
PM2.5-Induced Cardiac Structural Modifications and Declined Pro-Survival Signalling Pathways Are Responsible for the Inefficiency of GSK-3β Inhibitor in Attenuating Myocardial Ischemia-Reperfusion Injury in Rats


 , , ,
, , ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Animals
2.3. IR Induction and Hemodynamics Assessment
2.4. Experimental Protocol
2.5. Cardiac Injury Assessment
2.6. Western Blot
2.7. Mitochondrial Isolation, Bioenergetics, and Oxidative Stress Assessment
2.8. ELISA Assay
2.9. Statistical Analysis
3. Results
3.1. GSK3β Inhibitor Effectively Attenuated IR-Induced Cardiac Injury in Rat Hearts Exposed to PM2.5 for Shorter Duration
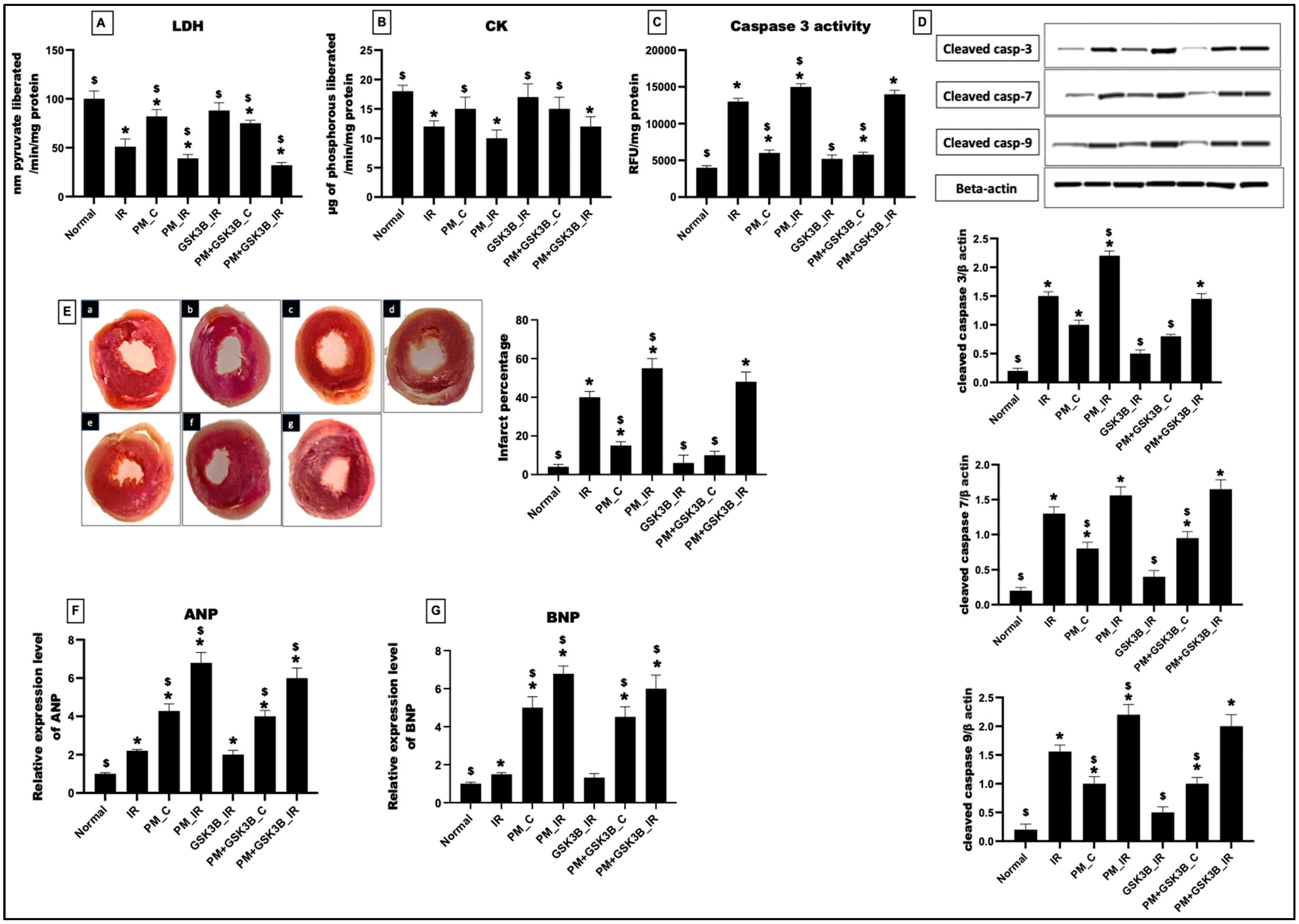
3.2. Long-Term Exposure to PM2.5 Abrogated the Potential of GSK3β Inhibitor to Ameliorate IR Injury in Rat Hearts
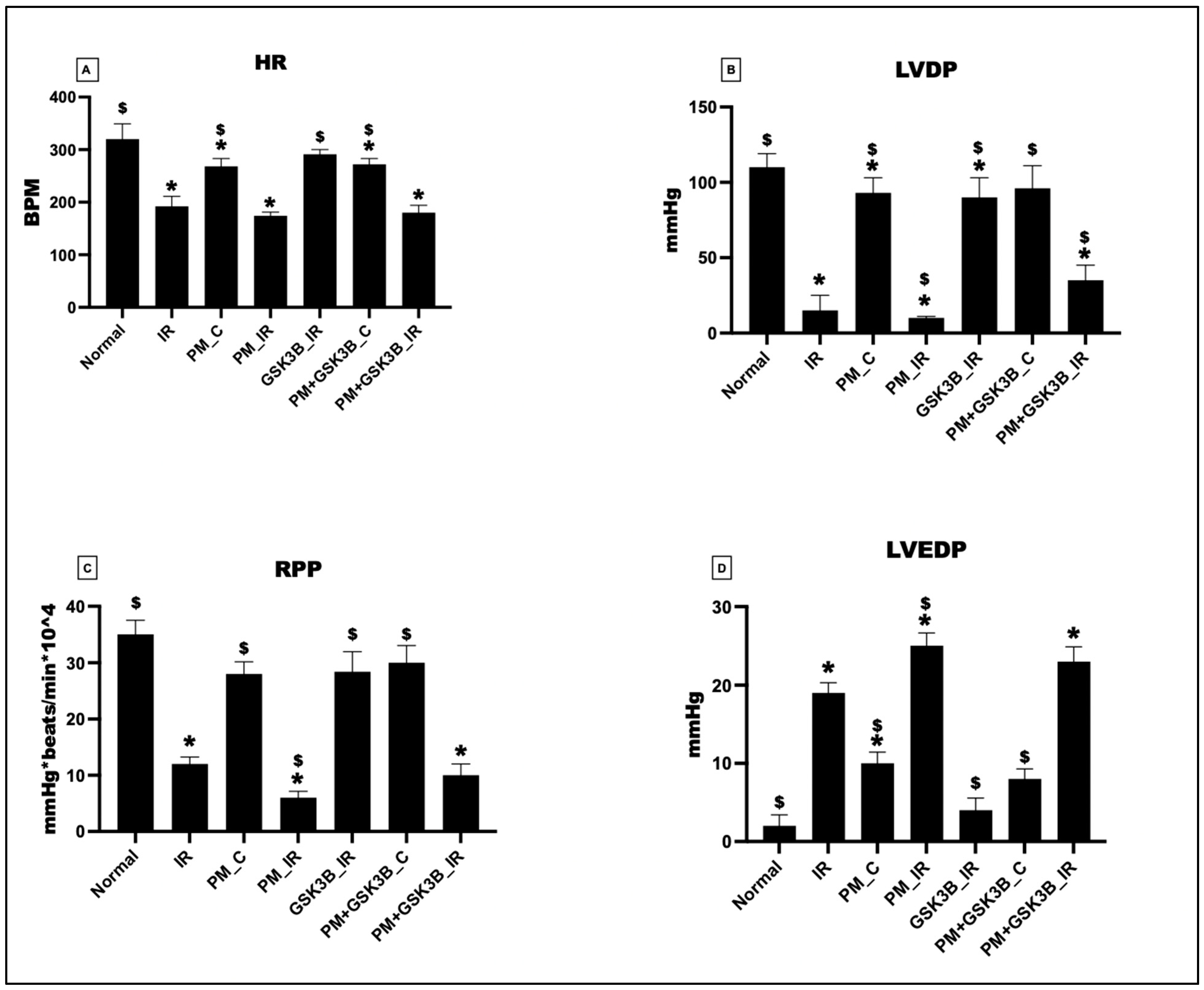
3.3. Long-Term Exposure to PM2.5 Abrogated the Potential of GSK3β Inhibitor to Ameliorate Cardiac Hemodynamics in Rat Hearts
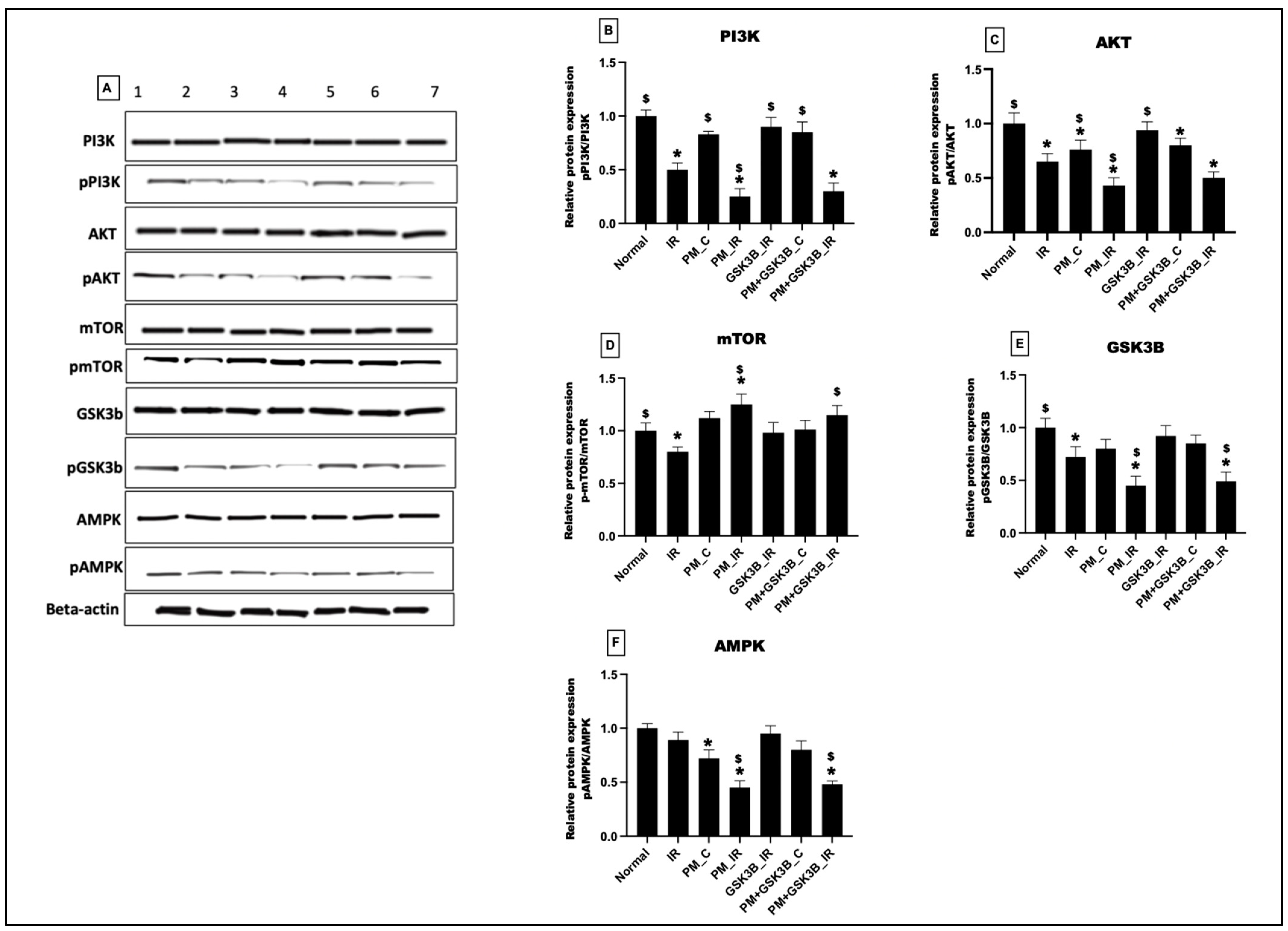
3.4. Long-Term Exposure to PM2.5 Inactivated the PI3K/AKT/mTOR/GSK3B/AMPK Signalling Axis in IR Rat Hearts Even in the Presence of GSK3β Inhibitor
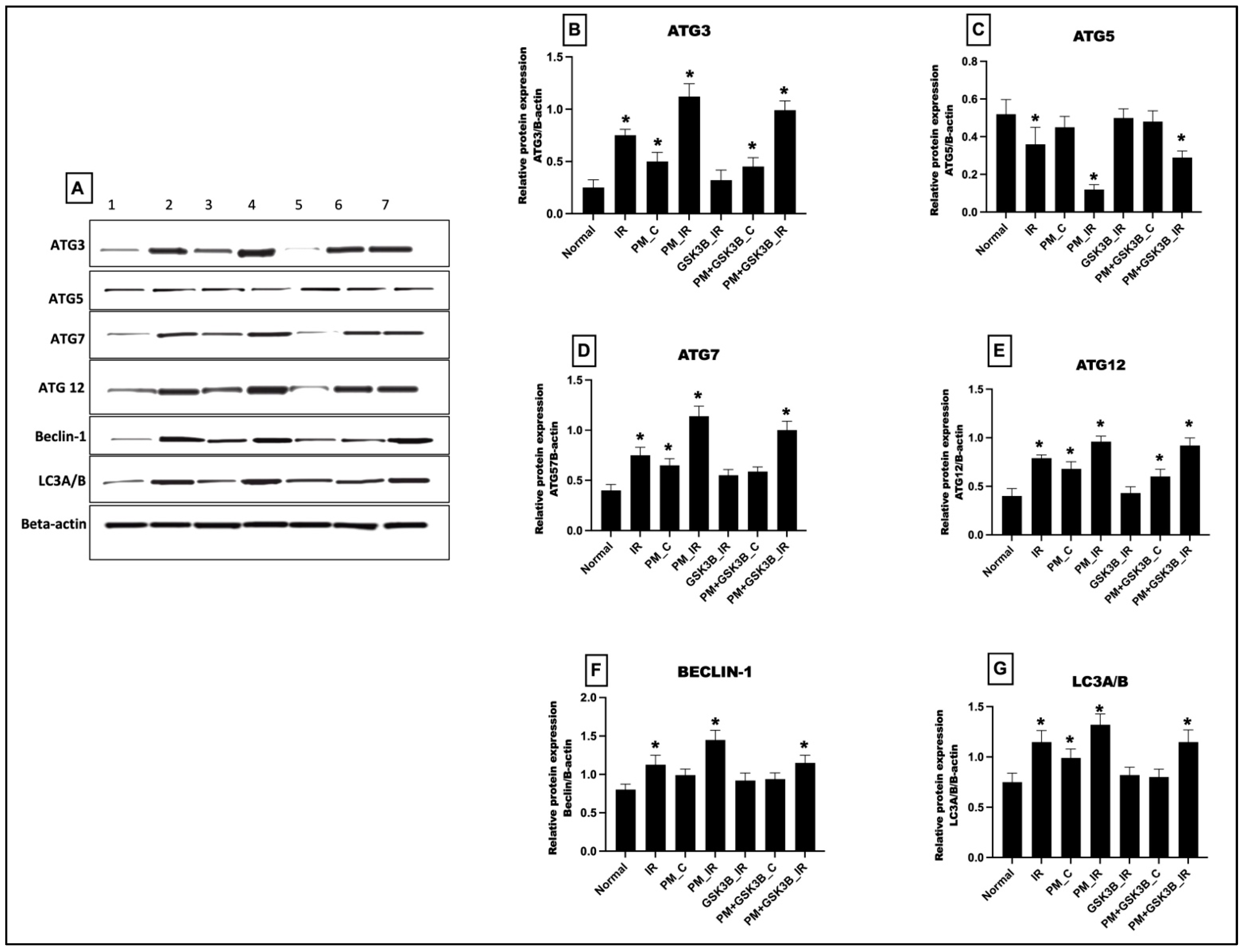
3.5. Long-Term Exposure to PM2.5 Activated the mTOR Dependent Autophagy in IR Rat Hearts Even in the Presence of GSK3β Inhibitor
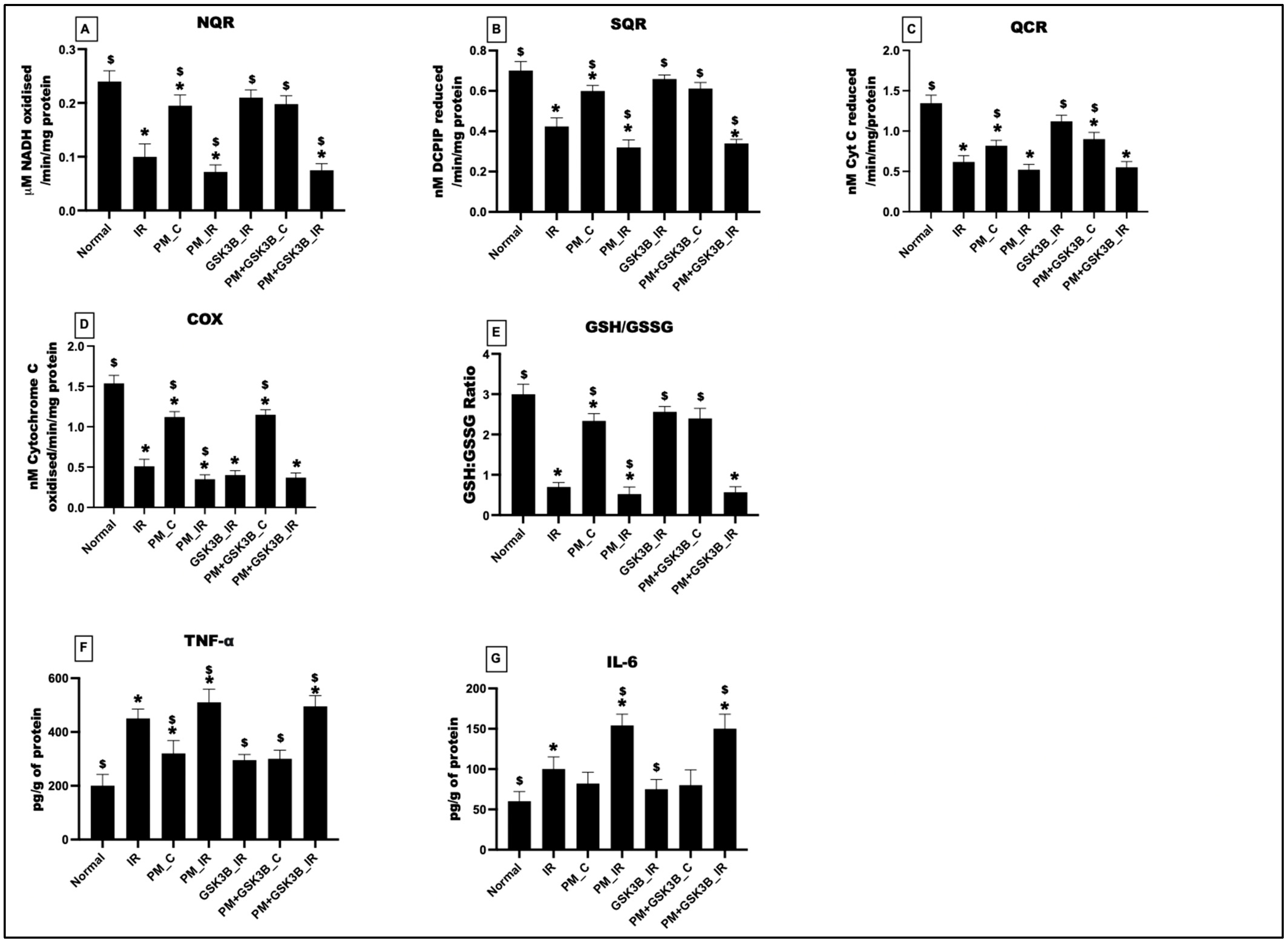
3.6. Long-Term Exposure to PM2.5 Induced Oxidative Stress, Mitochondrial Dysfunction and Inflammation in IR Rat Hearts Even in the Presence of GSK3β Inhibitor
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
References
- Basith, S.; Manavalan, B.; Shin, T.H.; Park, C.B.; Lee, W.-S.; Kim, J.; Lee, G. The Impact of Fine Particulate Matter 2.5 on the Cardiovascular System: A Review of the Invisible Killer. Nanomaterials 2022, 12, 2656. [Google Scholar] [CrossRef]
- Li, T.; Yu, Y.; Sun, Z.; Duan, J. A comprehensive understanding of ambient particulate matter and its components on the adverse health effects based from epidemiological and laboratory evidence. Part. Fibre Toxicol. 2022, 19, 67. [Google Scholar] [CrossRef]
- Liu, C.; Chan, K.H.; Lv, J.; Lam, H.; Newell, K.; Meng, X.; Liu, Y.; Chen, R.; Kartsonaki, C.; Wright, N.; et al. Long-Term Exposure to Ambient Fine Particulate Matter and Incidence of Major Cardiovascular Diseases: A Prospective Study of 0.5 Million Adults in China. Environ. Sci. Technol. 2022, 56, 13200–13211. [Google Scholar] [CrossRef]
- Li, T.; Zhang, Y.; Jiang, N.; Du, H.; Chen, C.; Wang, J.; Li, Q.; Feng, D.; Shi, X. Ambient fine particulate matter and cardiopulmonary health risks in China. Chin. Med. J. 2023, 136, 287–294. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, Z.; Hu, B.; Rangarajan, S.; Tse, L.A.; Wang, J.; Hu, L.; Wang, Y.; Xiang, Q.; Lin, Y.; et al. Associations of outdoor fine particulate air pollution and cardiovascular disease: Results from the Prospective Urban and Rural Epidemiology Study in China (PURE-China). Environ. Int. 2023, 174, 107829. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.; Evtyugina, M.; Vicente, E.; Vicente, A.; Rienda, I.C.; de la Campa, A.S.; Tomé, M.; Duarte, I. PM2.5 chemical composition and health risks by inhalation near a chemical complex. J. Environ. Sci. 2023, 124, 860–874. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Xu, X.; Chu, M.; Guo, Y.; Wang, J. Air particulate matter and cardiovascular disease: The epidemiological, biomedical and clinical evidence. J. Thorac. Dis. 2016, 8, E8–E19. [Google Scholar] [CrossRef]
- Pryor, J.T.; Cowley, L.O.; Simonds, S.E. The Physiological Effects of Air Pollution: Particulate Matter, Physiology and Disease. Front. Public Health 2022, 10, 882569. [Google Scholar] [CrossRef]
- Juhaszova, M.; Zorov, D.B.; Yaniv, Y.; Nuss, H.B.; Wang, S.; Sollott, S.J. Role of Glycogen synthase kinase-3β in cardioprotection. Circ. Res. 2009, 104, 1240–1252. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.M.; Bhave, S.R.; Ferraro, D.J.; Jaboin, J.J.; Hallahan, D.E.; Thotala, D. GSK-3β: A Bifunctional Role in Cell Death Pathways. Int. J. Cell Biol. 2012, 2012, 930710. [Google Scholar] [CrossRef]
- Phukan, S.; Babu, V.; Kannoji, A.; Hariharan, R.; Balaji, V. GSK3β: Role in therapeutic landscape and development of modulators. Br. J. Pharmacol. 2010, 160, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, J.; Di, L. Glycogen synthesis and beyond, a comprehensive review of GSK3 as a key regulator of metabolic pathways and a therapeutic target for treating metabolic diseases. Med. Res. Rev. 2021, 42, 946–982. [Google Scholar] [CrossRef]
- Rayasam, G.V.; Tulasi, V.K.; Sodhi, R.; Davis, J.A.; Ray, A. Glycogen synthase kinase 3: More than a namesake. Br. J. Pharmacol. 2009, 156, 885–898. [Google Scholar] [CrossRef]
- Sugden, P.H.; Fuller, S.J.; Weiss, S.C.; Clerk, A. Glycogen synthase kinase 3 (GSK3) in the heart: A point of integration in hypertrophic signalling and a therapeutic target? A critical analysis. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S137–S153. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, B.; Kurian, G.A. Diesel particulate matter exposure deteriorates cardiovascular health and increases the sensitivity of rat heart towards ischemia reperfusion injury via suppressing mitochondrial bioenergetics function. Chem. Interact. 2022, 351, 109769. [Google Scholar] [CrossRef]
- Sivakumar, B.; AlAsmari, A.F.; Ali, N.; Waseem, M.; Kurian, G.A. Consequential Impact of Particulate Matter Linked Inter-Fibrillar Mitochondrial Dysfunction in Rat Myocardium Subjected to Ischemia Reperfusion Injury. Biology 2022, 11, 1811. [Google Scholar] [CrossRef]
- Soares, R.O.S.; Losada, D.M.; Jordani, M.C.; Évora, P.; Castro-E-Silva, O. Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies. Int. J. Mol. Sci. 2019, 20, 5034. [Google Scholar] [CrossRef]
- Sivakumar, B.; Kurian, G.A. Inhalation of PM2.5 from diesel exhaust promote impairment of mitochondrial bioenergetics and dysregulate mitochondrial quality in rat heart: Implications in isoproterenol-induced myocardial infarction model. Inhal. Toxicol. 2022, 34, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, T.; Yang, P.C. Western blot: Technique, theory, and trouble shooting. N. Am. J. Med. Sci. 2012, 4, 429–434. [Google Scholar] [CrossRef]
- Barrientos, A.; Fontanesi, F.; Díaz, F. Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays. Curr. Protoc. Hum. Genet. 2009, 63, 19.3.1–19.3.14. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef]
- Lal, H.; Ahmad, F.; Woodgett, J.; Force, T. The GSK-3 family as therapeutic target for myocardial diseases. Circ. Res. 2015, 116, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Ramirez, A.; Waddell, D.S.; Li, Z.; Liu, X.; Wang, X.-F. Axin and GSK3-β control Smad3 protein stability and modulate TGF-β signaling. Genes Dev. 2008, 22, 106–120. [Google Scholar] [CrossRef]
- Badimon, L.; Casaní, L.; Camino-Lopez, S.; Juan-Babot, O.; Borrell-Pages, M. GSK3β inhibition and canonical Wnt signaling in mice hearts after myocardial ischemic damage. PLoS ONE 2019, 14, e0218098. [Google Scholar] [CrossRef]
- Perrelli, M.G.; Pagliaro, P.; Penna, C. Ischemia/reperfusion injury and cardioprotective mechanisms: Role of mitochondria and reactive oxygen species. World J. Cardiol. 2011, 3, 186–200. [Google Scholar] [CrossRef]
- Song, J.-Q.; Teng, X.; Cai, Y.; Tang, C.-S.; Qi, Y.-F. Activation of Akt/GSK-3β signaling pathway is involved in intermedin1–53 protection against myocardial apoptosis induced by ischemia/reperfusion. Apoptosis 2009, 14, 1299–1307. [Google Scholar] [CrossRef]
- Henriksen, E.J.; Dokken, B.B. Role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Curr. Drug Targets 2006, 7, 1435–1441. [Google Scholar] [CrossRef]
- Yadav, H.N.; Singh, M.; Sharma, P.L. Pharmacological inhibition of GSK-3β produces late phase of cardioprotection in hyperlipidemic rat: Possible involvement of HSP 72. Mol. Cell. Biochem. 2012, 369, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Chen, Z.; Gao, J.; Shi, W.; Li, L.; Jiang, S.; Hu, H.; Liu, Z.; Xu, D.; Wu, L. The Key Roles of GSK-3β in Regulating Mitochondrial Activity. Cell. Physiol. Biochem. 2017, 44, 1445–1459. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Clarke, S.J.; Javadov, S.A. Mitochondrial permeability transition pore opening during myocardial reperfusion—A target for cardioprotection. Cardiovasc. Res. 2004, 61, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Singh, A.P.; Tomar, D.; Rahmani, M.; Zhang, Q.; Woodgett, J.R.; Tilley, D.G.; Lal, H.; Force, T. Cardiomyocyte-GSK-3α promotes mPTP opening and heart failure in mice with chronic pressure overload. J. Mol. Cell. Cardiol. 2019, 130, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhao, T.; Feng, L.; Shi, Y.; Jiang, J.; Liang, S.; Sun, B.; Xu, Q.; Duan, J.; Sun, Z. PM2.5-induced ADRB2 hypermethylation contributed to cardiac dysfunction through cardiomyocytes apoptosis via PI3K/Akt pathway. Environ. Int. 2019, 127, 601–614. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhao, X.; Chen, J.; Aniagu, S.; Chen, T. PM2.5 induces cardiac malformations via PI3K/akt2/mTORC1 signaling pathway in zebrafish larvae. Environ. Pollut. 2023, 323, 121306. [Google Scholar] [CrossRef]
- Liu, Q.; Han, B.; Zhang, Y.; Jiang, T.; Ning, J.; Kang, A.; Huang, X.; Zhang, H.; Pang, Y.; Zhang, B.; et al. Potential molecular mechanism of cardiac hypertrophy in mice induced by exposure to ambient PM2.5. Ecotoxicol. Environ. Saf. 2021, 224, 112659. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Xia, W.; Liu, J.-C.; Yang, J.-Y.; Lee, D.-F.; Xia, J.; Bartholomeusz, G.; Li, Y.; Pan, Y.; Li, Z.; et al. Erk Associates with and Primes GSK-3β for Its Inactivation Resulting in Upregulation of β-Catenin. Mol. Cell 2005, 19, 159–170. [Google Scholar] [CrossRef]
- Chiarella, S.E.; Soberanes, S.; Urich, D.; Morales-Nebreda, L.; Nigdelioglu, R.; Green, D.; Young, J.B.; Gonzalez, A.; Rosario, C.; Misharin, A.V.; et al. β2-Adrenergic agonists augment air pollution–induced IL-6 release and thrombosis. J. Clin. Investig. 2014, 124, 2935–2946. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sivakumar, B.; Ali, N.; Ahmad, S.F.; Nadeem, A.; Waseem, M.; Kurian, G.A. PM2.5-Induced Cardiac Structural Modifications and Declined Pro-Survival Signalling Pathways Are Responsible for the Inefficiency of GSK-3β Inhibitor in Attenuating Myocardial Ischemia-Reperfusion Injury in Rats. Cells 2023, 12, 2064. https://doi.org/10.3390/cells12162064
Sivakumar B, Ali N, Ahmad SF, Nadeem A, Waseem M, Kurian GA. PM2.5-Induced Cardiac Structural Modifications and Declined Pro-Survival Signalling Pathways Are Responsible for the Inefficiency of GSK-3β Inhibitor in Attenuating Myocardial Ischemia-Reperfusion Injury in Rats. Cells. 2023; 12(16):2064. https://doi.org/10.3390/cells12162064
Chicago/Turabian StyleSivakumar, Bhavana, Nemat Ali, Sheikh F. Ahmad, Ahmed Nadeem, Mohammad Waseem, and Gino A. Kurian. 2023. "PM2.5-Induced Cardiac Structural Modifications and Declined Pro-Survival Signalling Pathways Are Responsible for the Inefficiency of GSK-3β Inhibitor in Attenuating Myocardial Ischemia-Reperfusion Injury in Rats" Cells 12, no. 16: 2064. https://doi.org/10.3390/cells12162064