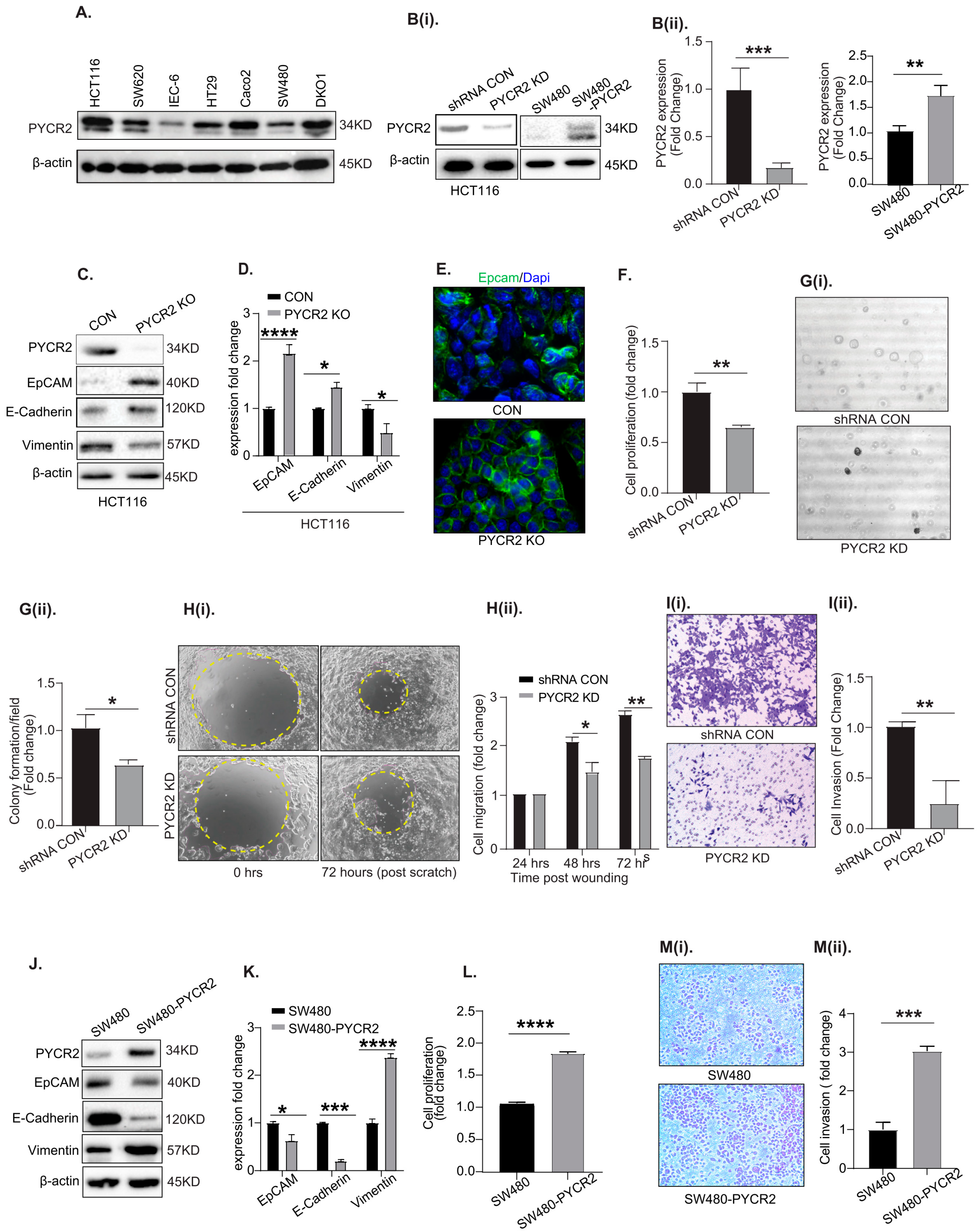
3.3. Genetic Manipulation of PYCR2 Expression in Colon Cancer Cells Modulates Epithelial-to-Mesenchymal Transition (EMT) and Their Tumorigenic and Invasive Abilities
3.4. Loss of PYCR2 Expression Decreases Intracellular Proline Content in CRC Cells
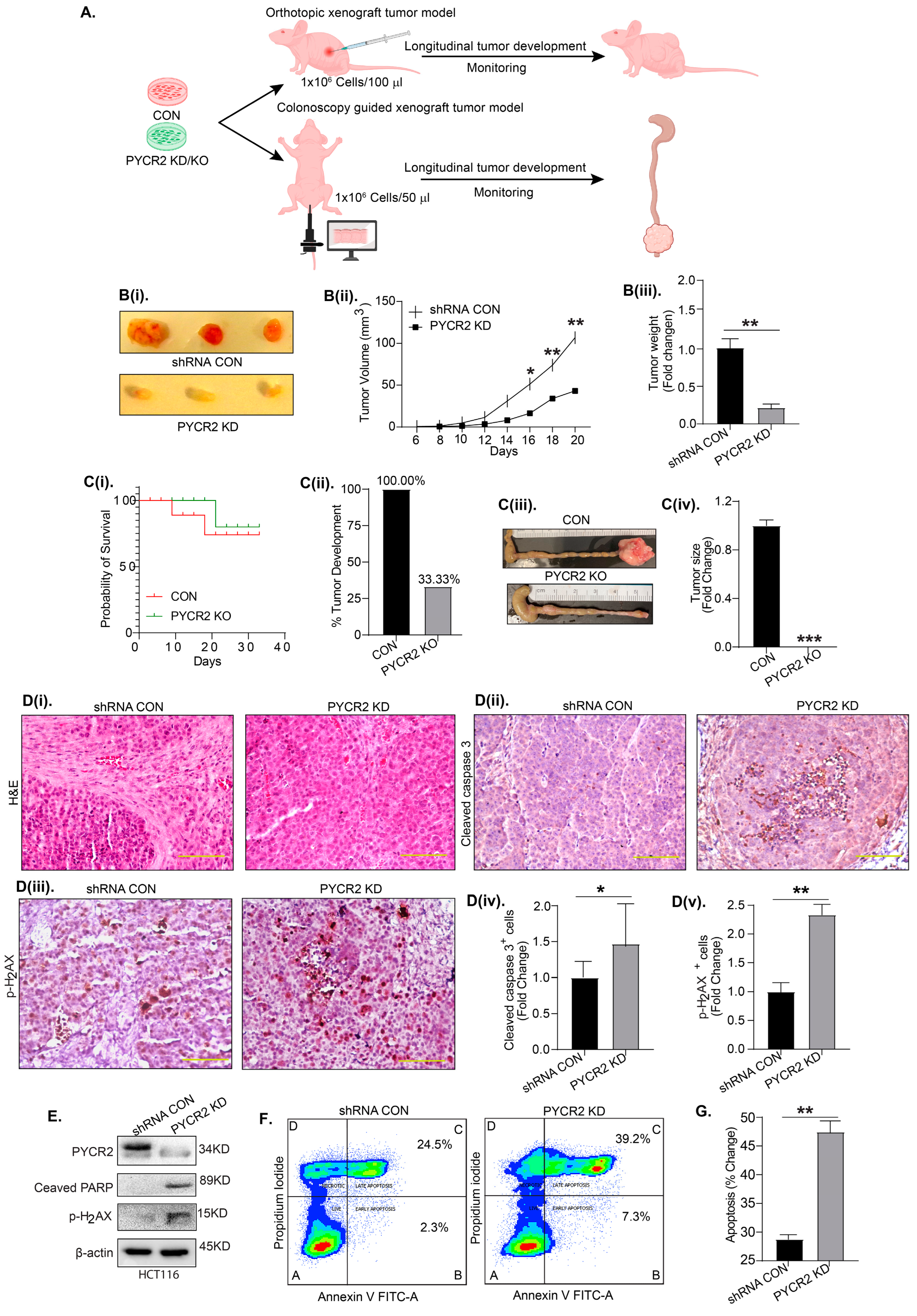
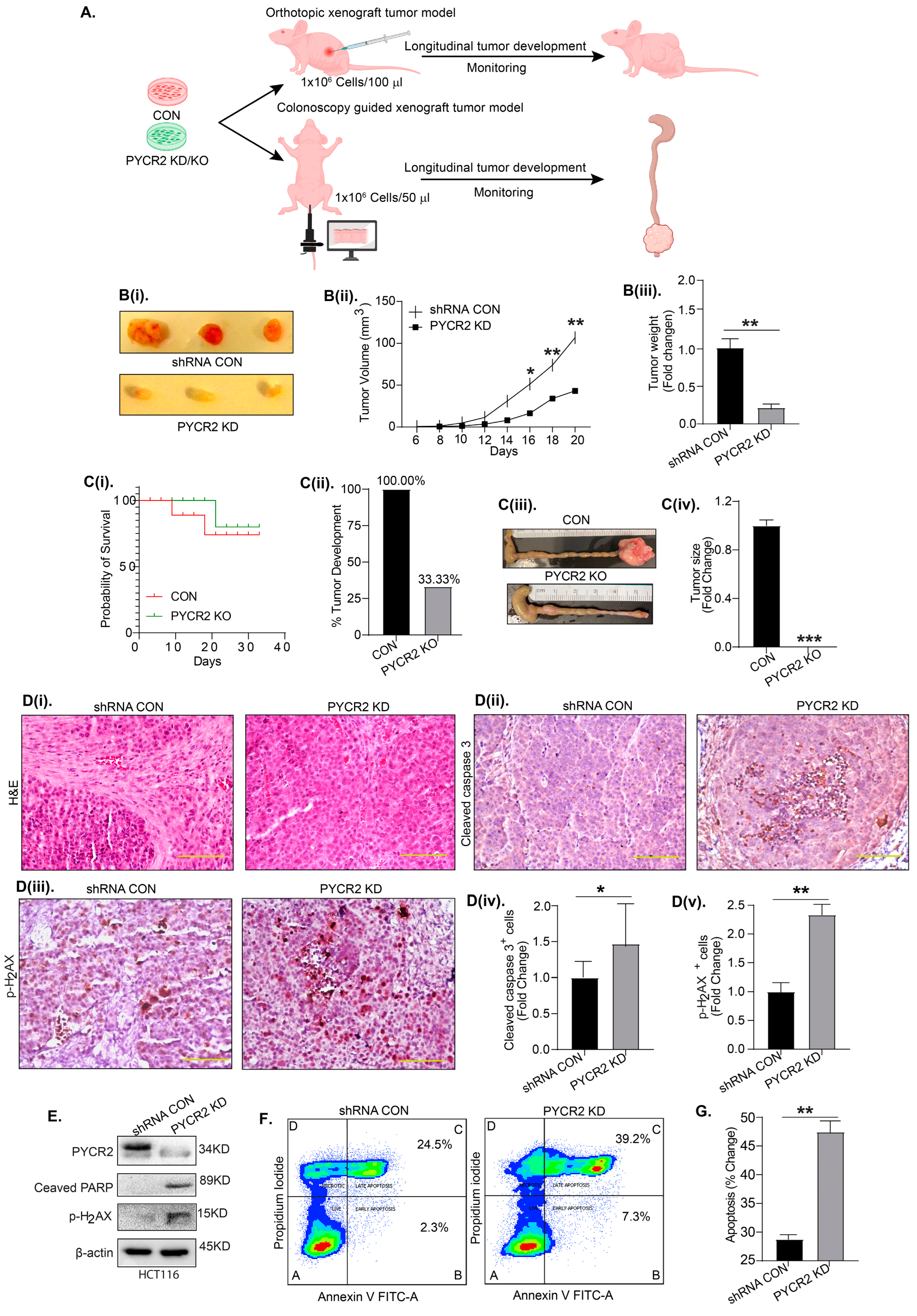
3.5. PYCR2 Inhibition Significantly Inhibits In Vivo Tumor Growth
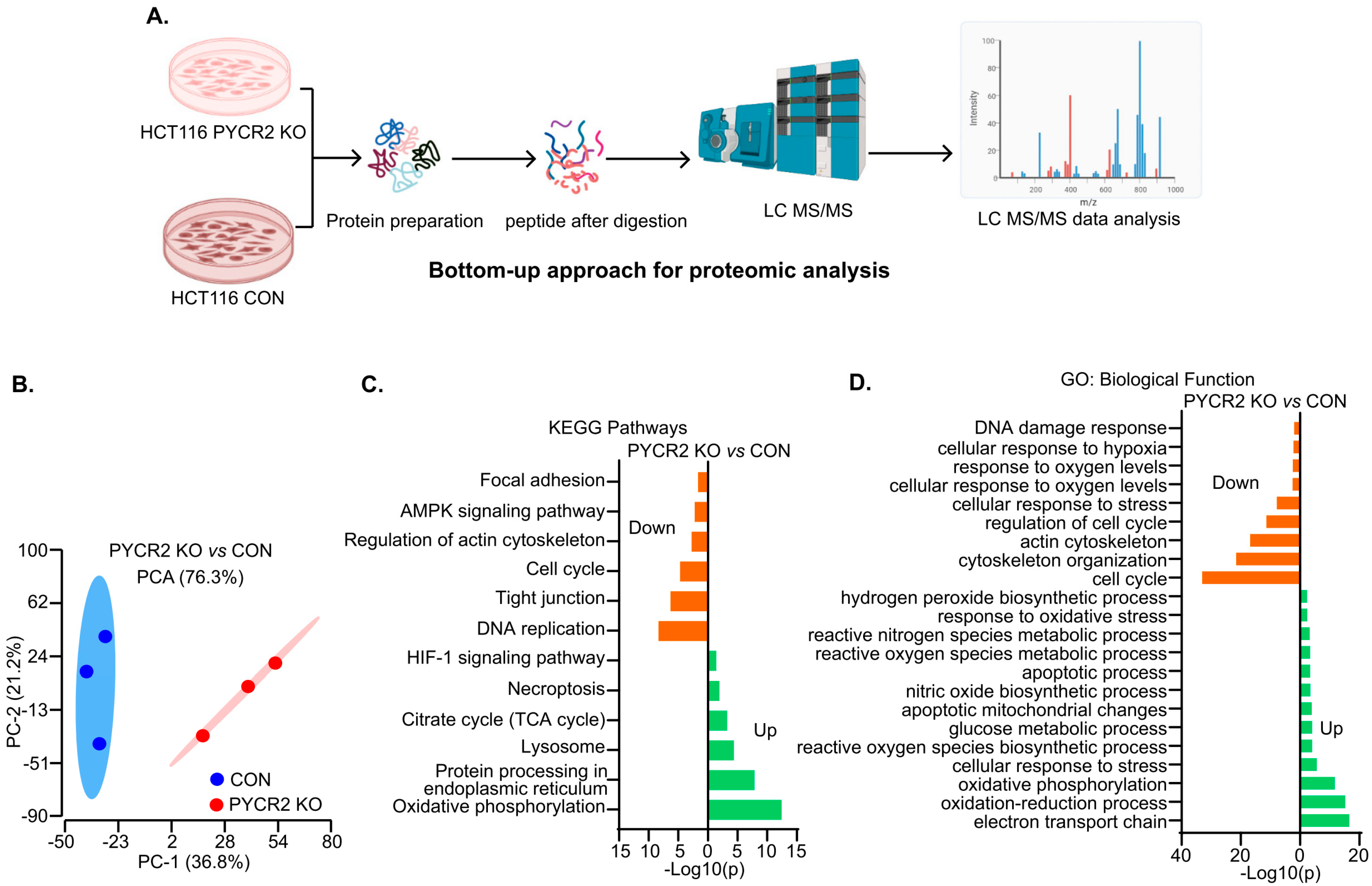
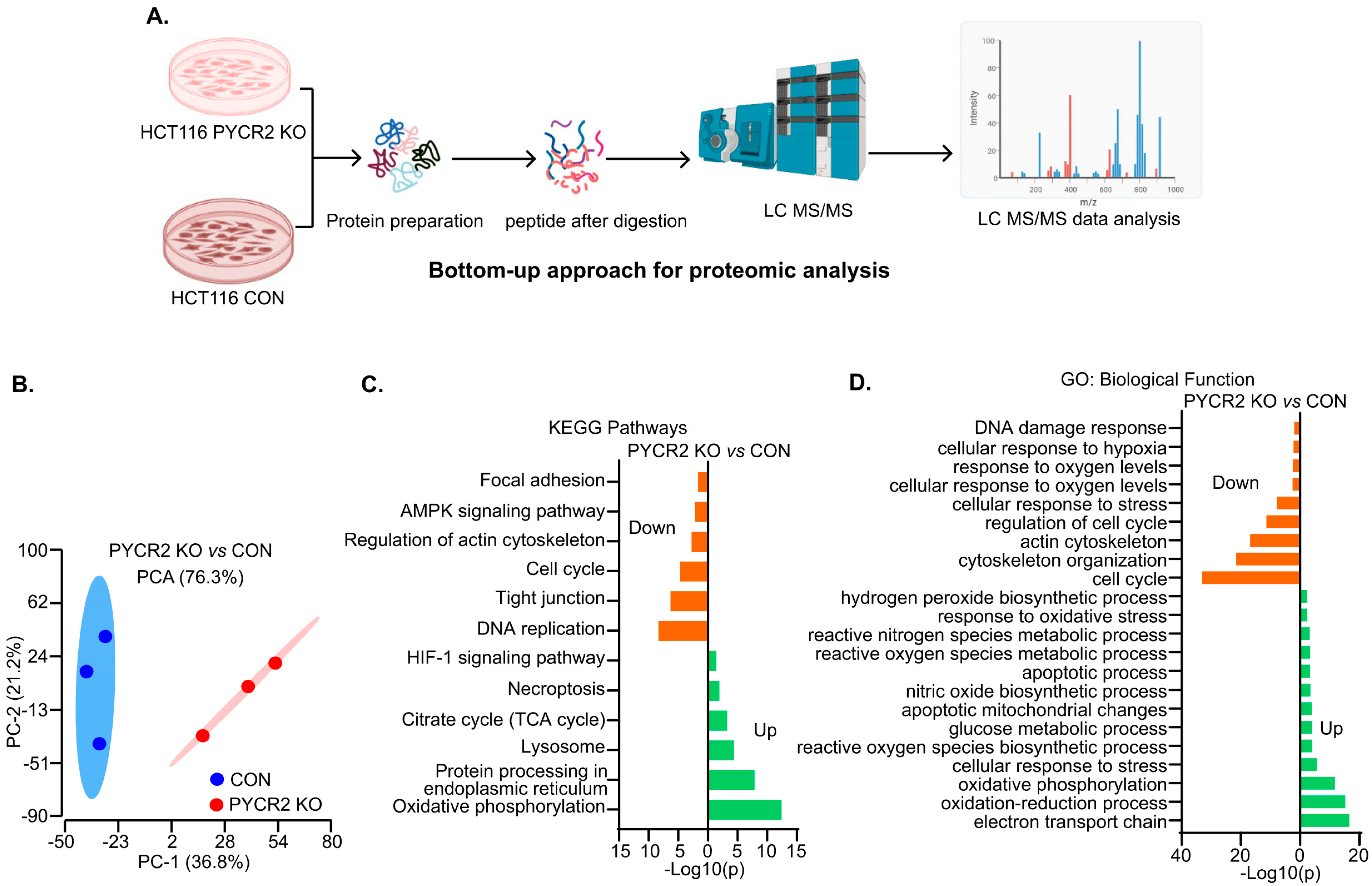
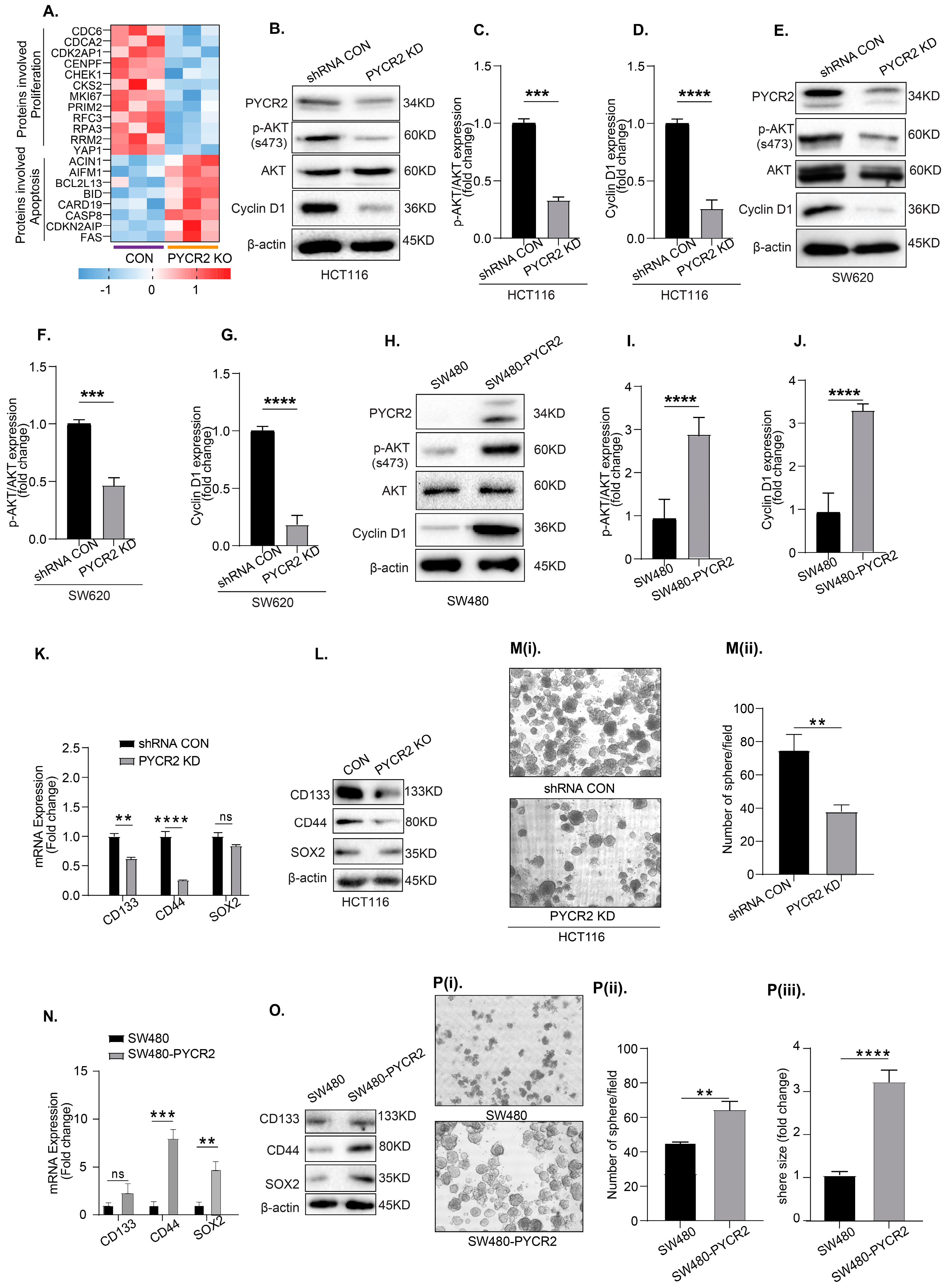
3.6. LC-MS/MS-Based Proteomics Analysis Showed the Profound Effects of PYCR2 Loss in Cell Survival and Proliferative and Metabolic Pathways
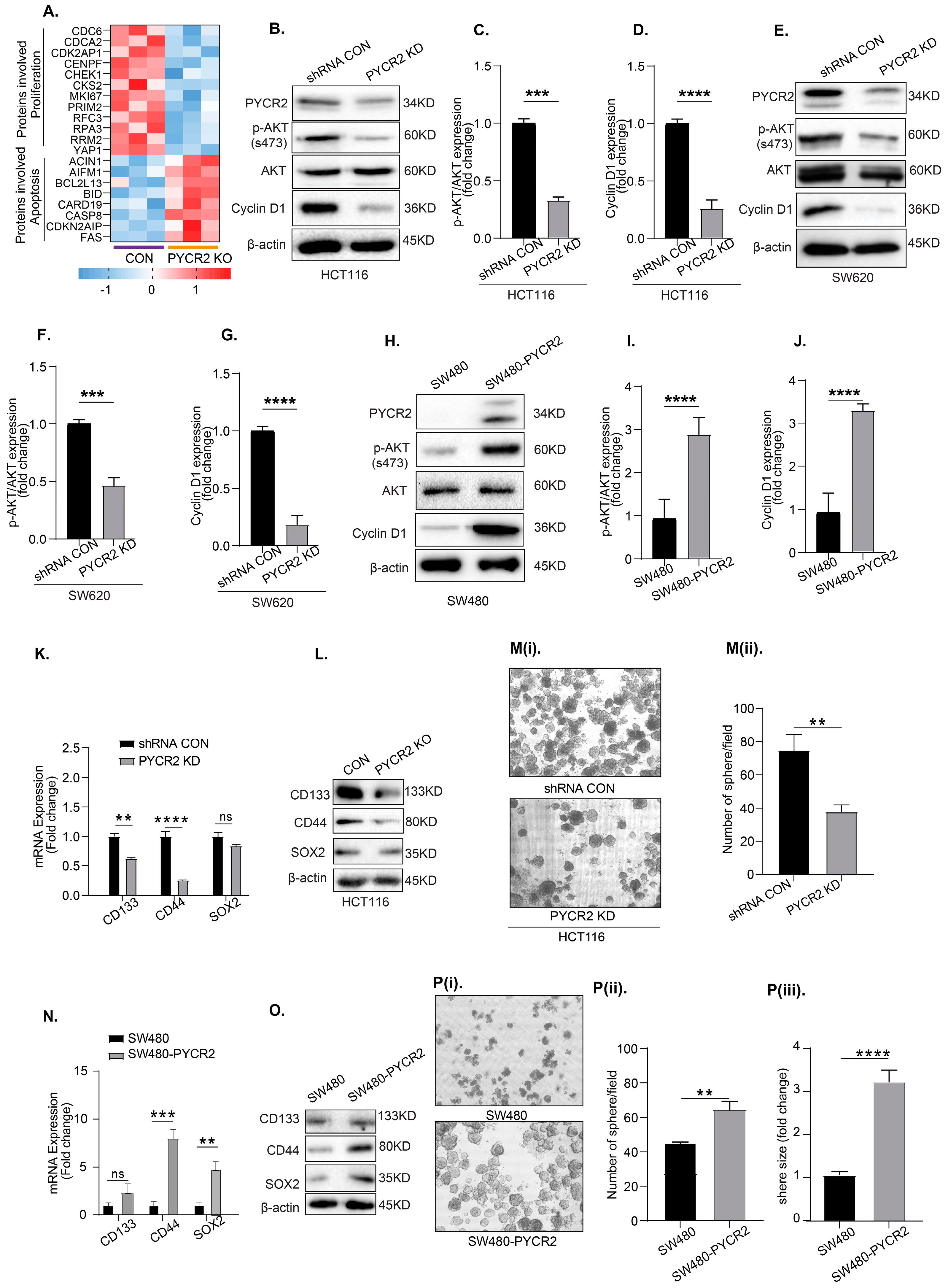
3.7. Loss of PYCR2 Reduces Cell Proliferation and Inhibits Cancer Stem Cell Populations
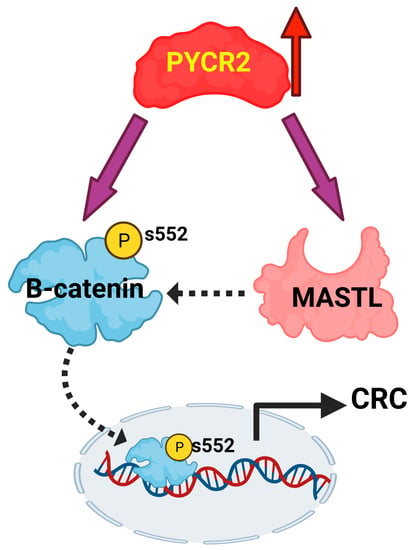
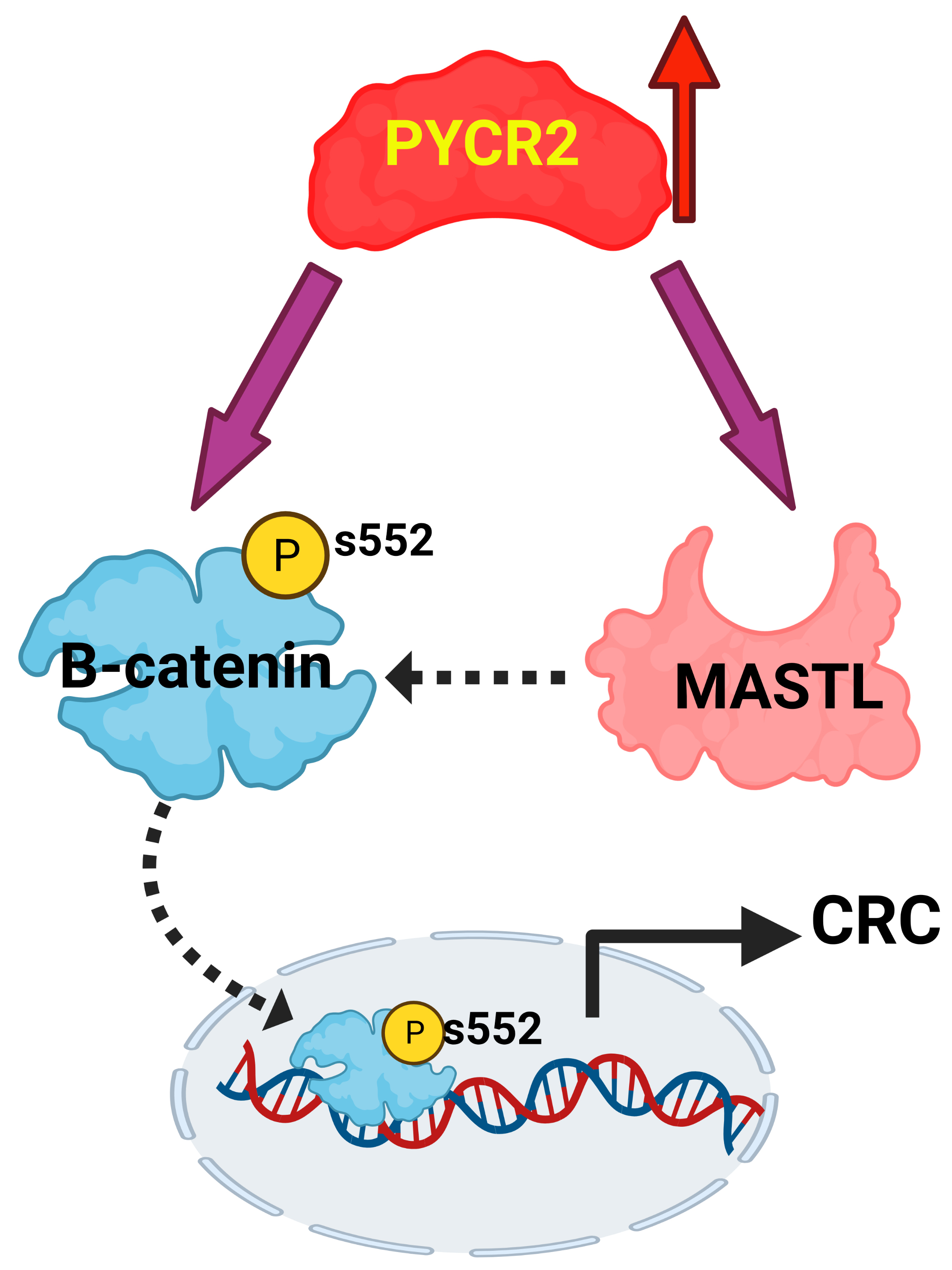
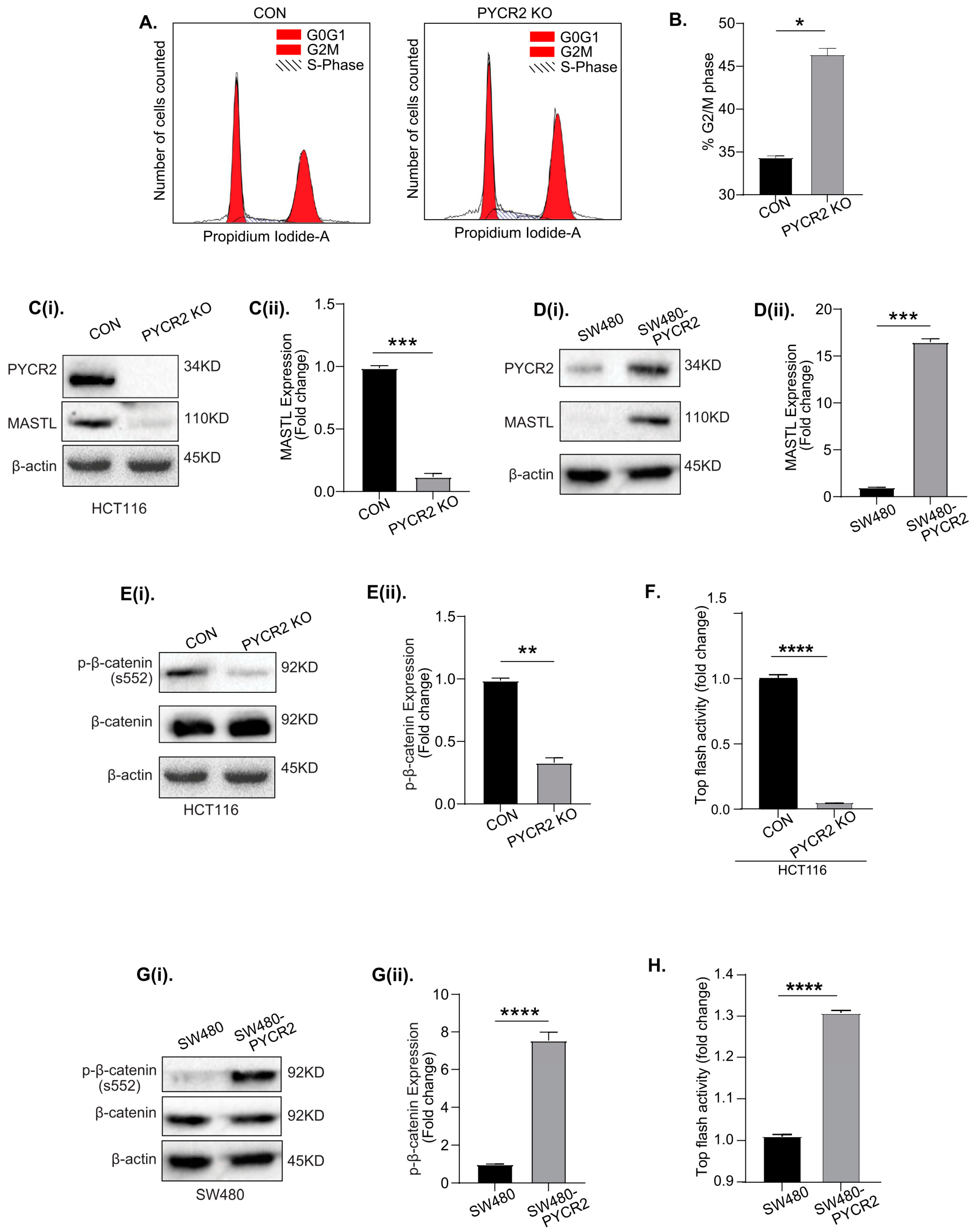
3.8. Loss of PYCR2 Inhibits MASTL/Wnt Signaling in Its Tumorigenic Promoting Effects
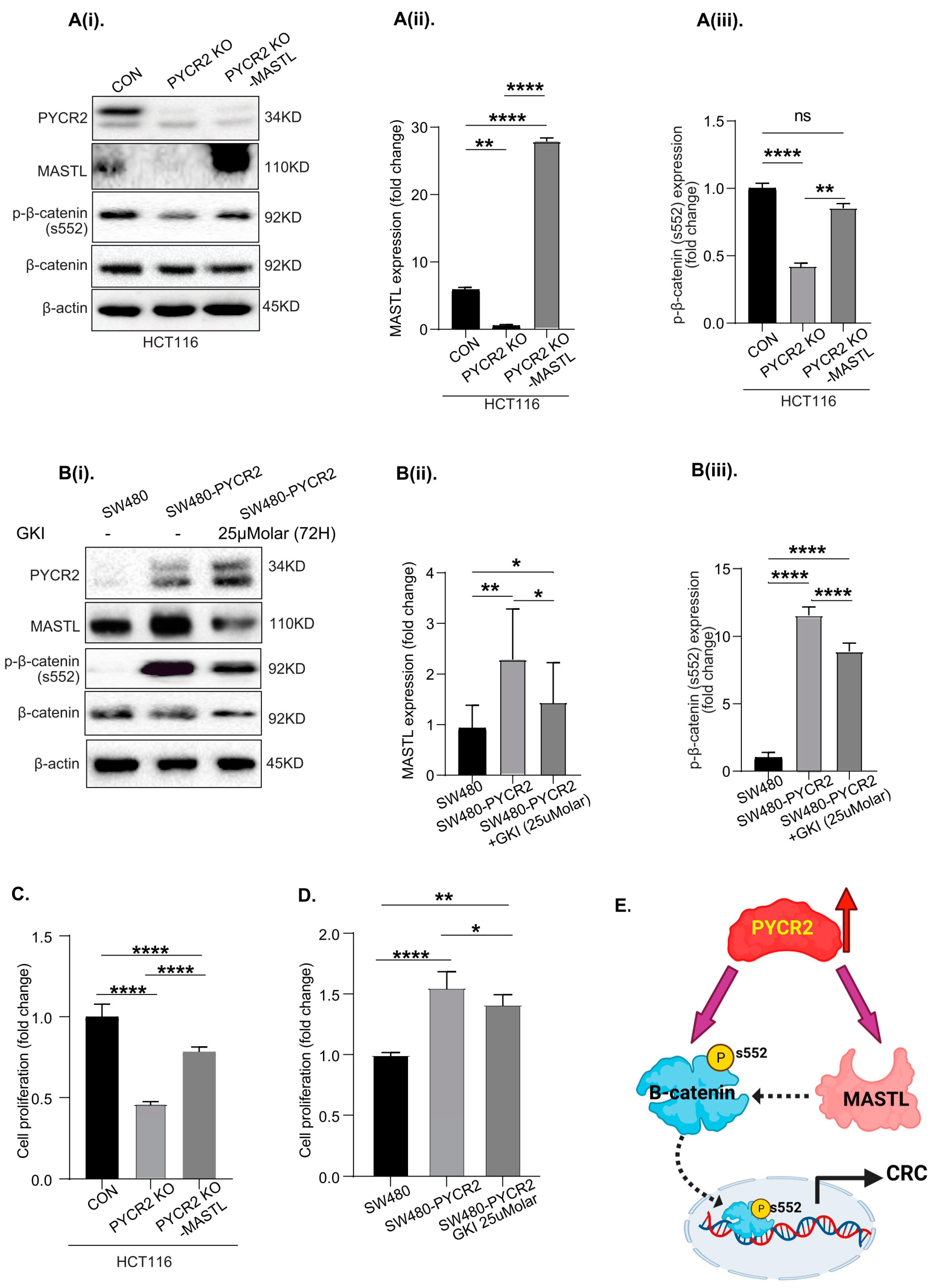
3.9. MASTL Mediates the CRC-Promoting Effects of PYCR2 Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [Green Version]
- Qiu, M.; Hu, J.; Yang, D.; Cosgrove, D.P.; Xu, R. Pattern of distant metastases in colorectal cancer: A SEER based study. Oncotarget 2015, 6, 38658–38666. [Google Scholar] [CrossRef] [Green Version]
- Adorno-Cruz, V.; Kibria, G.; Liu, X.; Doherty, M.; Junk, D.J.; Guan, D.; Hubert, C.; Venere, M.; Mulkearns-Hubert, E.; Sinyuk, M.; et al. Cancer stem cells: Targeting the roots of cancer, seeds of metastasis, and sources of therapy resistance. Cancer Res. 2015, 75, 924–929. [Google Scholar] [CrossRef] [Green Version]
- Endo, H.; Owada, S.; Inagaki, Y.; Shida, Y.; Tatemichi, M. Metabolic reprogramming sustains cancer cell survival following extracellular matrix detachment. Redox Biol. 2020, 36, 101643. [Google Scholar] [CrossRef]
- D’Aniello, C.; Patriarca, E.J.; Phang, J.M.; Minchiotti, G. Proline Metabolism in Tumor Growth and Metastatic Progression. Front. Oncol. 2020, 10, 776. [Google Scholar] [CrossRef]
- Zheng, Y.; Cabassa-Hourton, C.; Planchais, S.; Lebreton, S.; Savoure, A. The proline cycle as an eukaryotic redox valve. J. Exp. Bot. 2021, 72, 6856–6866. [Google Scholar] [CrossRef]
- Chalecka, M.; Kazberuk, A.; Palka, J.; Surazynski, A. P5C as an Interface of Proline Interconvertible Amino Acids and Its Role in Regulation of Cell Survival and Apoptosis. Int. J. Mol. Sci. 2021, 22, 11763. [Google Scholar] [CrossRef]
- Perez-Arellano, I.; Carmona-Alvarez, F.; Martinez, A.I.; Rodriguez-Diaz, J.; Cervera, J. Pyrroline-5-carboxylate synthase and proline biosynthesis: From osmotolerance to rare metabolic disease. Protein Sci. 2010, 19, 372–382. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Wu, Y.; Wang, J. Pyrroline-5-carboxylate reductase 1 promotes cell proliferation via inhibiting apoptosis in human malignant melanoma. Cancer Manag. Res. 2018, 10, 6399–6407. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.A.; Bart Williams, D.; Zhaorigetu, S.; Khalil, S.; Wan, G.; Valle, D. Functional genomics and SNP analysis of human genes encoding proline metabolic enzymes. Amino Acids 2008, 35, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhang, X.; Wei, X.; Feng, H.; Hu, B.; Deng, Z.; Liu, B.; Luan, Y.; Ruan, Y.; Liu, X.; et al. A Mitochondrial Dysfunction and Oxidative Stress Pathway-Based Prognostic Signature for Clear Cell Renal Cell Carcinoma. Oxid. Med. Cell Longev. 2021, 2021, 9939331. [Google Scholar] [CrossRef]
- Wang, Q.; Xie, Z.; Li, C.; Xu, C.; Ding, C.; Ju, S.; Chen, J.; Cui, Z.; Chen, C.; Gu, B.; et al. CRIF1 promotes the progression of non-small-cell lung cancer by SIRT3- mediated deacetylation of PYCR1. J. Mol. Histol. 2022, 53, 657–667. [Google Scholar] [CrossRef]
- Xu, H.; He, Y.; Lin, L.; Li, M.; Zhou, Z.; Yang, Y. MiR-1207-5p targets PYCR1 to inhibit the progression of prostate cancer. Biochem. Biophys. Res. Commun. 2021, 575, 56–64. [Google Scholar] [CrossRef]
- Liu, W.; Wang, Q.; Chang, J. Global metabolomic profiling of trastuzumab resistant gastric cancer cells reveals major metabolic pathways and metabolic signatures based on UHPLC-Q exactive-MS/MS. RSC Adv. 2019, 9, 41192–41208. [Google Scholar] [CrossRef]
- Keeratichamroen, S.; Subhasitanont, P.; Chokchaichamnankit, D.; Weeraphan, C.; Saharat, K.; Sritana, N.; Kantathavorn, N.; Wiriyaukaradecha, K.; Sricharunrat, T.; Paricharttanakul, N.M.; et al. Identification of potential cervical cancer serum biomarkers in Thai patients. Oncol. Lett. 2020, 19, 3815–3826. [Google Scholar] [CrossRef] [Green Version]
- Gao, Q.; Zhu, H.; Dong, L.; Shi, W.; Chen, R.; Song, Z.; Huang, C.; Li, J.; Dong, X.; Zhou, Y.; et al. Integrated Proteogenomic Characterization of HBV-Related Hepatocellular Carcinoma. Cell 2019, 179, 561–577.e22. [Google Scholar] [CrossRef]
- Nakayama, T.; Al-Maawali, A.; El-Quessny, M.; Rajab, A.; Khalil, S.; Stoler, J.M.; Tan, W.H.; Nasir, R.; Schmitz-Abe, K.; Hill, R.S.; et al. Mutations in PYCR2, Encoding Pyrroline-5-Carboxylate Reductase 2, Cause Microcephaly and Hypomyelination. Am. J. Hum. Genet. 2015, 96, 709–719. [Google Scholar] [CrossRef] [Green Version]
- Zaki, M.S.; Bhat, G.; Sultan, T.; Issa, M.; Jung, H.J.; Dikoglu, E.; Selim, L.; Mahmoud, I.G.; Abdel-Hamid, M.S.; Abdel-Salam, G.; et al. PYCR2 Mutations cause a lethal syndrome of microcephaly and failure to thrive. Ann. Neurol. 2016, 80, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.J.; Ni, W.; Wei, Q.; Wu, Z.Y. Spastic paraplegia as the only symptom in two adult-onset patients carrying a novel pathogenic variant in PYCR2. Eur. J. Neurol. 2021, 28, e17–e19. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Karthikeyan, S.K.; Korla, P.K.; Patel, H.; Shovon, A.R.; Athar, M.; Netto, G.J.; Qin, Z.S.; Kumar, S.; Manne, U.; et al. UALCAN: An update to the integrated cancer data analysis platform. Neoplasia 2022, 25, 18–27. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Kim, S.K.; Kim, S.Y.; Kim, J.H.; Roh, S.A.; Cho, D.H.; Kim, Y.S.; Kim, J.C. A nineteen gene-based risk score classifier predicts prognosis of colorectal cancer patients. Mol. Oncol. 2014, 8, 1653–1666. [Google Scholar] [CrossRef]
- Dhawan, P.; Singh, A.B.; Deane, N.G.; No, Y.; Shiou, S.R.; Schmidt, C.; Neff, J.; Washington, M.K.; Beauchamp, R.D. Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. J. Clin. Investig. 2005, 115, 1765–1776. [Google Scholar] [CrossRef] [Green Version]
- Thomas, E.; Thankan, R.S.; Purushottamachar, P.; Huang, W.; Kane, M.A.; Zhang, Y.; Ambulos, N.; Weber, D.J.; Njar, V.C.O. Transcriptome profiling reveals that VNPP433-3beta, the lead next-generation galeterone analog inhibits prostate cancer stem cells by downregulating epithelial-mesenchymal transition and stem cell markers. Mol. Carcinog. 2022, 61, 643–654. [Google Scholar] [CrossRef]
- Bahmad, H.F.; Cheaito, K.; Chalhoub, R.M.; Hadadeh, O.; Monzer, A.; Ballout, F.; El-Hajj, A.; Mukherji, D.; Liu, Y.N.; Daoud, G.; et al. Sphere-Formation Assay: Three-Dimensional in vitro Culturing of Prostate Cancer Stem/Progenitor Sphere-Forming Cells. Front. Oncol. 2018, 8, 347. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.; Ahmad, R.; Sharma, S.; Gowrikumar, S.; Primeaux, M.; Rana, S.; Natarajan, A.; Oupicky, D.; Hopkins, C.R.; Dhawan, P.; et al. PIK3C3 Inhibition Promotes Sensitivity to Colon Cancer Therapy by Inhibiting Cancer Stem Cells. Cancers 2021, 13, 2168. [Google Scholar] [CrossRef]
- Uppada, S.B.; Gowrikumar, S.; Ahmad, R.; Kumar, B.; Szeglin, B.; Chen, X.; Smith, J.J.; Batra, S.K.; Singh, A.B.; Dhawan, P. MASTL induces Colon Cancer progression and Chemoresistance by promoting Wnt/beta-catenin signaling. Mol. Cancer 2018, 17, 111. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, R.; Chaturvedi, R.; Olivares-Villagomez, D.; Habib, T.; Asim, M.; Shivesh, P.; Polk, D.B.; Wilson, K.T.; Washington, M.K.; Van Kaer, L.; et al. Targeted colonic claudin-2 expression renders resistance to epithelial injury, induces immune suppression, and protects from colitis. Mucosal Immunol. 2014, 7, 1340–1353. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, R.; Kumar, B.; Tamang, R.L.; Xu, W.; Talmon, G.A.; Mohs, A.M.; Dhawan, P.; Singh, A.B. Colonoscopy-based intramucosal transplantation of cancer cells for mouse modeling of colon cancer and lung metastasis. Biotechniques 2021, 71, 456–464. [Google Scholar] [CrossRef]
- Klupczynska, A.; Misiura, M.; Miltyk, W.; Oscilowska, I.; Palka, J.; Kokot, Z.J.; Matysiak, J. Development of an LC-MS Targeted Metabolomics Methodology to Study Proline Metabolism in Mammalian Cell Cultures. Molecules 2020, 25, 4639. [Google Scholar] [CrossRef]
- Williams, I.; Frank, L. Improved chemical synthesis and enzymatic assay of delta-1-pyrroline-5-carboxylic acid. Anal. Biochem. 1975, 64, 85–97. [Google Scholar] [CrossRef]
- Forlani, G.; Funck, D. A Specific and Sensitive Enzymatic Assay for the Quantitation of L-Proline. Front. Plant Sci. 2020, 11, 582026. [Google Scholar] [CrossRef]
- Yin, F.; Huang, X.; Xuan, Y. Pyrroline-5-Carboxylate Reductase-2 Promotes Colorectal Cancer Progression via Activating PI3K/AKT/mTOR Pathway. Dis. Markers 2021, 2021, 9950663. [Google Scholar] [CrossRef]
- Stastna, M.; Janeckova, L.; Hrckulak, D.; Kriz, V.; Korinek, V. Human Colorectal Cancer from the Perspective of Mouse Models. Genes 2019, 10, 788. [Google Scholar] [CrossRef] [Green Version]
- Pope, J.L.; Ahmad, R.; Bhat, A.A.; Washington, M.K.; Singh, A.B.; Dhawan, P. Claudin-1 overexpression in intestinal epithelial cells enhances susceptibility to adenamatous polyposis coli-mediated colon tumorigenesis. Mol. Cancer 2014, 13, 167. [Google Scholar] [CrossRef] [Green Version]
- Shussman, N.; Wexner, S.D. Colorectal polyps and polyposis syndromes. Gastroenterol. Rep. 2014, 2, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Scurrah, C.R.; McKinley, E.T.; Simmons, A.J.; Ramirez-Solano, M.A.; Zhu, X.; Markham, N.O.; Heiser, C.N.; Vega, P.N.; Rolong, A.; et al. Differential pre-malignant programs and microenvironment chart distinct paths to malignancy in human colorectal polyps. Cell 2021, 184, 6262–6280.e26. [Google Scholar] [CrossRef]
- Depciuch, J.; Klebowski, B.; Stec, M.; Szatanek, R.; Weglarczyk, K.; Baj-Krzyworzeka, M.; Parlinska-Wojtan, M.; Baran, J. Similarities in the General Chemical Composition of Colon Cancer Cells and Their Microvesicles Investigated by Spectroscopic Methods-Potential Clinical Relevance. Int. J. Mol. Sci. 2020, 21, 1826. [Google Scholar] [CrossRef] [Green Version]
- Gires, O.; Pan, M.; Schinke, H.; Canis, M.; Baeuerle, P.A. Expression and function of epithelial cell adhesion molecule EpCAM: Where are we after 40 years? Cancer Metastasis Rev. 2020, 39, 969–987. [Google Scholar] [CrossRef]
- Niknami, Z.; Muhammadnejad, A.; Ebrahimi, A.; Harsani, Z.; Shirkoohi, R. Significance of E-cadherin and Vimentin as epithelial-mesenchymal transition markers in colorectal carcinoma prognosis. EXCLI J. 2020, 19, 917–926. [Google Scholar] [CrossRef]
- Liu, C.Y.; Lin, H.H.; Tang, M.J.; Wang, Y.K. Vimentin contributes to epithelial-mesenchymal transition cancer cell mechanics by mediating cytoskeletal organization and focal adhesion maturation. Oncotarget 2015, 6, 15966–15983. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, N.; Dickman, M.B.; Becker, D.F. Proline modulates the intracellular redox environment and protects mammalian cells against oxidative stress. Free Radic. Biol. Med. 2008, 44, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Zareba, I.; Huynh, T.Y.L.; Kazberuk, A.; Teul, J.; Klupczynska, A.; Matysiak, J.; Surazynski, A.; Palka, J. Overexpression of Prolidase Induces Autophagic Death in MCF-7 Breast Cancer Cells. Cell Physiol. Biochem. 2020, 54, 875–887. [Google Scholar] [CrossRef]
- Shimura, T.; Noma, N.; Oikawa, T.; Ochiai, Y.; Kakuda, S.; Kuwahara, Y.; Takai, Y.; Takahashi, A.; Fukumoto, M. Activation of the AKT/cyclin D1/Cdk4 survival signaling pathway in radioresistant cancer stem cells. Oncogenesis 2012, 1, e12. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Xia, L.; Wang, H.; Oyang, L.; Su, M.; Liu, Q.; Lin, J.; Tan, S.; Tian, Y.; Liao, Q.; et al. Cancer stem cells in progression of colorectal cancer. Oncotarget 2018, 9, 33403–33415. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.; Wyatt, D.; Baker, A.T.; Simms, P.; Peiffer, D.S.; Fernandez, M.; Rakha, E.; Green, A.; Filipovic, A.; Miele, L.; et al. Inhibition of HER2 Increases JAGGED1-dependent Breast Cancer Stem Cells: Role for Membrane JAGGED1. Clin. Cancer Res. 2018, 24, 4566–4578. [Google Scholar] [CrossRef] [Green Version]
- Raghavan, S.; Mehta, P.; Xie, Y.; Lei, Y.L.; Mehta, G. Ovarian cancer stem cells and macrophages reciprocally interact through the WNT pathway to promote pro-tumoral and malignant phenotypes in 3D engineered microenvironments. J. Immunother. Cancer 2019, 7, 190. [Google Scholar] [CrossRef] [Green Version]
- Leng, Z.; Xia, Q.; Chen, J.; Li, Y.; Xu, J.; Zhao, E.; Zheng, H.; Ai, W.; Dong, J. Lgr5+CD44+EpCAM+ Strictly Defines Cancer Stem Cells in Human Colorectal Cancer. Cell Physiol. Biochem. 2018, 46, 860–872. [Google Scholar] [CrossRef]
- Takeda, K.; Mizushima, T.; Yokoyama, Y.; Hirose, H.; Wu, X.; Qian, Y.; Ikehata, K.; Miyoshi, N.; Takahashi, H.; Haraguchi, N.; et al. Sox2 is associated with cancer stem-like properties in colorectal cancer. Sci. Rep. 2018, 8, 17639. [Google Scholar] [CrossRef] [Green Version]
- Oshima, N.; Yamada, Y.; Nagayama, S.; Kawada, K.; Hasegawa, S.; Okabe, H.; Sakai, Y.; Aoi, T. Induction of cancer stem cell properties in colon cancer cells by defined factors. PLoS ONE 2014, 9, e101735. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.L.; Sun, Y.F.; Wang, B.L.; Shen, M.N.; Zhou, Y.; Chen, J.W.; Hu, B.; Gong, Z.J.; Zhang, X.; Cao, Y.; et al. Sphere-forming culture enriches liver cancer stem cells and reveals Stearoyl-CoA desaturase 1 as a potential therapeutic target. BMC Cancer 2019, 19, 760. [Google Scholar] [CrossRef] [Green Version]
- Schatoff, E.M.; Leach, B.I.; Dow, L.E. Wnt Signaling and Colorectal Cancer. Curr. Colorectal Cancer Rep. 2017, 13, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Fang, D.; Hawke, D.; Zheng, Y.; Xia, Y.; Meisenhelder, J.; Nika, H.; Mills, G.B.; Kobayashi, R.; Hunter, T.; Lu, Z. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J. Biol. Chem. 2007, 282, 11221–11229. [Google Scholar] [CrossRef] [Green Version]
- Phan, L.M.; Yeung, S.C.; Lee, M.H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [CrossRef]
- Wang, S.; Gu, L.; Huang, L.; Fang, J.; Liu, Z.; Xu, Q. The upregulation of PYCR2 is associated with aggressive colon cancer progression and a poor prognosis. Biochem. Biophys. Res. Commun. 2021, 572, 20–26. [Google Scholar] [CrossRef]
- Cespedes, M.V.; Espina, C.; Garcia-Cabezas, M.A.; Trias, M.; Boluda, A.; Gomez del Pulgar, M.T.; Sancho, F.J.; Nistal, M.; Lacal, J.C.; Mangues, R. Orthotopic microinjection of human colon cancer cells in nude mice induces tumor foci in all clinically relevant metastatic sites. Am. J. Pathol. 2007, 170, 1077–1085. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Yu, H.; Tan, X.F.; Lim, T.K.; Zubaidah, R.M.; Tan, H.T.; Chung, M.C.; Lin, Q. Identification of key players for colorectal cancer metastasis by iTRAQ quantitative proteomics profiling of isogenic SW480 and SW620 cell lines. J. Proteome Res. 2011, 10, 4373–4387. [Google Scholar] [CrossRef]
- Qvarnstrom, O.F.; Simonsson, M.; Eriksson, V.; Turesson, I.; Carlsson, J. gammaH2AX and cleaved PARP-1 as apoptotic markers in irradiated breast cancer BT474 cellular spheroids. Int. J. Oncol. 2009, 35, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Tao, F.; Zhang, X.; Zhang, Y.; Sun, X.; Wu, D. Role of Wnt/beta-Catenin Signaling in the Chemoresistance Modulation of Colorectal Cancer. BioMed Res. Int. 2020, 2020, 9390878. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lama Tamang, R.; Kumar, B.; Patel, S.M.; Thapa, I.; Ahmad, A.; Kumar, V.; Ahmad, R.; Becker, D.F.; Bastola, D.; Dhawan, P.; et al. Pyrroline-5-Carboxylate Reductase-2 Promotes Colorectal Carcinogenesis by Modulating Microtubule-Associated Serine/Threonine Kinase-like/Wnt/β-Catenin Signaling. Cells 2023, 12, 1883. https://doi.org/10.3390/cells12141883
Lama Tamang R, Kumar B, Patel SM, Thapa I, Ahmad A, Kumar V, Ahmad R, Becker DF, Bastola D, Dhawan P, et al. Pyrroline-5-Carboxylate Reductase-2 Promotes Colorectal Carcinogenesis by Modulating Microtubule-Associated Serine/Threonine Kinase-like/Wnt/β-Catenin Signaling. Cells. 2023; 12(14):1883. https://doi.org/10.3390/cells12141883
Chicago/Turabian StyleLama Tamang, Raju, Balawant Kumar, Sagar M. Patel, Ishwor Thapa, Alshomrani Ahmad, Vikas Kumar, Rizwan Ahmad, Donald F. Becker, Dundy (Kiran) Bastola, Punita Dhawan, and et al. 2023. "Pyrroline-5-Carboxylate Reductase-2 Promotes Colorectal Carcinogenesis by Modulating Microtubule-Associated Serine/Threonine Kinase-like/Wnt/β-Catenin Signaling" Cells 12, no. 14: 1883. https://doi.org/10.3390/cells12141883
APA StyleLama Tamang, R., Kumar, B., Patel, S. M., Thapa, I., Ahmad, A., Kumar, V., Ahmad, R., Becker, D. F., Bastola, D., Dhawan, P., & Singh, A. B. (2023). Pyrroline-5-Carboxylate Reductase-2 Promotes Colorectal Carcinogenesis by Modulating Microtubule-Associated Serine/Threonine Kinase-like/Wnt/β-Catenin Signaling. Cells, 12(14), 1883. https://doi.org/10.3390/cells12141883