

A Diagnostic Gene-Expression Signature in Fibroblasts of Amyotrophic Lateral Sclerosis


 ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Primary Fibroblast Isolation and Culture
2.3. RNA Isolation, Microarray Processing, and Data Extraction
2.4. Gene Expression Profiling and Class Prediction Modeling
2.5. Functional Enrichment and Network Analysis
3. Results
3.1. Transcriptome Profiles Reveal a Molecular Signature for sALS Fibroblasts
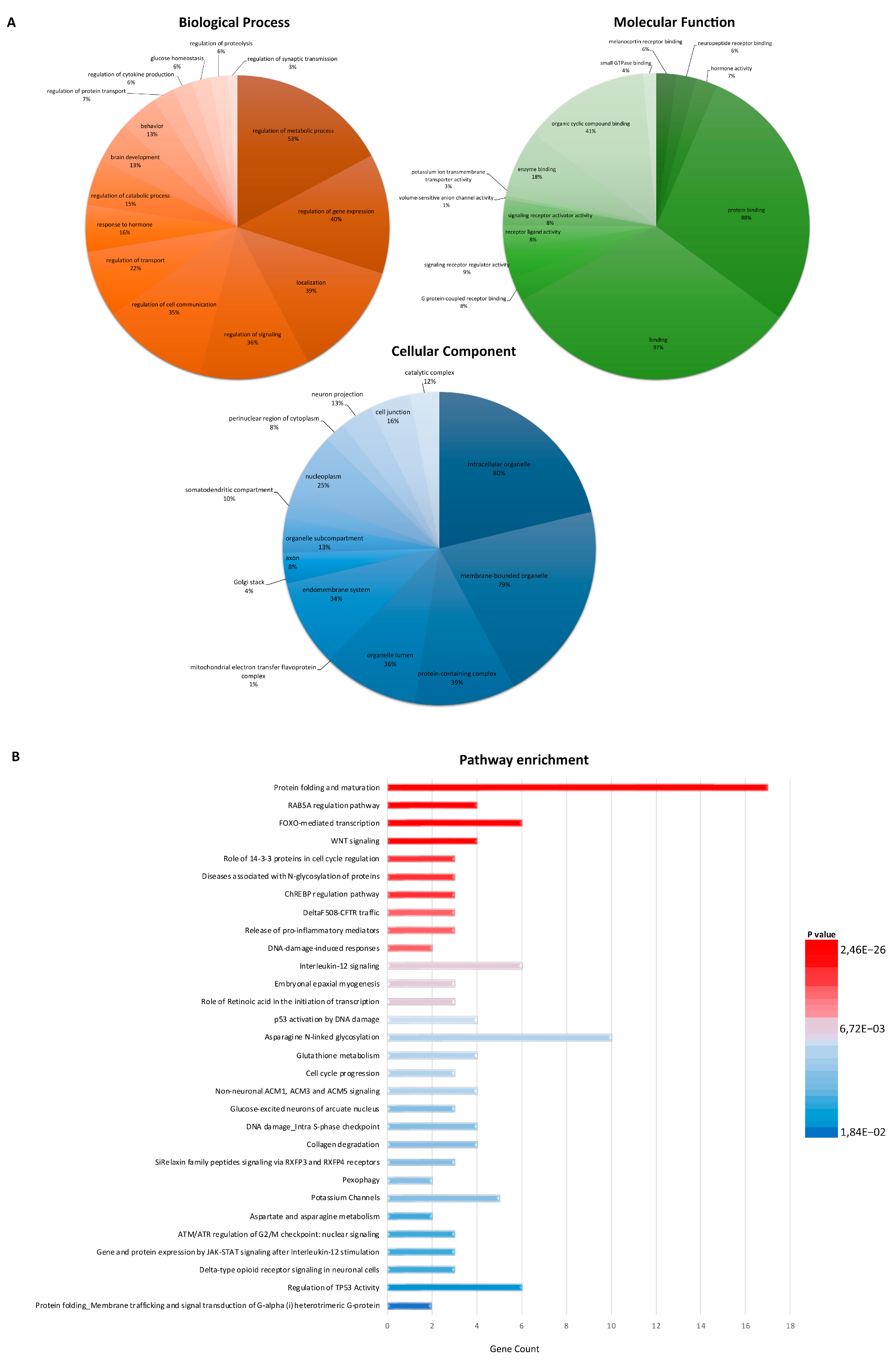
3.2. Functional Enrichment Analysis Defines Key Factors and Processes Perturbed in sALS Fibroblasts
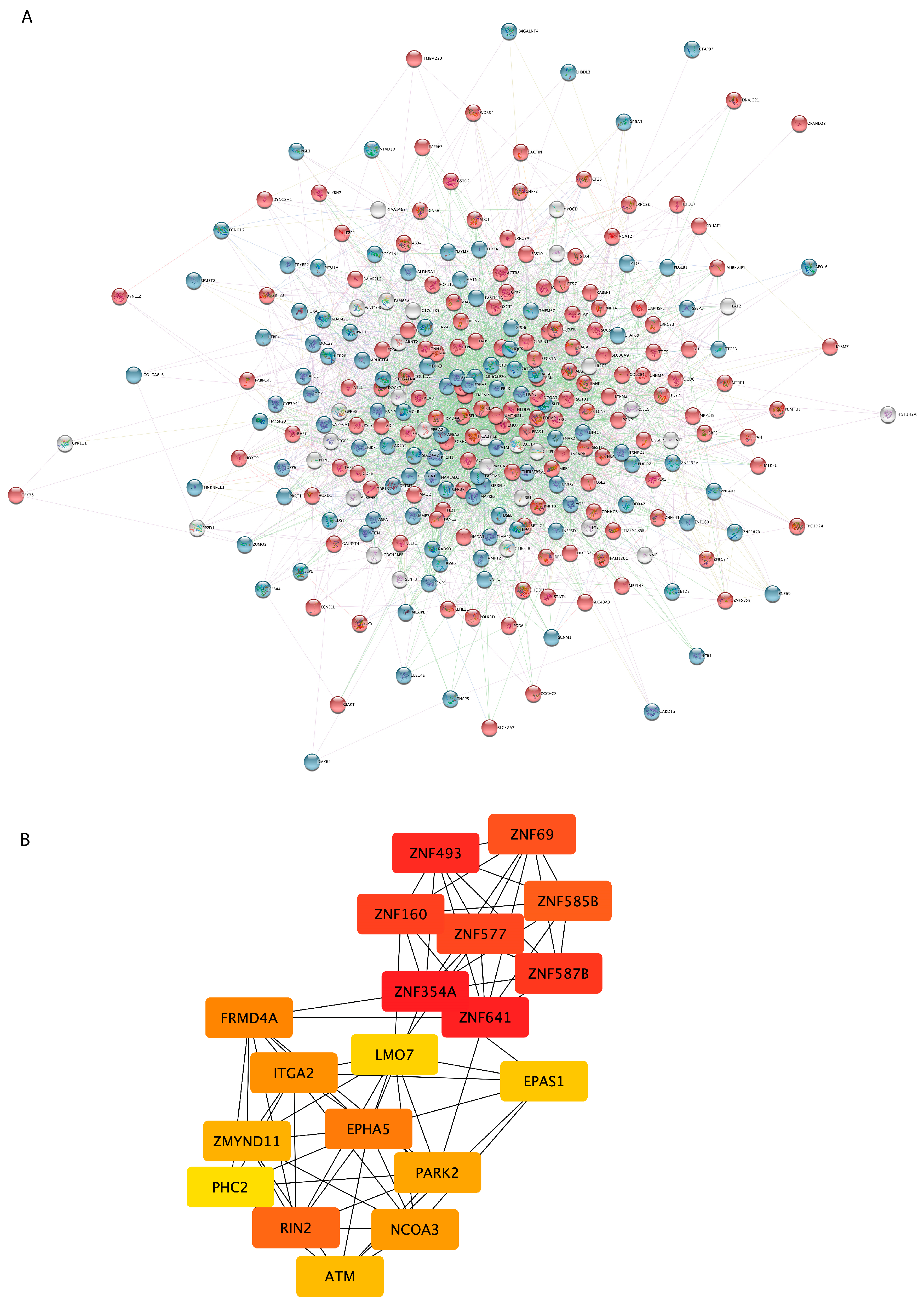
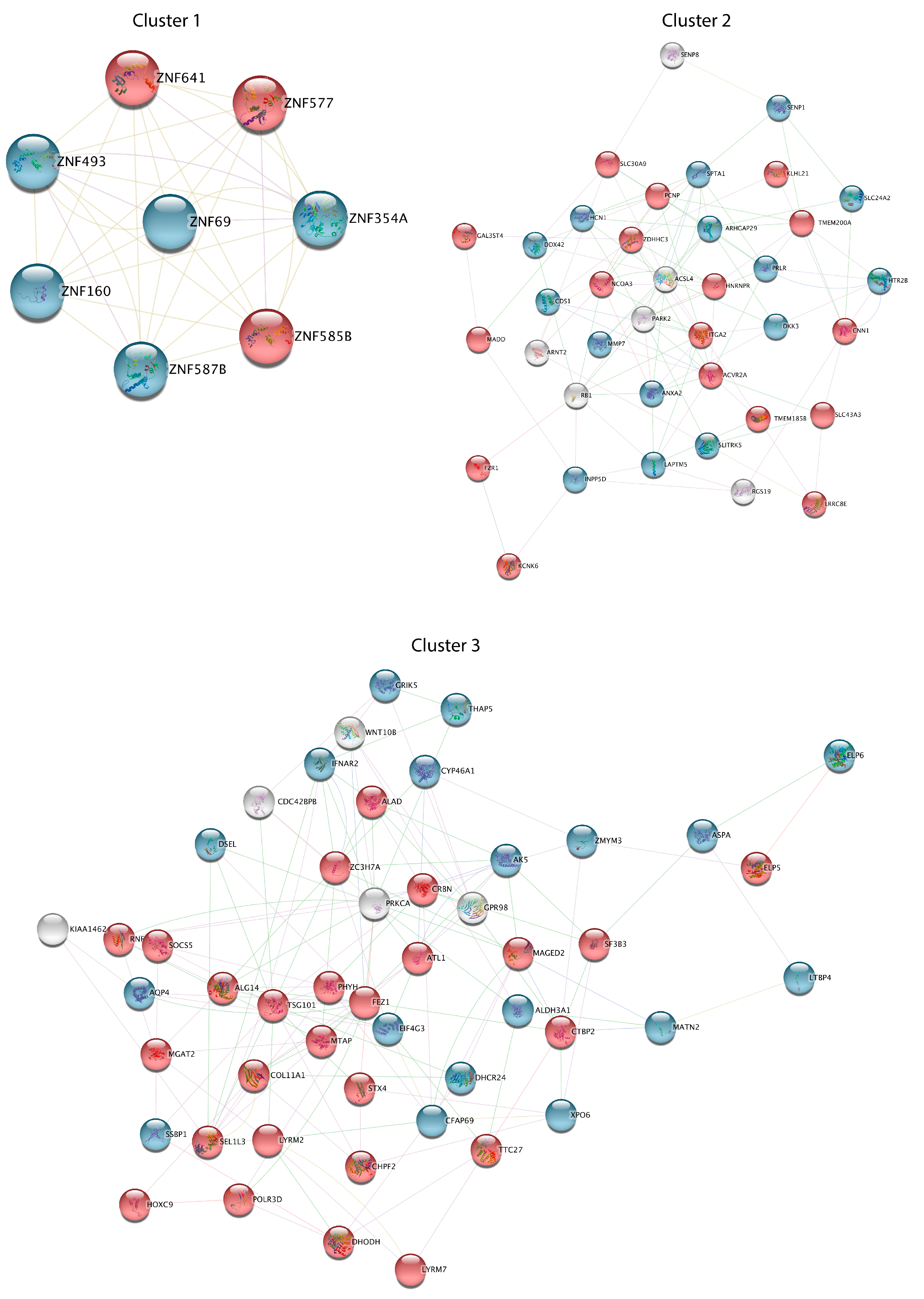
3.3. PPI Network Analysis Reveals Important Hub Proteins and Sub-Network Modules
3.4. Class Prediction Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Brenner, D.; Freischmidt, A. Update on genetics of amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2022, 35, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Smukowski, S.N.; Maioli, H.; Latimer, C.S.; Bird, T.D.; Jayadev, S.; Valdmanis, P.N. Progress in Amyotrophic Lateral Sclerosis Gene Discovery. Neurol. Genet. 2022, 8, e669. [Google Scholar] [CrossRef]
- Moura, M.C.; Novaes, M.R.C.G.; Eduardo, E.J.; Zago, Y.S.S.P.; Freitas, R.D.N.B.; Casulari, L.A. Prognostic Factors in Amyotrophic Lateral Sclerosis: A Population-Based Study. PLoS ONE 2015, 10, e0141500. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R. El escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Sub-committee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J. Neurol. Sci. 1994, 124, 96–107. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, M.; Dengler, R.; Eisen, A.; England, J.D.; Kaji, R.; Kimura, J.; Mills, K.; Mitsumoto, H.; Nodera, H.; Shefner, J.; et al. Electrodiagnostic criteria for diagnosis of ALS. Clin. Neurophysiol. 2008, 119, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.G.; Munsat, T.L.; Swash, M.; Brooks, B.R. Consensus guidelines for the design and implementation of clinical trials in ALS. J. Neurol. Sci. 1999, 169, 2–12. [Google Scholar] [CrossRef]
- Shefner, J.M.; Al-Chalabi, A.; Baker, M.R.; Cui, L.-Y.; de Carvalho, M.; Eisen, A.; Grosskreutz, J.; Hardiman, O.; Henderson, R.; Matamala, J.M.; et al. A proposal for new diagnostic criteria for ALS. Clin. Neurophysiol. 2020, 131, 1975–1978. [Google Scholar] [CrossRef]
- Kadena, K.; Vlamos, P. The Importance of Diagnostic and Prognostic Biomarker Identification and Classification towards Understanding ALS Pathogenesis; Springer International Publishing: Berlin/Heidelberg, Germany, 2021; pp. 119–120. [Google Scholar] [CrossRef]
- Ruffo, P.; Cavallaro, S.; Conforti, F.L. The Advent of Omics Sciences in Clinical Trials of Motor Neuron Diseases. J. Pers. Med. 2022, 12, 758. [Google Scholar] [CrossRef]
- Mitropoulos, K.; Katsila, T.; Patrinos, G.P.; Pampalakis, G. Multi-Omics for Biomarker Discovery and Target Validation in Biofluids for Amyotrophic Lateral Sclerosis Diagnosis. OMICS A J. Integr. Biol. 2018, 22, 52–64. [Google Scholar] [CrossRef]
- Hedl, T.J.; Gil, R.S.; Cheng, F.; Rayner, S.L.; Davidson, J.M.; De Luca, A.; Villalva, M.D.; Ecroyd, H.; Walker, A.K.; Lee, A. Proteomics Approaches for Biomarker and Drug Target Discovery in ALS and FTD. Front. Neurosci. 2019, 13, 548. [Google Scholar] [CrossRef] [Green Version]
- Mazón, M.; Costa, J.F.V.; Ten-Esteve, A.; Martí-Bonmatí, L. Imaging Biomarkers for the Diagnosis and Prognosis of Neurodegenerative Diseases. The Example of Amyotrophic Lateral Sclerosis. Front. Neurosci. 2018, 12, 784. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.G.; Oeckl, P.; Feneberg, E.; Bowser, R.; Otto, M.; Fischer, R.; Kessler, B.; Turner, M.R. Advancing mechanistic understanding and biomarker development in amyotrophic lateral sclerosis. Expert Rev. Proteom. 2021, 18, 977–994. [Google Scholar] [CrossRef]
- Swindell, W.R.; Kruse, C.P.S.; List, E.O.; Berryman, D.E.; Kopchick, J.J. ALS blood expression profiling identifies new biomarkers, patient subgroups, and evidence for neutrophilia and hypoxia. J. Transl. Med. 2019, 17, 170. [Google Scholar] [CrossRef] [Green Version]
- van Rheenen, W.; Diekstra, F.P.; Harschnitz, O.; Westeneng, H.-J.; van Eijk, K.R.; Saris, C.G.J.; Groen, E.J.N.; van Es, M.A.; Blauw, H.M.; van Vught, P.W.J.; et al. Whole blood transcriptome analysis in amyotrophic lateral sclerosis: A biomarker study. PLoS ONE 2018, 13, e0198874. [Google Scholar] [CrossRef] [Green Version]
- Tam, O.H.; Rozhkov, N.V.; Shaw, R.; Kim, D.; Hubbard, I.; Fennessey, S.; Propp, N.; Fagegaltier, D.; Harris, B.T.; Ostrow, L.W.; et al. Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia. Cell Rep. 2019, 29, 1164–1177.e5. [Google Scholar] [CrossRef] [Green Version]
- Baxi, E.G.; Thompson, T.; Li, J.; Kaye, J.A.; Lim, R.G.; Wu, J.; Ramamoorthy, D.; Lima, L.; Vaibhav, V.; Matlock, A.; et al. Answer ALS, a large-scale resource for sporadic and familial ALS combining clinical and multi-omics data from induced pluripotent cell lines. Nat. Neurosci. 2022, 25, 226–237. [Google Scholar] [CrossRef] [PubMed]
- D’Erchia, A.M.; Gallo, A.; Manzari, C.; Raho, S.; Horner, D.S.; Chiara, M.; Valletti, A.; Aiello, I.; Mastropasqua, F.; Ciaccia, L.; et al. Massive transcriptome sequencing of human spinal cord tissues provides new insights into motor neuron degeneration in ALS. Sci. Rep. 2017, 7, 10046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morello, G.; Salomone, S.; D’agata, V.; Conforti, F.L.; Cavallaro, S. From Multi-Omics Approaches to Precision Medicine in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2020, 14, 577755. [Google Scholar] [CrossRef]
- Alharbi, F.; Vakanski, A. Machine Learning Methods for Cancer Classification Using Gene Expression Data: A Review. Bioengineering 2023, 10, 173. [Google Scholar] [CrossRef]
- Tran, K.A.; Kondrashova, O.; Bradley, A.; Williams, E.D.; Pearson, J.V.; Waddell, N. Deep learning in cancer diagnosis, prognosis and treatment selection. Genome Med. 2021, 13, 152. [Google Scholar] [CrossRef]
- Guo, D.; Zhang, S.; Tang, Z.; Wang, H. Construction of gene-classifier and co-expression network analysis of genes in association with major depressive disorder. Psychiatry Res. 2020, 293, 113387. [Google Scholar] [CrossRef] [PubMed]
- Falchetti, M.; Prediger, R.D.; Zanotto-Filho, A. Classification algorithms applied to blood-based transcriptome meta-analysis to predict idiopathic Parkinson’s disease. Comput. Biol. Med. 2020, 124, 103925. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, X.; Lin, J.; Li, S.; Deng, G.; Wei, J. Classifiers for Predicting Coronary Artery Disease Based on Gene Expression Profiles in Peripheral Blood Mononuclear Cells. Int. J. Gen. Med. 2021, 14, 5651–5663. [Google Scholar] [CrossRef] [PubMed]
- Caballé-Cervigón, N.; Castillo-Sequera, J.L.; Gómez-Pulido, J.A.; Gómez-Pulido, J.M.; Polo-Luque, M.L. Machine Learning Applied to Diagnosis of Human Diseases: A Systematic Review. Appl. Sci. 2020, 10, 5135. [Google Scholar] [CrossRef]
- Ashton, J.J.; Young, A.; Johnson, M.J.; Beattie, R.M. Using machine learning to impact on long-term clinical care: Principles, challenges, and practicalities. Pediatr. Res. 2022, 93, 324–333. [Google Scholar] [CrossRef]
- Perez, G.A.; Castillo, R. Identification of Systemic Sclerosis through Machine Learning Algorithms and Gene Expression. Mathematics 2022, 10, 4632. [Google Scholar] [CrossRef]
- Wagh, V.V.; Agrawal, S.; Purohit, S.; Pachpor, T.; Narlikar, L.; Paralikar, V.; Khare, S. Ensemble of machine learning based prediction methods for diagnosis of schizophrenia using peripheral blood gene expression profile. medRxiv 2023. [Google Scholar] [CrossRef]
- Apolloni, S.; Fabbrizio, P.; Amadio, S.; Napoli, G.; Verdile, V.; Morello, G.; Iemmolo, R.; Aronica, E.; Cavallaro, S.; Volonté, C. Histamine Regulates the Inflammatory Profile of SOD1-G93A Microglia and the Histaminergic System Is Dysregulated in Amyotrophic Lateral Sclerosis. Front. Immunol. 2017, 8, 1689. [Google Scholar] [CrossRef] [Green Version]
- Morello, G.; Guarnaccia, M.; Spampinato, A.G.; La Cognata, V.; D’agata, V.; Cavallaro, S. Copy Number Variations in Amyotrophic Lateral Sclerosis: Piecing the Mosaic Tiles Together through a Systems Biology Approach. Mol. Neurobiol. 2017, 55, 1299–1322. [Google Scholar] [CrossRef] [Green Version]
- Morello, G.; Spampinato, A.G.; Conforti, F.L.; D’agata, V.; Cavallaro, S. Selection and Prioritization of Candidate Drug Targets for Amyotrophic Lateral Sclerosis Through a Meta-Analysis Approach. J. Mol. Neurosci. 2017, 61, 563–580. [Google Scholar] [CrossRef] [Green Version]
- Morello, G.; Spampinato, A.G.; Cavallaro, S. Molecular Taxonomy of Sporadic Amyotrophic Lateral Sclerosis Using Disease-Associated Genes. Front. Neurol. 2017, 8, 152. [Google Scholar] [CrossRef] [Green Version]
- Morello, G.; Spampinato, A.G.; Cavallaro, S. Neuroinflammation and ALS: Transcriptomic Insights into Molecular Disease Mechanisms and Therapeutic Targets. Mediat. Inflamm. 2017, 2017, 7070469. [Google Scholar] [CrossRef] [Green Version]
- Morello, G.; Cavallaro, S. Transcriptional analysis reveals distinct subtypes in amyotrophic lateral sclerosis: Implications for personalized therapy. Futur. Med. Chem. 2015, 7, 1335–1359. [Google Scholar] [CrossRef] [Green Version]
- Gentile, G.; Morello, G.; La Cognata, V.; Guarnaccia, M.; Conforti, F.L.; Cavallaro, S. Dysregulated miRNAs as Biomarkers and Therapeutical Targets in Neurodegenerative Diseases. J. Pers. Med. 2022, 12, 770. [Google Scholar] [CrossRef]
- La Cognata, V.; Golini, E.; Iemmolo, R.; Balletta, S.; Morello, G.; De Rosa, C.; Villari, A.; Marinelli, S.; Vacca, V.; Bonaventura, G.; et al. CXCR2 increases in ALS cortical neurons and its inhibition prevents motor neuron degeneration in vitro and improves neuromuscular function in SOD1G93A mice. Neurobiol. Dis. 2021, 160, 105538. [Google Scholar] [CrossRef] [PubMed]
- La Cognata, V.; Morello, G.; Cavallaro, S. Omics Data and Their Integrative Analysis to Support Stratified Medicine in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 4820. [Google Scholar] [CrossRef] [PubMed]
- Volonté, C.; Morello, G.; Spampinato, A.G.; Amadio, S.; Apolloni, S.; D’agata, V.; Cavallaro, S. Omics-based exploration and functional validation of neurotrophic factors and histamine as therapeutic targets in ALS. Ageing Res. Rev. 2020, 62, 101121. [Google Scholar] [CrossRef] [PubMed]
- La Cognata, V.; Gentile, G.; Aronica, E.; Cavallaro, S. Splicing Players Are Differently Expressed in Sporadic Amyotrophic Lateral Sclerosis Molecular Clusters and Brain Regions. Cells 2020, 9, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aronica, E.; Baas, F.; Iyer, A.; Asbroek, A.L.T.; Morello, G.; Cavallaro, S. Molecular classification of amyotrophic lateral sclerosis by unsupervised clustering of gene expression in motor cortex. Neurobiol. Dis. 2014, 74, 359–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apolloni, S.; Amadio, S.; Fabbrizio, P.; Morello, G.; Spampinato, A.G.; Latagliata, E.C.; Salvatori, I.; Proietti, D.; Ferri, A.; Madaro, L.; et al. Histaminergic transmission slows progression of amyotrophic lateral sclerosis. J. Cachex- Sarcopenia Muscle 2019, 10, 872–893. [Google Scholar] [CrossRef] [Green Version]
- Morello, G.; Spampinato, A.G.; Conforti, F.L.; Cavallaro, S. Taxonomy Meets Neurology, the Case of Amyotrophic Lateral Sclerosis. Front. Neurosci. 2018, 12, 673. [Google Scholar] [CrossRef] [PubMed]
- Morello, G.; Guarnaccia, M.; Spampinato, A.G.; Salomone, S.; D’agata, V.; Conforti, F.L.; Aronica, E.; Cavallaro, S. Integrative multi-omic analysis identifies new drivers and pathways in molecularly distinct subtypes of ALS. Sci. Rep. 2019, 9, 9968. [Google Scholar] [CrossRef] [Green Version]
- Ungaro, C.; Sprovieri, T.; Morello, G.; Perrone, B.; Spampinato, A.G.; Simone, I.L.; Trojsi, F.; Monsurrò, M.R.; Spataro, R.; La Bella, V.; et al. Genetic investigation of amyotrophic lateral sclerosis patients in south Italy: A two-decade analysis. Neurobiol. Aging 2020, 99, 99.e7–99.e14. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, C.J.; Dariolli, R.; Jorge, F.M.; Monteiro, M.R.; Maximino, J.R.; Martins, R.S.; Strauss, B.E.; Krieger, J.E.; Callegaro, D.; Chadi, G. Gene expression profiling for human iPS-derived motor neurons from sporadic ALS patients reveals a strong association between mitochondrial functions and neurodegeneration. Front. Cell. Neurosci. 2015, 9, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raman, R.; Allen, S.P.; Goodall, E.F.; Kramer, S.; Ponger, L.-L.; Heath, P.R.; Milo, M.; Hollinger, H.C.; Walsh, T.; Highley, J.R.; et al. Gene expression signatures in motor neurone disease fibroblasts reveal dysregulation of metabolism, hypoxia-response and RNA processing functions. Neuropathol. Appl. Neurobiol. 2015, 41, 201–226. [Google Scholar] [CrossRef] [Green Version]
- Lincecum, J.M.; Vieira, F.G.; Wang, M.Z.; Thompson, K.; De Zutter, G.S.; Kidd, J.; Moreno, A.; Sanchez, R.; Carrion, I.J.; A Levine, B.; et al. From transcriptome analysis to therapeutic anti-CD40L treatment in the SOD1 model of amyotrophic lateral sclerosis. Nat. Genet. 2010, 42, 392–399. [Google Scholar] [CrossRef]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef]
- Cedarbaum, J.M.; Stambler, N.; Malta, E.; Fuller, C.; Hilt, D.; Thurmond, B.; Nakanishi, A. The ALSFRS-R: A revised ALS functional rating scale that incorporates assessments of respiratory function. J. Neurol. Sci. 1999, 169, 13–21. [Google Scholar] [CrossRef]
- Kimura, F.; Fujimura, C.; Ishida, S.; Nakajima, H.; Furutama, D.; Uehara, H.; Shinoda, K.; Sugino, M.; Hanafusa, T. Progression rate of ALSFRS-R at time of diagnosis predicts survival time in ALS. Neurology 2006, 66, 265–267. [Google Scholar] [CrossRef]
- Bello, M.L.; Di Fini, F.; Notaro, A.; Spataro, R.; Conforti, F.L.; La Bella, V. ALS-Related Mutant FUS Protein Is Mislocalized to Cytoplasm and Is Recruited into Stress Granules of Fibroblasts from Asymptomatic FUS P525L Mutation Carriers. Neurodegener. Dis. 2017, 17, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Fetoni, A.R.; Zorzi, V.; Paciello, F.; Ziraldo, G.; Peres, C.; Raspa, M.; Scavizzi, F.; Salvatore, A.M.; Crispino, G.; Tognola, G.; et al. Cx26 partial loss causes accelerated presbycusis by redox imbalance and dysregulation of Nfr2 pathway. Redox Biol. 2018, 19, 301–317. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2020, 49, D605–D612. [Google Scholar] [CrossRef]
- Von Mering, C.; Huynen, M.; Jaeggi, D.; Schmidt, S.; Bork, P.; Snel, B. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 2003, 31, 258–261. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. S4), S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maere, S.; Heymans, K.; Kuiper, M. BiNGO: A Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics 2005, 21, 3448–3449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Won, Y.H.; Lee, M.-Y.; Choi, Y.-C.; Ha, Y.; Kim, H.; Kim, D.-Y.; Kim, M.-S.; Yu, J.H.; Seo, J.H.; Kim, M.; et al. Elucidation of Relevant Neuroinflammation Mechanisms Using Gene Expression Profiling in Patients with Amyotrophic Lateral Sclerosis. PLoS ONE 2016, 11, e0165290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Månberg, A.; Skene, N.; Sanders, F.; Trusohamn, M.; Remnestål, J.; Szczepińska, A.; Aksoylu, I.S.; Lönnerberg, P.; Ebarasi, L.; Wouters, S.; et al. Altered perivascular fibroblast activity precedes ALS disease onset. Nat. Med. 2021, 27, 640–646. [Google Scholar] [CrossRef]
- Konrad, C.; Kawamata, H.; Bredvik, K.G.; Arreguin, A.J.; Cajamarca, S.A.; Hupf, J.C.; Ravits, J.M.; Miller, T.M.; Maragakis, N.J.; Hales, C.M.; et al. Fibroblast bioenergetics to classify amyotrophic lateral sclerosis patients. Mol. Neurodegener. 2017, 12, 76. [Google Scholar] [CrossRef] [Green Version]
- A Fels, J.; Casalena, G.; Konrad, C.; E Holmes, H.; Dellinger, R.W.; Manfredi, G. Gene expression profiles in sporadic ALS fibroblasts define disease subtypes and the metabolic effects of the investigational drug EH301. Hum. Mol. Genet. 2022, 31, 3458–3477. [Google Scholar] [CrossRef]
- Kumbier, K.; Roth, M.; Li, Z.; Lazzari-Dean, J.; Waters, C.; Huang, P.; Korobeynikov, V.; York, N.; Consortium, G.C.; Phatnani, H.; et al. A scalable screening platform for phenotypic subtyping of ALS patient-derived fibroblasts. bioRxiv 2022. [Google Scholar] [CrossRef]
- van Blitterswijk, M.; Mullen, B.; Wojtas, A.; Heckman, M.G.; Diehl, N.N.; Baker, M.C.; DeJesus-Hernandez, M.; Brown, P.H.; E Murray, M.; Hsiung, G.-Y.R.; et al. Genetic modifiers in carriers of repeat expansions in the C9ORF72 gene. Mol. Neurodegener. 2014, 9, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamel, F.; Umbach, D.; Hu, H.; Munsat, T.; Shefner, J.; Taylor, J.; Sandler, D. Lead Exposure as a Risk Factor for Amyotrophic Lateral Sclerosis. Neurodegener. Dis. 2005, 2, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Kamel, F.; Umbach, D.M.; A Lehman, T.; Park, L.P.; Munsat, T.L.; Shefner, J.M.; Sandler, D.P.; Hu, H.; Taylor, J.A. Amyotrophic lateral sclerosis, lead, and genetic susceptibility: Polymorphisms in the delta-aminolevulinic acid dehydratase and vitamin D receptor genes. Environ. Health Perspect. 2003, 111, 1335–1339. [Google Scholar] [CrossRef] [PubMed]
- Calvo, A.C.; Manzano, R.; Atencia-Cibreiro, G.; Oliván, S.; Muñoz, M.J.; Zaragoza, P.; Cordero-Vázquez, P.; Esteban-Pérez, J.; García-Redondo, A.; Osta, R. Genetic Biomarkers for ALS Disease in Transgenic SOD1G93A Mice. PLoS ONE 2012, 7, e32632. [Google Scholar] [CrossRef] [Green Version]
- Léger, B.; Vergani, L.; Sorarù, G.; Hespel, P.; Derave, W.; Gobelet, C.; D’Ascenzio, C.; Angelini, C.; Russell, A.P. Human skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a reduction in Akt and an increase in atrogin-1. FASEB J. 2006, 20, 583–585. [Google Scholar] [CrossRef] [Green Version]
- Hao, Z.; Liu, L.; Tao, Z.; Wang, R.; Ren, H.; Sun, H.; Lin, Z.; Zhang, Z.; Mu, C.; Zhou, J.; et al. Motor dysfunction and neurodegeneration in a C9orf72 mouse line expressing poly-PR. Nat. Commun. 2019, 10, 2906. [Google Scholar] [CrossRef] [Green Version]
- Kaliszewska, A.; Allison, J.; Col, T.-T.; Shaw, C.; Arias, N. Elucidating the Role of Cerebellar Synaptic Dysfunction in C9orf72-ALS/FTD—A Systematic Review and Meta-Analysis. Cerebellum 2021, 21, 681–714. [Google Scholar] [CrossRef]
- Blauw, H.M.; Al-Chalabi, A.; Andersen, P.M.; van Vught, P.W.; Diekstra, F.P.; van Es, M.A.; Saris, C.G.; Groen, E.J.; van Rheenen, W.; Koppers, M.; et al. A large genome scan for rare CNVs in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2010, 19, 4091–4099. [Google Scholar] [CrossRef] [Green Version]
- Locke, D.P.; Sharp, A.J.; McCarroll, S.A.; McGrath, S.D.; Newman, T.L.; Cheng, Z.; Schwartz, S.; Albertson, D.G.; Pinkel, D.; Altshuler, D.M.; et al. Linkage Disequilibrium and Heritability of Copy-Number Polymorphisms within Duplicated Regions of the Human Genome. Am. J. Hum. Genet. 2006, 79, 275–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A van Es, M.; van Vught, P.W.; Blauw, H.M.; Franke, L.; Saris, C.G.; Bosch, L.V.D.; de Jong, S.W.; de Jong, V.; Baas, F.; Slot, R.V.; et al. Genetic variation in DPP6 is associated with susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 2007, 40, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Myszczynska, M.; Ferraiuolo, L. New In Vitro Models to Study Amyotrophic Lateral Sclerosis. Brain Pathol. 2016, 26, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Andrés-Benito, P.; Moreno, J.; Aso, E.; Povedano, M.; Ferrer, I. Amyotrophic lateral sclerosis, gene deregulation in the anterior horn of the spinal cord and frontal cortex area 8: Implications in frontotemporal lobar degeneration. Aging 2017, 9, 823–851. [Google Scholar] [CrossRef] [Green Version]
- Palomo, G.M.; Granatiero, V.; Kawamata, H.; Konrad, C.; Kim, M.; Arreguin, A.; Zhao, D.; A Milner, T.; Manfredi, G. Parkin is a disease modifier in the mutant SOD 1 mouse model of ALS. EMBO Mol. Med. 2018, 10, e8888. [Google Scholar] [CrossRef]
- Quinn, P.M.J.; Moreira, P.I.; Ambrósio, A.F.; Alves, C.H. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 189. [Google Scholar] [CrossRef]
- Butti, Z.; Patten, S.A. RNA Dysregulation in Amyotrophic Lateral Sclerosis. Front. Genet. 2019, 9, 712. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Li, M.; Ji, Y.; Zhu, J.; Chen, Z.; Zhang, L.; Deng, C.; Cheng, Q.; Wang, W.; Shen, Y.; et al. Identification of Regulatory Factors and Prognostic Markers in Amyotrophic Lateral Sclerosis. Antioxidants 2022, 11, 303. [Google Scholar] [CrossRef]
- Barmada, S.J. Linking RNA Dysfunction and Neurodegeneration in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 340–351. [Google Scholar] [CrossRef] [Green Version]
- Volkening, K.; Strong, M.J. Perturbed RNA Metabolism in Amyotrophic Lateral Sclerosis. Oxford University Press: Oxford, UK, 2012; pp. 353–368. [Google Scholar] [CrossRef]
- Laneve, P.; Tollis, P.; Caffarelli, E. RNA Deregulation in Amyotrophic Lateral Sclerosis: The Noncoding Perspective. Int. J. Mol. Sci. 2021, 22, 10285. [Google Scholar] [CrossRef]
- Anderson, D.M.; Cannavino, J.; Li, H.; Anderson, K.M.; Nelson, B.R.; McAnally, J.; Bezprozvannaya, S.; Liu, Y.; Lin, W.; Liu, N.; et al. Severe muscle wasting and denervation in mice lacking the RNA-binding protein ZFP106. Proc. Natl. Acad. Sci. USA 2016, 113, 201608423-503. [Google Scholar] [CrossRef] [PubMed]
- Joyce, P.I.; Fratta, P.; Landman, A.S.; Mcgoldrick, P.; Wackerhage, H.; Groves, M.; Busam, B.S.; Galino, J.; Corrochano, S.; Beskina, O.A.; et al. Deficiency of the zinc finger protein ZFP106 causes motor and sensory neurodegeneration. Hum. Mol. Genet. 2015, 25, 291–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, B.P.; Naumann, M.; Sterneckert, J.; Hermann, A. Genome Wide Analysis Points towards Subtype-Specific Diseases in Different Genetic Forms of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 6938. [Google Scholar] [CrossRef]
- Bu, S.; Lv, Y.; Liu, Y.; Qiao, S.; Wang, H. Zinc Finger Proteins in Neuro-Related Diseases Progression. Front. Neurosci. 2021, 15, 760567. [Google Scholar] [CrossRef]
- Rué, L.; Oeckl, P.; Timmers, M.; Lenaerts, A.; van der Vos, J.; Smolders, S.; Poppe, L.; de Boer, A.; Bosch, L.V.D.; Van Damme, P.; et al. Reduction of ephrin-A5 aggravates disease progression in amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2019, 7, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Pang, D.; Li, C.; Gu, X.; Chen, Y.; Ou, R.; Wei, Q.; Shang, H. The expression discrepancy and characteristics of long non-coding RNAs in peripheral blood leukocytes from amyotrophic lateral sclerosis patients. Mol. Neurobiol. 2022, 59, 3678–3689. [Google Scholar] [CrossRef]
- Gagliardi, S.; Zucca, S.; Pandini, C.; Diamanti, L.; Bordoni, M.; Sproviero, D.; Arigoni, M.; Olivero, M.; Pansarasa, O.; Ceroni, M.; et al. Long non-coding and coding RNAs characterization in Peripheral Blood Mononuclear Cells and Spinal Cord from Amyotrophic Lateral Sclerosis patients. Sci. Rep. 2018, 8, 2378. [Google Scholar] [CrossRef] [Green Version]
- Joilin, G.; Gray, E.; Thompson, A.G.; Talbot, K.; Leigh, P.N.; Newbury, S.F.; Turner, M.R.; Hafezparast, M. Profiling non-coding RNA expression in cerebrospinal fluid of amyotrophic lateral sclerosis patients. Ann. Med. 2022, 54, 3068–3077. [Google Scholar] [CrossRef]
- Joilin, G.; Gray, E.; Thompson, A.G.; Bobeva, Y.; Talbot, K.; Weishaupt, J.; Ludolph, A.; Malaspina, A.; Leigh, P.N.; Newbury, S.F.; et al. Identification of a potential non-coding RNA biomarker signature for amyotrophic lateral sclerosis. Brain Commun. 2020, 2, fcaa053. [Google Scholar] [CrossRef]
- Chen, K.-W.; Chen, J.-A. Functional Roles of Long Non-coding RNAs in Motor Neuron Development and Disease. J. Biomed. Sci. 2020, 27, 38. [Google Scholar] [CrossRef]
- Mamoor, S. Differential expression of PSMD5-AS1 in amyotrophic lateral sclerosis. OSF 2022. preprint. [Google Scholar] [CrossRef]
- Henderson, A.R.; Wang, Q.; Meechoovet, B.; Siniard, A.L.; Naymik, M.; De Both, M.; Huentelman, M.J.; Caselli, R.J.; Driver-Dunckley, E.; Dunckley, T. DNA Methylation and Expression Profiles of Whole Blood in Parkinson’s Disease. Front. Genet. 2021, 12, 640266. [Google Scholar] [CrossRef]
- Tan, X.; Liu, Y.; Zhang, T.; Cong, S. Integrated analysis of differentially expressed genes and construction of a competing endogenous RNA network in human Huntington neural progenitor cells. BMC Med. Genom. 2021, 14, 48. [Google Scholar] [CrossRef]
- Burk, K.; Pasterkamp, R.J. Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. 2019, 137, 859–877. [Google Scholar] [CrossRef] [Green Version]
- Jovičić, A.; Paul, J.W.; Gitler, A.D. Nuclear transport dysfunction: A common theme in amyotrophic lateral sclerosis and frontotemporal dementia. J. Neurochem. 2016, 138, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefera, T.W.; Steyn, F.J.; Ngo, S.T.; Borges, K. CNS glucose metabolism in Amyotrophic Lateral Sclerosis: A therapeutic target? Cell Biosci. 2021, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Tefera, T.W.; Borges, K. Metabolic Dysfunctions in Amyotrophic Lateral Sclerosis Pathogenesis and Potential Metabolic Treatments. Front. Neurosci. 2017, 10, 611. [Google Scholar] [CrossRef] [Green Version]
- MacLean, M.; López-Díez, R.; Vasquez, C.; Gugger, P.F.; Schmidt, A.M. Neuronal–glial communication perturbations in murine SOD1G93A spinal cord. Commun. Biol. 2022, 5, 177. [Google Scholar] [CrossRef]
- Rabin, S.J.; Kim, J.M.H.; Baughn, M.; Libby, R.T.; Kim, Y.J.; Fan, Y.; Libby, R.T.; La Spada, A.; Stone, B.; Ravits, J. Sporadic ALS has compartment-specific aberrant exon splicing and altered cell–matrix adhesion biology. Hum. Mol. Genet. 2010, 19, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Brockington, A.; Heath, P.R.; Holden, H.; Kasher, P.; Bender, F.L.; Claes, F.; Lambrechts, D.; Sendtner, M.; Carmeliet, P.; Shaw, P.J. Downregulation of genes with a function in axon outgrowth and synapse formation in motor neurones of the VEGFδ/δ mouse model of amyotrophic lateral sclerosis. BMC Genom. 2010, 11, 203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laszlo, Z.I.; Hindley, N.; Avila, A.S.; Kline, R.A.; Eaton, S.L.; Lamont, D.J.; Smith, C.; Spires-Jones, T.L.; Wishart, T.M.; Henstridge, C.M. Synaptic proteomics reveal distinct molecular signatures of cognitive change and C9ORF72 repeat expansion in the human ALS cortex. Acta Neuropathol. Commun. 2022, 10, 156. [Google Scholar] [CrossRef] [PubMed]
- Bendotti, C.; Marino, M.; Cheroni, C.; Fontana, E.; Crippa, V.; Poletti, A.; De Biasi, S. Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: Implication for protein aggregation and immune response. Prog. Neurobiol. 2012, 97, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Blokhuis, A.M.; Groen, E.J.N.; Koppers, M.; Berg, L.H.v.D.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yerbury, J.; Chisholm, C.; Lum, J.; Farrawell, N. Ubiquitin homeostasis disruption, a common cause of proteostasis collapse in amyotrophic lateral sclerosis? Neural Regen. Res. 2022, 17, 2218–2220. [Google Scholar] [CrossRef]
- Kirk, S.E.; Tracey, T.J.; Steyn, F.; Ngo, S.T. Biomarkers of Metabolism in Amyotrophic Lateral Sclerosis. Front. Neurol. 2019, 10, 191. [Google Scholar] [CrossRef]
- Silbernagel, N.; Walecki, M.; Schäfer, M.K.-H.; Kessler, M.; Zobeiri, M.; Rinné, S.; Kiper, A.K.; Komadowski, M.A.; Vowinkel, K.S.; Wemhöner, K.; et al. The VAMP-associated protein VAPB is required for cardiac and neuronal pacemaker channel function. FASEB J. 2018, 32, 6159–6173. [Google Scholar] [CrossRef] [Green Version]
- Nolan, M.; Malleret, G.; Dudman, J.; Buhl, D.; Santoro, B.; Gibbs, E.; Vronskaya, S.; Buzsaki, G.; Siegelbaum, S.; Kandel, E. A Behavioral Role for Dendritic IntegrationHCN1 Channels Constrain Spatial Memory and Plasticity at Inputs to Distal Dendrites of CA1 Pyramidal Neurons. Cell 2004, 119, 719–732. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Venugopal, S.; Majid, S.; Ahn, I.S.; Diamante, G.; Hong, J.; Yang, X.; Chandler, S.H. Single-cell RNA-seq analysis of the brainstem of mutant SOD1 mice reveals perturbed cell types and pathways of amyotrophic lateral sclerosis. Neurobiol. Dis. 2020, 141, 104877. [Google Scholar] [CrossRef]
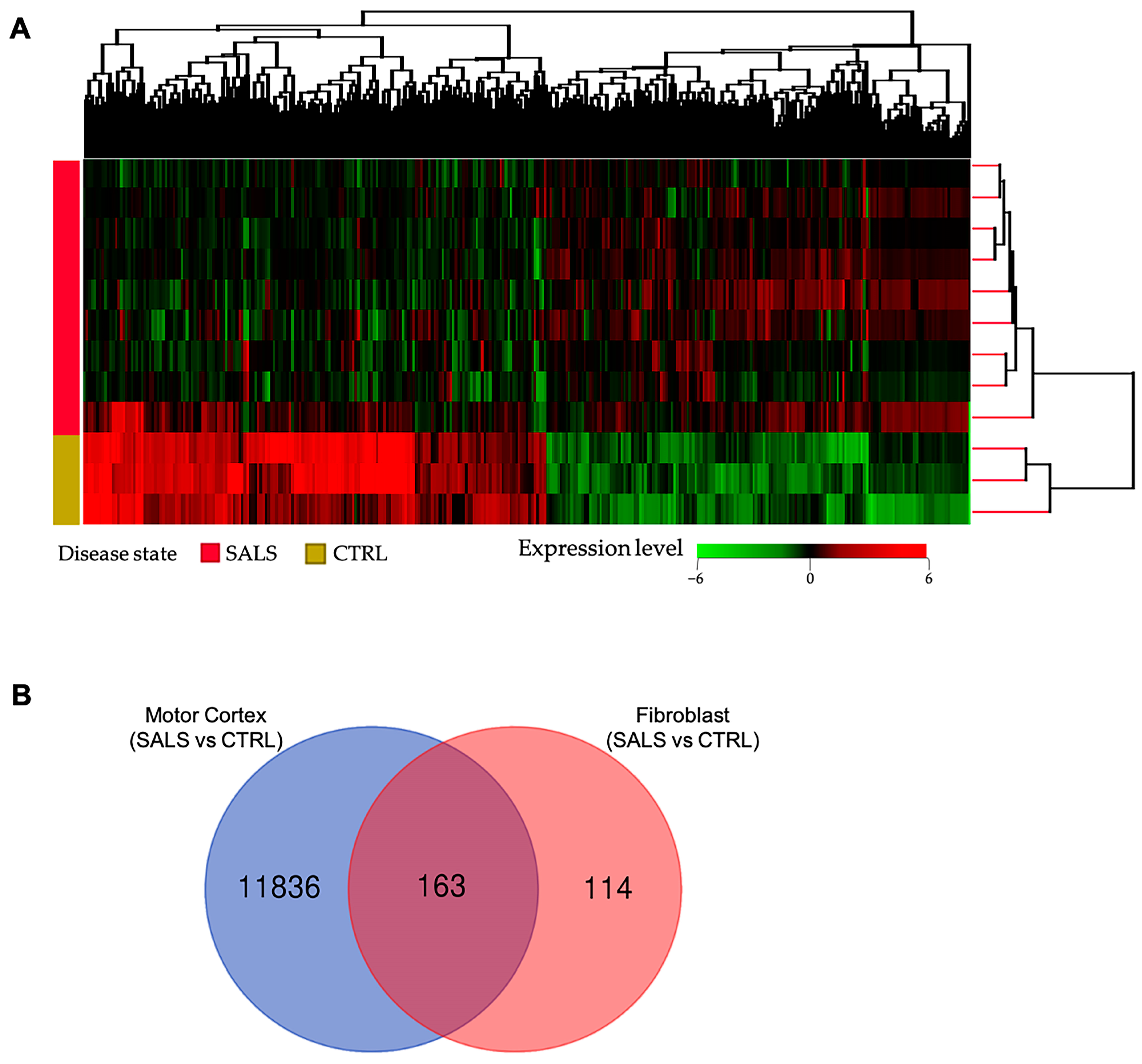
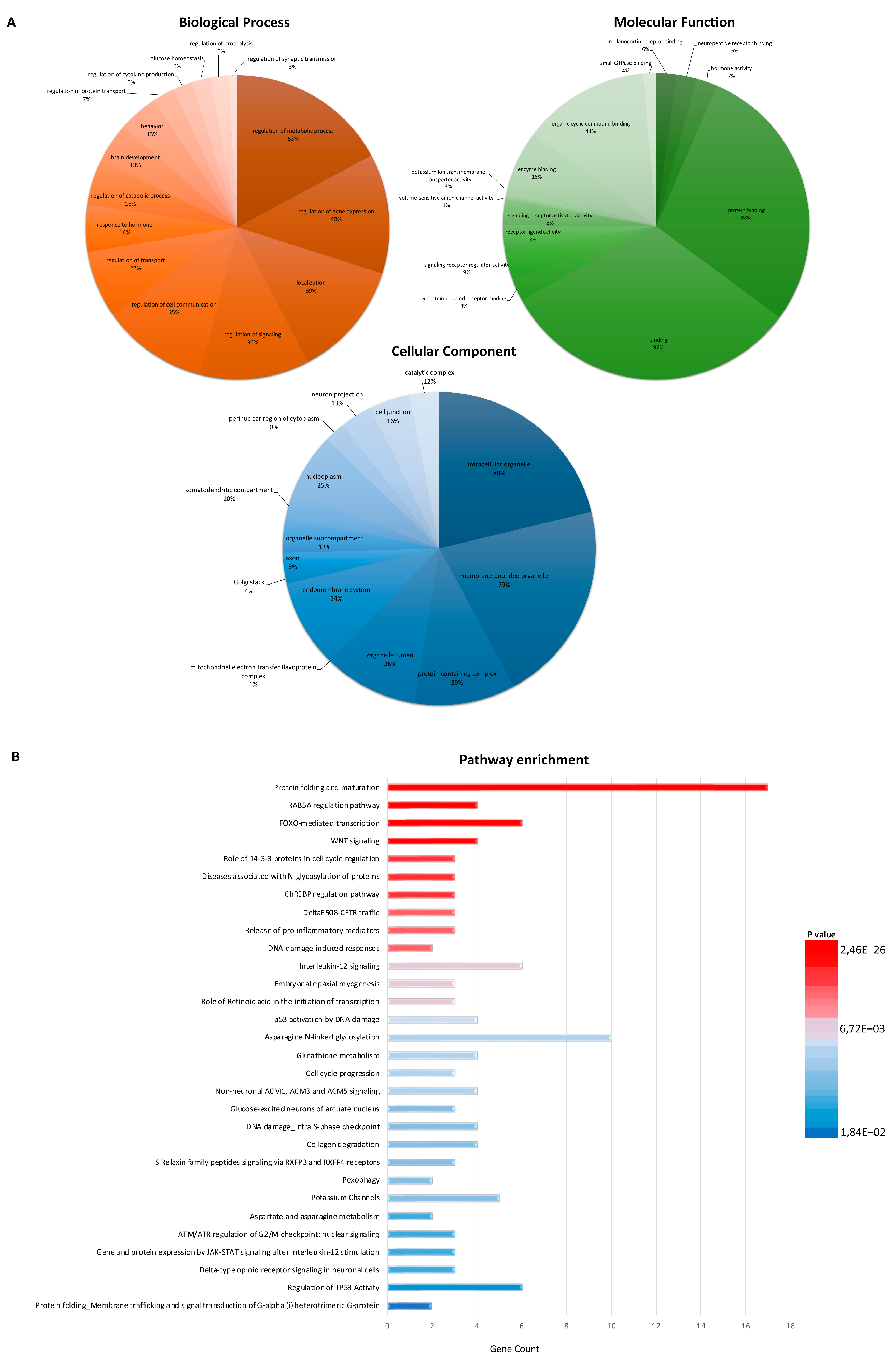
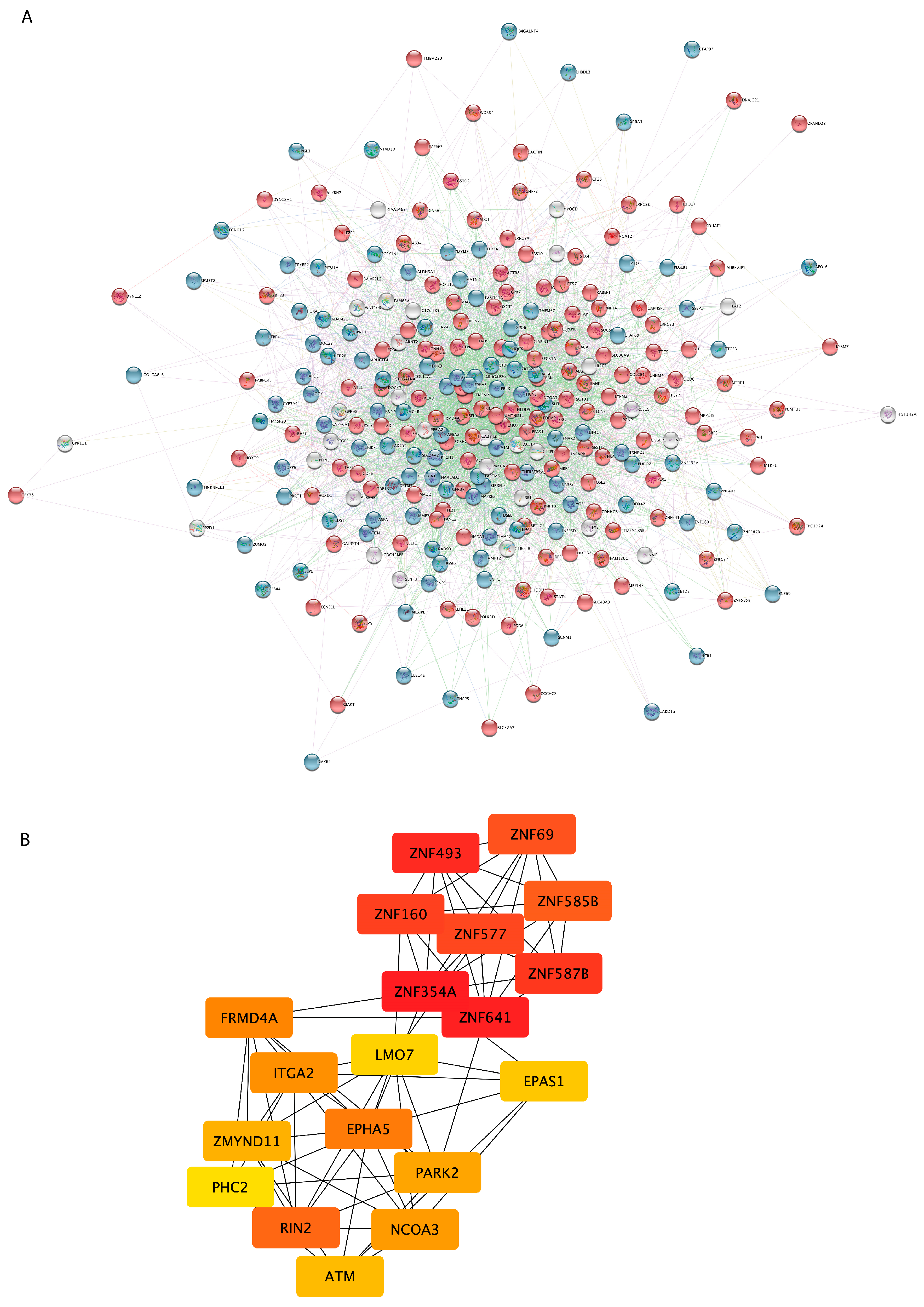


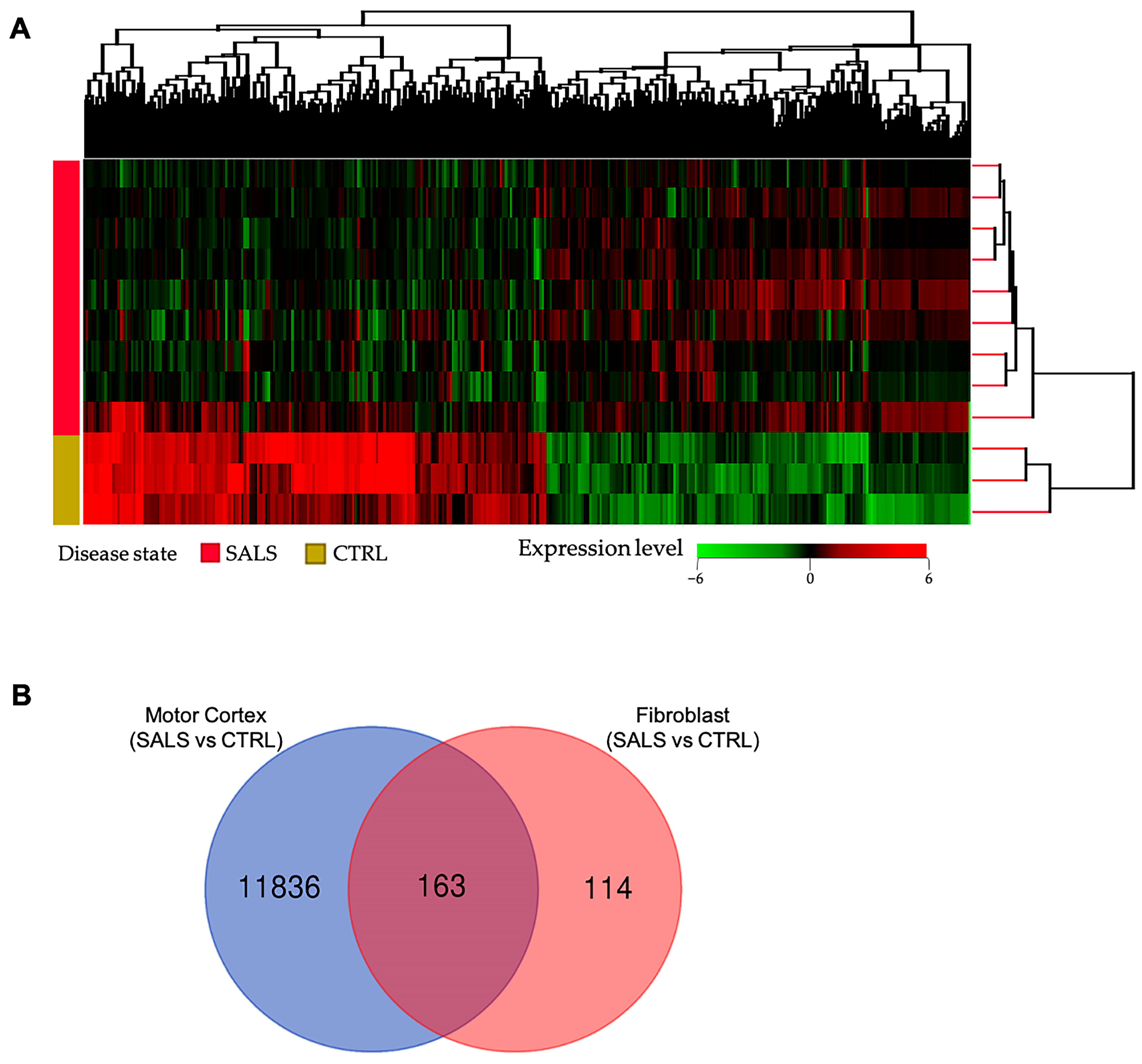{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | ALS | Healthy Controls | p |
|---|---|---|---|
| (n = 9) | (n = 3) | ||
| Age at onset | 63 (42–77) | N.A. | |
| Age at skin biopsy | 64 (47–79) | 64 (56–72) | 0.85 * |
| Sex (M/F) | 3/6 | 1/2 | 0.90 ** |
| ΔFS | 0.52 (0.39–1.11) | ||
| Site of onset (n,%) | |||
| Spinal | 6 (66.6) | ||
| Bulbar | 3 (33.4) |
| Accession Number | Repository | Platform | Sample Type | Number of Samples (ALS/Controls) | References |
|---|---|---|---|---|---|
| GSE56808 | GEO | Affymetrix Human Genome U133 Plus 2.0 Array | Fibroblasts | 12 (6/6) | [47] |
| GSE68240 | GEO | Agilent-028004 SurePrint G3 Human GE 8x60 K Microarray | Fibroblasts | 6 (3/3) | [46] |
| GSE112680 | GEO | Illumina HumanHT-12 V4.0 expression beadchip | Whole blood | 301 (164/137) | [15] |
| GSE112676 | GEO | Illumina HumanHT-12 V3.0 expression beadchip | Whole blood | 741 (233/508) | [14,15] |
| E-TABM-940 | ArrayExpress | Affymetrix GeneChip Human Genome U133 Plus 2.0 | Whole blood | 85 (57/28) | [48] |
| E-MTAB-2325 | ArrayExpress | Agilent-014850 Whole Human Genome Microarray 4x44 K | Motor cortex | 41 (31/10) | [40] |
| Training Set (GSE233881) | Test Set 1 (GSE56808) | Test Set 2 (GSE68240) | Test Set 3 (GSE112680) | Test Set 4 (GSE112676) | Test Set 5 (E-TABM-940) | Test Set 6 (E-MTAB-2325) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALS (n = 9) | CTRL (n = 3) | ALS (n = 6) | CTRL (n = 6) | ALS (n = 3) | CTRL (n = 3) | ALS (n = 164) | CTRL (n = 137) | ALS (n = 233) | CTRL (n = 508) | ALS (n = 57) | CTRL (n = 28) | ALS (n = 31) | CTRL (n = 10) | |
| Correct number of patients | 9 | 3 | 5 | 5 | 2 | 3 | 144 | 113 | 142 | 399 | 55 | 19 | 28 | 6 |
| Incorrect number of patients | 0 | 0 | 1 | 1 | 1 | 0 | 20 | 24 | 91 | 109 | 2 | 9 | 3 | 4 |
| Accuracy * | 100% | 83% | 83% | 85% | 73% | 87% | 83% | |||||||
| Sensitivity ** | 100% | 83% | 67% | 88% | 61% | 96% | 90% | |||||||
| Specificity *** | 100% | 83% | 100% | 82% | 78% | 68% | 60% | |||||||
| Training Set (GSE233881) | Test Set 1 (GSE56808) | Test Set 2 (GSE68240) | Test Set 3 (GSE112680) | Test Set 4 (GSE112676) | Test Set 5 (E-TABM-940) | Test Set 6 (E-MTAB-2325) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALS (n = 9) | CTRL (n = 3) | ALS (n = 6) | CTRL (n = 6) | ALS (n = 3) | CTRL (n = 3) | ALS (n = 164) | CTRL (n = 137) | ALS (n = 233) | CTRL (n = 508) | ALS (n = 57) | CTRL (n = 28) | ALS (n = 31) | CTRL (n = 10) | |
| Correct number of patients | 9 | 3 | 5 | 5 | 2 | 3 | 128 | 96 | 115 | 437 | 50 | 20 | 26 | 6 |
| Incorrect number of patients | 0 | 0 | 1 | 1 | 1 | 0 | 36 | 41 | 118 | 71 | 7 | 8 | 5 | 4 |
| Accuracy * | 100% | 83% | 83% | 75% | 75% | 83% | 83% | |||||||
| Sensitivity ** | 100% | 83% | 67% | 78% | 50% | 88% | 84% | |||||||
| Specificity *** | 100% | 83% | 100% | 70% | 86% | 72% | 60% | |||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morello, G.; La Cognata, V.; Guarnaccia, M.; La Bella, V.; Conforti, F.L.; Cavallaro, S. A Diagnostic Gene-Expression Signature in Fibroblasts of Amyotrophic Lateral Sclerosis. Cells 2023, 12, 1884. https://doi.org/10.3390/cells12141884
Morello G, La Cognata V, Guarnaccia M, La Bella V, Conforti FL, Cavallaro S. A Diagnostic Gene-Expression Signature in Fibroblasts of Amyotrophic Lateral Sclerosis. Cells. 2023; 12(14):1884. https://doi.org/10.3390/cells12141884
Chicago/Turabian StyleMorello, Giovanna, Valentina La Cognata, Maria Guarnaccia, Vincenzo La Bella, Francesca Luisa Conforti, and Sebastiano Cavallaro. 2023. "A Diagnostic Gene-Expression Signature in Fibroblasts of Amyotrophic Lateral Sclerosis" Cells 12, no. 14: 1884. https://doi.org/10.3390/cells12141884
APA StyleMorello, G., La Cognata, V., Guarnaccia, M., La Bella, V., Conforti, F. L., & Cavallaro, S. (2023). A Diagnostic Gene-Expression Signature in Fibroblasts of Amyotrophic Lateral Sclerosis. Cells, 12(14), 1884. https://doi.org/10.3390/cells12141884