
Tripartite Motif-Containing Protein 32 (TRIM32): What Does It Do for Skeletal Muscle?


Abstract
:1. Introduction
2. Skeletal Muscle
2.1. Regenerative Skeletal Muscle
2.2. Types of Skeletal Muscle Fibers
2.3. Contraction and Relaxation of Skeletal Muscle
3. Tripartite Motif-Containing Protein 32 (TRIM32)
3.1. TRIM Family
3.2. TRIM32 and Its Domains
3.3. Autoubiquitination of TRIM32
4. TRIM32-Binding Proteins in Skeletal Muscle
4.1. Actin
4.2. Desmin
4.3. c-Myc
4.4. Dysbindin
4.5. SERCA1a
5. TRIM32 in Skeletal Muscle
5.1. Various Expression Levels of TRIM32 in Skeletal Muscle
5.2. TRIM32 Plays a Positive Role in Terminal Differentiation
6. TRIM32 and Limb–Girdle Muscular Dystrophies (LGMDs)
6.1. LGMDs
6.2. LGMD2H/LGMDR8
6.3. LGMD2H-Causing Genetic Variants of TRIM32 in Humans and Disease Symptoms
7. Animal Models of LGMD2H
7.1. A TRIM32-Knockout Mouse Model
7.2. A TRIM32 Knock-In Mouse Model
7.3. Drosophila Models
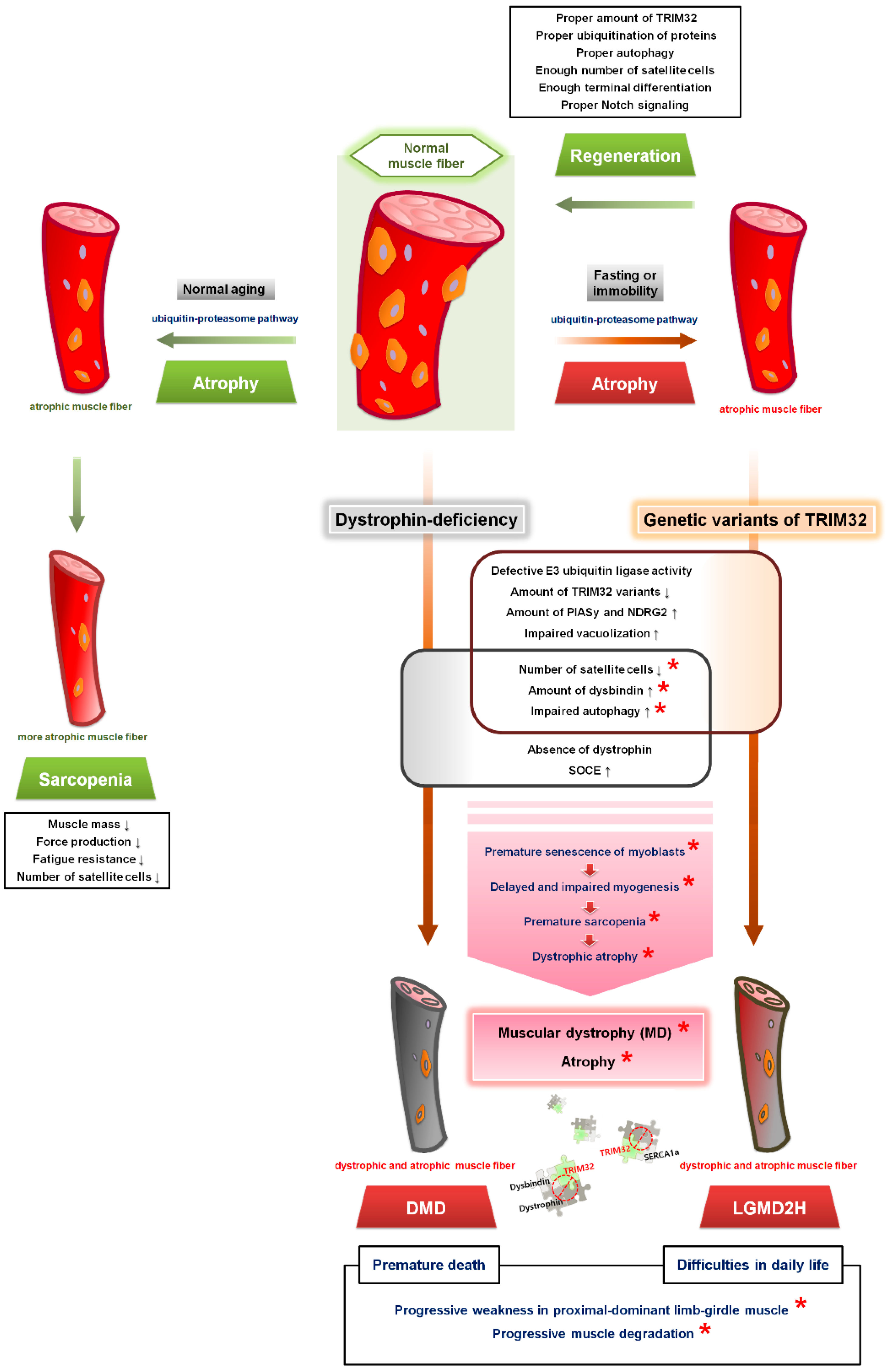
8. TRIM32 and Skeletal Muscle Atrophy
8.1. Skeletal Muscle Atrophy
8.2. Correlation of TRIM32 with Metabolic Proteins in Skeletal Muscle Atrophy
8.3. TRIM32-Mediated Autophagy in Skeletal Muscle Atrophy
8.4. Premature Senescence of Satellite Cells and Skeletal Muscle Atrophy in LGMD2H
9. TRIM32 and DMD
9.1. DMD
9.2. TRIM32 in DMD
10. TRIM32 in Non-Muscle Cells or Tissues or Other Organs
11. Points to Be Considered When Studying TRIM32 in Skeletal Muscle
12. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Frontera, W.R.; Ochala, J. Skeletal muscle: A brief review of structure and function. Calcif. Tissue Int. 2015, 96, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Fridell, R.A.; Harding, L.S.; Bogerd, H.P.; Cullen, B.R. Identification of a novel human zinc finger protein that specifically interacts with the activation domain of lentiviral Tat proteins. Virology 1995, 209, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Frosk, P.; Weiler, T.; Nylen, E.; Sudha, T.; Greenberg, C.R.; Morgan, K.; Fujiwara, T.M.; Wrogemann, K. Limb-girdle muscular dystrophy type 2H associated with mutation in TRIM32, a putative E3-ubiquitin-ligase gene. Am. J. Hum. Genet. 2002, 70, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Bawa, S.; Piccirillo, R.; Geisbrecht, E.R. TRIM32: A Multifunctional Protein Involved in Muscle Homeostasis, Glucose Metabolism, and Tumorigenesis. Biomolecules 2021, 11, 408. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, B.M.; Stanton, B.A. Berne & Levy Physiology, 7th ed.; Elsevier: Philadelphia, PA, USA, 2018; p. xii. 867p. [Google Scholar]
- Collins, C.A.; Olsen, I.; Zammit, P.S.; Heslop, L.; Petrie, A.; Partridge, T.A.; Morgan, J.E. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell 2005, 122, 289–301. [Google Scholar] [CrossRef]
- Yang, D.; Morris, S.F.; Sigurdson, L. The sartorius muscle: Anatomic considerations for reconstructive surgeons. Surg. Radiol. Anat. 1998, 20, 307–310. [Google Scholar] [CrossRef]
- Szent-Gyorgyi, A.G. The early history of the biochemistry of muscle contraction. J. Gen. Physiol. 2004, 123, 631–641. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef]
- Essen, B.; Jansson, E.; Henriksson, J.; Taylor, A.W.; Saltin, B. Metabolic characteristics of fibre types in human skeletal muscle. Acta Physiol. Scand. 1975, 95, 153–165. [Google Scholar] [CrossRef]
- Peter, J.B.; Barnard, R.J.; Edgerton, V.R.; Gillespie, C.A.; Stempel, K.E. Metabolic profiles of three fiber types of skeletal muscle in guinea pigs and rabbits. Biochemistry 1972, 11, 2627–2633. [Google Scholar] [CrossRef]
- Hargreaves, M.; Spriet, L.L. Skeletal muscle energy metabolism during exercise. Nat. Metab. 2020, 2, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Tarnopolsky, M.A. Metabolic Myopathies. Continuum 2016, 22, 1829–1851. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, D.T.; MacIntyre, R.; Fuda, N.; Fiori, J.; Barrilla, J.; Ramizel, L. Analysis of glycolytic enzyme co-localization in Drosophila flight muscle. J. Exp. Biol. 2003, 206, 2031–2038. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, W.; Gizak, A.; Rakus, D. Phosphoglycerate mutase in mammalian striated muscles: Subcellular localization and binding partners. FEBS Lett. 2009, 583, 1841–1845. [Google Scholar] [CrossRef] [PubMed]
- Foucault, G.; Vacher, M.; Merkulova, T.; Keller, A.; Arrio-Dupont, M. Presence of enolase in the M-band of skeletal muscle and possible indirect interaction with the cytosolic muscle isoform of creatine kinase. Biochem. J. 1999, 338 Pt 1, 115–121. [Google Scholar] [CrossRef]
- Calderon, J.C.; Bolanos, P.; Caputo, C. The excitation-contraction coupling mechanism in skeletal muscle. Biophys. Rev. 2014, 6, 133–160. [Google Scholar] [CrossRef]
- Woo, J.S.; Jeong, S.Y.; Park, J.H.; Choi, J.H.; Lee, E.H. Calsequestrin: A well-known but curious protein in skeletal muscle. Exp. Mol. Med. 2020, 52, 1908–1925. [Google Scholar] [CrossRef]
- Cho, C.H.; Woo, J.S.; Perez, C.F.; Lee, E.H. A focus on extracellular Ca2+ entry into skeletal muscle. Exp. Mol. Med. 2017, 49, e378. [Google Scholar] [CrossRef]
- Lee, E.H. Ca2+ channels and skeletal muscle diseases. Prog. Biophys. Mol. Biol. 2010, 103, 35–43. [Google Scholar] [CrossRef]
- Cho, C.H.; Lee, K.J.; Lee, E.H. With the greatest care, stromal interaction molecule (STIM) proteins verify what skeletal muscle is doing. BMB Rep. 2018, 51, 378–387. [Google Scholar] [CrossRef]
- Choi, J.H.; Jeong, S.Y.; Oh, M.R.; Allen, P.D.; Lee, E.H. TRPCs: Influential Mediators in Skeletal Muscle. Cells 2020, 9, 850. [Google Scholar] [CrossRef] [PubMed]
- Baylor, S.M.; Hollingworth, S. Sarcoplasmic reticulum calcium release compared in slow-twitch and fast-twitch fibres of mouse muscle. J. Physiol. 2003, 551, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Launikonis, B.S.; Murphy, R.M.; Edwards, J.N. Toward the roles of store-operated Ca2+ entry in skeletal muscle. Pflug. Arch. 2010, 460, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Tibbits, G.F.; Thomas, M.J. Ca2+ transport across the plasma membrane of striated muscle. Med. Sci. Sports Exerc. 1989, 21, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Pardo, J.V.; Siliciano, J.D.; Craig, S.W. A vinculin-containing cortical lattice in skeletal muscle: Transverse lattice elements (“costameres”) mark sites of attachment between myofibrils and sarcolemma. Proc. Natl. Acad. Sci. USA 1983, 80, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Craig, S.W.; Pardo, J.V. Gamma actin, spectrin, and intermediate filament proteins colocalize with vinculin at costameres, myofibril-to-sarcolemma attachment sites. Cell Motil. 1983, 3, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Peter, A.K.; Cheng, H.; Ross, R.S.; Knowlton, K.U.; Chen, J. The costamere bridges sarcomeres to the sarcolemma in striated muscle. Prog. Pediatr. Cardiol. 2011, 31, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Bloch, R.J.; Capetanaki, Y.; O’Neill, A.; Reed, P.; Williams, M.W.; Resneck, W.G.; Porter, N.C.; Ursitti, J.A. Costameres: Repeating structures at the sarcolemma of skeletal muscle. Clin. Orthop. Relat. Res. 2002, S203–S210. [Google Scholar] [CrossRef]
- Ervasti, J.M. Costameres: The Achilles’ heel of Herculean muscle. J. Biol. Chem. 2003, 278, 13591–13594. [Google Scholar] [CrossRef]
- Sardiello, M.; Cairo, S.; Fontanella, B.; Ballabio, A.; Meroni, G. Genomic analysis of the TRIM family reveals two groups of genes with distinct evolutionary properties. BMC Evol. Biol. 2008, 8, 225. [Google Scholar] [CrossRef]
- Overa, K.S.; Garcia-Garcia, J.; Bhujabal, Z.; Jain, A.; Overvatn, A.; Larsen, K.B.; Deretic, V.; Johansen, T.; Lamark, T.; Sjottem, E. TRIM32, but not its muscular dystrophy-associated mutant, positively regulates and is targeted to autophagic degradation by p62/SQSTM1. J. Cell Sci. 2019, 132, jcs236596. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Hatakeyama, S. TRIM proteins and diseases. J. Biochem. 2017, 161, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Tocchini, C.; Ciosk, R. TRIM-NHL proteins in development and disease. Semin. Cell Dev. Biol. 2015, 47–48, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, E.; Meroni, G. TRIM32 ubiquitin E3 ligase, one enzyme for several pathologies: From muscular dystrophy to tumours. Int. J. Biochem. Cell Biol. 2016, 79, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, L.M.; Jaffray, E.G.; Hay, R.T.; Meroni, G. Functional interactions between ubiquitin E2 enzymes and TRIM proteins. Biochem. J. 2011, 434, 309–319. [Google Scholar] [CrossRef]
- Kudryashova, E.; Wu, J.; Havton, L.A.; Spencer, M.J. Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum. Mol. Genet. 2009, 18, 1353–1367. [Google Scholar] [CrossRef] [PubMed]
- Borden, K.L.; Freemont, P.S. The RING finger domain: A recent example of a sequence-structure family. Curr. Opin. Struct. Biol. 1996, 6, 395–401. [Google Scholar] [CrossRef]
- Saurin, A.J.; Borden, K.L.; Boddy, M.N.; Freemont, P.S. Does this have a familiar RING? Trends Biochem. Sci. 1996, 21, 208–214. [Google Scholar] [CrossRef]
- Budhidarmo, R.; Nakatani, Y.; Day, C.L. RINGs hold the key to ubiquitin transfer. Trends Biochem. Sci. 2012, 37, 58–65. [Google Scholar] [CrossRef]
- Tocchini, C.; Keusch, J.J.; Miller, S.B.; Finger, S.; Gut, H.; Stadler, M.B.; Ciosk, R. The TRIM-NHL protein LIN-41 controls the onset of developmental plasticity in Caenorhabditis elegans. PLoS Genet. 2014, 10, e1004533. [Google Scholar] [CrossRef] [PubMed]
- Koliopoulos, M.G.; Esposito, D.; Christodoulou, E.; Taylor, I.A.; Rittinger, K. Functional role of TRIM E3 ligase oligomerization and regulation of catalytic activity. EMBO J. 2016, 35, 1204–1218. [Google Scholar] [CrossRef] [PubMed]
- Lazzari, E.; El-Halawany, M.S.; De March, M.; Valentino, F.; Cantatore, F.; Migliore, C.; Onesti, S.; Meroni, G. Analysis of the Zn-Binding Domains of TRIM32, the E3 Ubiquitin Ligase Mutated in Limb Girdle Muscular Dystrophy 2H. Cells 2019, 8, 254. [Google Scholar] [CrossRef] [PubMed]
- Reymond, A.; Meroni, G.; Fantozzi, A.; Merla, G.; Cairo, S.; Luzi, L.; Riganelli, D.; Zanaria, E.; Messali, S.; Cainarca, S.; et al. The tripartite motif family identifies cell compartments. EMBO J. 2001, 20, 2140–2151. [Google Scholar] [CrossRef] [PubMed]
- Massiah, M.A.; Simmons, B.N.; Short, K.M.; Cox, T.C. Solution structure of the RBCC/TRIM B-box1 domain of human MID1: B-box with a RING. J. Mol. Biol. 2006, 358, 532–545. [Google Scholar] [CrossRef]
- Slack, F.J.; Ruvkun, G. A novel repeat domain that is often associated with RING finger and B-box motifs. Trends Biochem. Sci. 1998, 23, 474–475. [Google Scholar] [CrossRef]
- LaBeau-DiMenna, E.M.; Clark, K.A.; Bauman, K.D.; Parker, D.S.; Cripps, R.M.; Geisbrecht, E.R. Thin, a Trim32 ortholog, is essential for myofibril stability and is required for the integrity of the costamere in Drosophila. Proc. Natl. Acad. Sci. USA 2012, 109, 17983–17988. [Google Scholar] [CrossRef] [PubMed]
- Domsch, K.; Ezzeddine, N.; Nguyen, H.T. Abba is an essential TRIM/RBCC protein to maintain the integrity of sarcomeric cytoarchitecture. J. Cell Sci. 2013, 126, 3314–3323. [Google Scholar] [CrossRef] [PubMed]
- Bawa, S.; Brooks, D.S.; Neville, K.E.; Tipping, M.; Sagar, M.A.; Kollhoff, J.A.; Chawla, G.; Geisbrecht, B.V.; Tennessen, J.M.; Eliceiri, K.W.; et al. Drosophila TRIM32 cooperates with glycolytic enzymes to promote cell growth. eLife 2020, 9, e52358. [Google Scholar] [CrossRef] [PubMed]
- Saccone, V.; Palmieri, M.; Passamano, L.; Piluso, G.; Meroni, G.; Politano, L.; Nigro, V. Mutations that impair interaction properties of TRIM32 associated with limb-girdle muscular dystrophy 2H. Hum. Mutat. 2008, 29, 240–247. [Google Scholar] [CrossRef]
- Choi, J.H.; Jeong, S.Y.; Kim, J.; Woo, J.S.; Lee, E.H. Tripartite motif-containing protein 32 regulates Ca2+ movement in skeletal muscle. Am. J. Physiol. Cell Physiol. 2022, 323, C1860–C1871. [Google Scholar] [CrossRef] [PubMed]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed]
- Hillje, A.L.; Worlitzer, M.M.; Palm, T.; Schwamborn, J.C. Neural stem cells maintain their stemness through protein kinase C zeta-mediated inhibition of TRIM32. Stem Cells 2011, 29, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, T.; Taoka, M.; Shoji, I.; Kato, H.; Sato, T.; Hatakeyama, S.; Isobe, T.; Hachiya, N. 14-3-3 proteins sequester a pool of soluble TRIM32 ubiquitin ligase to repress autoubiquitylation and cytoplasmic body formation. J. Cell Sci. 2013, 126, 2014–2026. [Google Scholar] [CrossRef]
- Cohen, S.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. Ubiquitylation by Trim32 causes coupled loss of desmin, Z-bands, and thin filaments in muscle atrophy. J. Cell Biol. 2012, 198, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Kudryashova, E.; Kudryashov, D.; Kramerova, I.; Spencer, M.J. Trim32 is a ubiquitin ligase mutated in limb girdle muscular dystrophy type 2H that binds to skeletal muscle myosin and ubiquitinates actin. J. Mol. Biol. 2005, 354, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Locke, M.; Tinsley, C.L.; Benson, M.A.; Blake, D.J. TRIM32 is an E3 ubiquitin ligase for dysbindin. Hum. Mol. Genet. 2009, 18, 2344–2358. [Google Scholar] [CrossRef]
- Nicklas, S.; Otto, A.; Wu, X.; Miller, P.; Stelzer, S.; Wen, Y.; Kuang, S.; Wrogemann, K.; Patel, K.; Ding, H.; et al. TRIM32 regulates skeletal muscle stem cell differentiation and is necessary for normal adult muscle regeneration. PLoS ONE 2012, 7, e30445. [Google Scholar] [CrossRef] [PubMed]
- Kudryashova, E.; Kramerova, I.; Spencer, M.J. Satellite cell senescence underlies myopathy in a mouse model of limb-girdle muscular dystrophy 2H. J. Clin. Investig. 2012, 122, 1764–1776. [Google Scholar] [CrossRef]
- Albor, A.; El-Hizawi, S.; Horn, E.J.; Laederich, M.; Frosk, P.; Wrogemann, K.; Kulesz-Martin, M. The interaction of Piasy with Trim32, an E3-ubiquitin ligase mutated in limb-girdle muscular dystrophy type 2H, promotes Piasy degradation and regulates UVB-induced keratinocyte apoptosis through NFκB. J. Biol. Chem. 2006, 281, 25850–25866. [Google Scholar] [CrossRef] [PubMed]
- Mokhonova, E.I.; Avliyakulov, N.K.; Kramerova, I.; Kudryashova, E.; Haykinson, M.J.; Spencer, M.J. The E3 ubiquitin ligase TRIM32 regulates myoblast proliferation by controlling turnover of NDRG2. Hum. Mol. Genet. 2015, 24, 2873–2883. [Google Scholar] [CrossRef] [PubMed]
- Di Rienzo, M.; Antonioli, M.; Fusco, C.; Liu, Y.; Mari, M.; Orhon, I.; Refolo, G.; Germani, F.; Corazzari, M.; Romagnoli, A.; et al. Autophagy induction in atrophic muscle cells requires ULK1 activation by TRIM32 through unanchored K63-linked polyubiquitin chains. Sci. Adv. 2019, 5, eaau8857. [Google Scholar] [CrossRef] [PubMed]
- Izumi, H.; Kaneko, Y. Trim32 facilitates degradation of MYCN on spindle poles and induces asymmetric cell division in human neuroblastoma cells. Cancer Res. 2014, 74, 5620–5630. [Google Scholar] [CrossRef]
- Kano, S.; Miyajima, N.; Fukuda, S.; Hatakeyama, S. Tripartite motif protein 32 facilitates cell growth and migration via degradation of Abl-interactor 2. Cancer Res. 2008, 68, 5572–5580. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Wang, X.L.; Ly, P.; Belyi, V.; Xu-Monette, Z.Y.; Young, K.H.; Hu, W.; Feng, Z. E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell Death Differ. 2014, 21, 1792–1804. [Google Scholar] [CrossRef]
- Luo, Q.; Wu, X.; Nan, Y.; Chang, W.; Zhao, P.; Zhang, Y.; Su, D.; Liu, Z. TRIM32/USP11 Balances ARID1A Stability and the Oncogenic/Tumor-Suppressive Status of Squamous Cell Carcinoma. Cell Rep. 2020, 30, 98–111.e5. [Google Scholar] [CrossRef]
- Ryu, Y.S.; Lee, Y.; Lee, K.W.; Hwang, C.Y.; Maeng, J.S.; Kim, J.H.; Seo, Y.S.; You, K.H.; Song, B.; Kwon, K.S. TRIM32 protein sensitizes cells to tumor necrosis factor (TNFα)-induced apoptosis via its RING domain-dependent E3 ligase activity against X-linked inhibitor of apoptosis (XIAP). J. Biol. Chem. 2011, 286, 25729–25738. [Google Scholar] [CrossRef] [PubMed]
- Bahnassawy, L.; Perumal, T.M.; Gonzalez-Cano, L.; Hillje, A.L.; Taher, L.; Makalowski, W.; Suzuki, Y.; Fuellen, G.; del Sol, A.; Schwamborn, J.C. TRIM32 modulates pluripotency entry and exit by directly regulating Oct4 stability. Sci. Rep. 2015, 5, 13456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hu, M.M.; Wang, Y.Y.; Shu, H.B. TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 2012, 287, 28646–28655. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Wang, L.; Ding, H.; Schwamborn, J.C.; Li, S.; Dorf, M.E. TRIM32 Senses and Restricts Influenza A Virus by Ubiquitination of PB1 Polymerase. PLoS Pathog. 2015, 11, e1004960. [Google Scholar] [CrossRef]
- Zhao, M.; Song, K.; Hao, W.; Wang, L.; Patil, G.; Li, Q.; Xu, L.; Hua, F.; Fu, B.; Schwamborn, J.C.; et al. Non-proteolytic ubiquitination of OTULIN regulates NF-kappaB signaling pathway. J. Mol. Cell Biol. 2020, 12, 163–175. [Google Scholar] [CrossRef]
- Hammell, C.M.; Lubin, I.; Boag, P.R.; Blackwell, T.K.; Ambros, V. nhl-2 Modulates microRNA activity in Caenorhabditis elegans. Cell 2009, 136, 926–938. [Google Scholar] [CrossRef] [PubMed]
- Schwamborn, J.C.; Berezikov, E.; Knoblich, J.A. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell 2009, 136, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Rybak, A.; Fuchs, H.; Hadian, K.; Smirnova, L.; Wulczyn, E.A.; Michel, G.; Nitsch, R.; Krappmann, D.; Wulczyn, F.G. The let-7 target gene mouse lin-41 is a stem cell specific E3 ubiquitin ligase for the miRNA pathway protein Ago2. Nat. Cell Biol. 2009, 11, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Neumuller, R.A.; Betschinger, J.; Fischer, A.; Bushati, N.; Poernbacher, I.; Mechtler, K.; Cohen, S.M.; Knoblich, J.A. Mei-P26 regulates microRNAs and cell growth in the Drosophila ovarian stem cell lineage. Nature 2008, 454, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Williams, F.P.; Haubrich, K.; Perez-Borrajero, C.; Hennig, J. Emerging RNA-binding roles in the TRIM family of ubiquitin ligases. Biol. Chem. 2019, 400, 1443–1464. [Google Scholar] [CrossRef]
- Goyani, S.; Roy, M.; Singh, R. TRIM-NHL as RNA Binding Ubiquitin E3 Ligase (RBUL): Implication in development and disease pathogenesis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166066. [Google Scholar] [CrossRef]
- Connacher, R.P.; Goldstrohm, A.C. Molecular and biological functions of TRIM-NHL RNA-binding proteins. Wiley Interdiscip. Rev. RNA 2021, 12, e1620. [Google Scholar] [CrossRef]
- Luther, P.K. The vertebrate muscle Z-disc: Sarcomere anchor for structure and signalling. J. Muscle Res. Cell Motil. 2009, 30, 171–185. [Google Scholar] [CrossRef]
- Prill, K.; Dawson, J.F. Assembly and Maintenance of Sarcomere Thin Filaments and Associated Diseases. Int. J. Mol. Sci. 2020, 21, 542. [Google Scholar] [CrossRef] [PubMed]
- Paulin, D.; Li, Z. Desmin: A major intermediate filament protein essential for the structural integrity and function of muscle. Exp. Cell Res. 2004, 301, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.H.; Wold, B.J. c-myc inhibition of MyoD and myogenin-initiated myogenic differentiation. Mol. Cell. Biol. 1991, 11, 2842–2851. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, Q.; Oiso, N.; Novak, E.K.; Gautam, R.; O’Brien, E.P.; Tinsley, C.L.; Blake, D.J.; Spritz, R.A.; Copeland, N.G.; et al. Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat. Genet. 2003, 35, 84–89. [Google Scholar] [CrossRef]
- Johnson, K.; De Ridder, W.; Topf, A.; Bertoli, M.; Phillips, L.; De Jonghe, P.; Baets, J.; Deconinck, T.; Rakocevic Stojanovic, V.; Peric, S.; et al. Extending the clinical and mutational spectrum of TRIM32-related myopathies in a non-Hutterite population. J. Neurol. Neurosurg. Psychiatry 2019, 90, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Liewluck, T.; Tracy, J.A.; Sorenson, E.J.; Engel, A.G. Scapuloperoneal muscular dystrophy phenotype due to TRIM32-sarcotubular myopathy in South Dakota Hutterite. Neuromuscul. Disord. 2013, 23, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Servian-Morilla, E.; Cabrera-Serrano, M.; Rivas-Infante, E.; Carvajal, A.; Lamont, P.J.; Pelayo-Negro, A.L.; Ravenscroft, G.; Junckerstorff, R.; Dyke, J.M.; Fletcher, S.; et al. Altered myogenesis and premature senescence underlie human TRIM32-related myopathy. Acta Neuropathol. Commun. 2019, 7, 30. [Google Scholar] [CrossRef]
- Frosk, P.; Del Bigio, M.R.; Wrogemann, K.; Greenberg, C.R. Hutterite brothers both affected with two forms of limb girdle muscular dystrophy: LGMD2H and LGMD2I. Eur. J. Hum. Genet. 2005, 13, 978–982. [Google Scholar] [CrossRef] [PubMed]
- Ntim, M.; Li, Q.F.; Zhang, Y.; Liu, X.D.; Li, N.; Sun, H.L.; Zhang, X.; Khan, B.; Wang, B.; Wu, Q.; et al. TRIM32 Deficiency Impairs Synaptic Plasticity by Excitatory-Inhibitory Imbalance via Notch Pathway. Cereb. Cortex 2020, 30, 4617–4632. [Google Scholar] [CrossRef]
- Roti, G.; Carlton, A.; Ross, K.N.; Markstein, M.; Pajcini, K.; Su, A.H.; Perrimon, N.; Pear, W.S.; Kung, A.L.; Blacklow, S.C.; et al. Complementary genomic screens identify SERCA as a therapeutic target in NOTCH1 mutated cancer. Cancer Cell 2013, 23, 390–405. [Google Scholar] [CrossRef]
- Pagliaro, L.; Marchesini, M.; Roti, G. Targeting oncogenic Notch signaling with SERCA inhibitors. J. Hematol. Oncol. 2021, 14, 8. [Google Scholar] [CrossRef]
- Vargas-Franco, D.; Kalra, R.; Draper, I.; Pacak, C.A.; Asakura, A.; Kang, P.B. The Notch signaling pathway in skeletal muscle health and disease. Muscle Nerve 2022, 66, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Kudryashova, E.; Struyk, A.; Mokhonova, E.; Cannon, S.C.; Spencer, M.J. The common missense mutation D489N in TRIM32 causing limb girdle muscular dystrophy 2H leads to loss of the mutated protein in knock-in mice resulting in a Trim32-null phenotype. Hum. Mol. Genet. 2011, 20, 3925–3932. [Google Scholar] [CrossRef] [PubMed]
- Guglieri, M.; Straub, V.; Bushby, K.; Lochmuller, H. Limb-girdle muscular dystrophies. Curr. Opin. Neurol. 2008, 21, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, O.A.; Jiang, X.M. Limb-girdle muscular dystrophies: Where next after six decades from the first proposal. Mol. Med. Rep. 2014, 9, 1515–1532. [Google Scholar] [CrossRef]
- Bushby, K.M. The limb-girdle muscular dystrophies-multiple genes, multiple mechanisms. Hum. Mol. Genet. 1999, 8, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.C. Neuromuscular disorders: Gene location. Neuromuscul. Disord. 2001, 11, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Theadom, A.; Rodrigues, M.; Roxburgh, R.; Balalla, S.; Higgins, C.; Bhattacharjee, R.; Jones, K.; Krishnamurthi, R.; Feigin, V. Prevalence of muscular dystrophies: A systematic literature review. Neuroepidemiology 2014, 43, 259–268. [Google Scholar] [CrossRef]
- Murphy, A.P.; Straub, V. The Classification, Natural History and Treatment of the Limb Girdle Muscular Dystrophies. J. Neuromuscul. Dis. 2015, 2, S7–S19. [Google Scholar] [CrossRef]
- Wicklund, M.P. The Limb-Girdle Muscular Dystrophies. Continuum 2019, 25, 1599–1618. [Google Scholar] [CrossRef]
- Barton, E.R.; Pacak, C.A.; Stoppel, W.L.; Kang, P.B. The ties that bind: Functional clusters in limb-girdle muscular dystrophy. Skelet. Muscle 2020, 10, 22. [Google Scholar] [CrossRef]
- Straub, V.; Murphy, A.; Udd, B.; LGMD Workshop Study Group. 229th ENMC international workshop: Limb girdle muscular dystrophies—Nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscul. Disord. 2018, 28, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Jokela, M.; Lehtinen, S.; Palmio, J.; Saukkonen, A.M.; Huovinen, S.; Vihola, A.; Udd, B. A novel COL6A2 mutation causing late-onset limb-girdle muscular dystrophy. J. Neurol. 2019, 266, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Beales, P.L.; Elcioglu, N.; Woolf, A.S.; Parker, D.; Flinter, F.A. New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J. Med. Genet. 1999, 36, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Chiang, A.P.; Beck, J.S.; Yen, H.J.; Tayeh, M.K.; Scheetz, T.E.; Swiderski, R.E.; Nishimura, D.Y.; Braun, T.A.; Kim, K.Y.; Huang, J.; et al. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11). Proc. Natl. Acad. Sci. USA 2006, 103, 6287–6292. [Google Scholar] [CrossRef]
- Muller-Felber, W.; Schlotter, B.; Topfer, M.; Ketelsen, U.P.; Muller-Hocker, J.; Pongratz, D. Phenotypic variability in two brothers with sarcotubular myopathy. J. Neurol. 1999, 246, 408–411. [Google Scholar] [CrossRef]
- Weiler, T.; Greenberg, C.R.; Zelinski, T.; Nylen, E.; Coghlan, G.; Crumley, M.J.; Fujiwara, T.M.; Morgan, K.; Wrogemann, K. A gene for autosomal recessive limb-girdle muscular dystrophy in Manitoba Hutterites maps to chromosome region 9q31–q33: Evidence for another limb-girdle muscular dystrophy locus. Am. J. Hum. Genet. 1998, 63, 140–147. [Google Scholar] [CrossRef]
- Shokeir, M.H.; Kobrinsky, N.L. Autosomal recessive muscular dystrophy in Manitoba Hutterites. Clin. Genet. 1976, 9, 197–202. [Google Scholar] [CrossRef]
- Schoser, B.G.; Frosk, P.; Engel, A.G.; Klutzny, U.; Lochmuller, H.; Wrogemann, K. Commonality of TRIM32 mutation in causing sarcotubular myopathy and LGMD2H. Ann. Neurol. 2005, 57, 591–595. [Google Scholar] [CrossRef]
- Chandrasekharan, S.V.; Sundaram, S.; Malaichamy, S.; Poyuran, R.; Nair, S.S. Myoneuropathic presentation of limb girdle muscular dystrophy R8 with a novel TRIM32 mutation. Neuromuscul. Disord. 2021, 31, 886–890. [Google Scholar] [CrossRef]
- Borg, K.; Stucka, R.; Locke, M.; Melin, E.; Ahlberg, G.; Klutzny, U.; Hagen, M.; Huebner, A.; Lochmuller, H.; Wrogemann, K.; et al. Intragenic deletion of TRIM32 in compound heterozygotes with sarcotubular myopathy/LGMD2H. Hum. Mutat. 2009, 30, E831–E844. [Google Scholar] [CrossRef]
- Cossee, M.; Lagier-Tourenne, C.; Seguela, C.; Mohr, M.; Leturcq, F.; Gundesli, H.; Chelly, J.; Tranchant, C.; Koenig, M.; Mandel, J.L. Use of SNP array analysis to identify a novel TRIM32 mutation in limb-girdle muscular dystrophy type 2H. Neuromuscul. Disord. 2009, 19, 255–260. [Google Scholar] [CrossRef]
- Panicucci, C.; Traverso, M.; Baratto, S.; Romeo, C.; Iacomino, M.; Gemelli, C.; Tagliafico, A.; Broda, P.; Zara, F.; Bruno, C.; et al. Novel TRIM32 mutation in sarcotubular myopathy. Acta Myol. 2019, 38, 8–12. [Google Scholar] [PubMed]
- Neri, M.; Selvatici, R.; Scotton, C.; Trabanelli, C.; Armaroli, A.; De Grandis, D.; Levy, N.; Gualandi, F.; Ferlini, A. A patient with limb girdle muscular dystrophy carries a TRIM32 deletion, detected by a novel CGH array, in compound heterozygosis with a nonsense mutation. Neuromuscul. Disord. 2013, 23, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Marchuk, M.; Dovbonos, T.; Makukh, H.; Semeryak, O.; Sharhorodska, Y. Sarcotubular Myopathy Due to Novel TRIM32 Mutation in Association with Multiple Sclerosis. Brain Sci. 2021, 11, 1020. [Google Scholar] [CrossRef] [PubMed]
- Bawa, S.; Gameros, S.; Baumann, K.; Brooks, D.S.; Kollhoff, J.A.; Zolkiewski, M.; Re Cecconi, A.D.; Panini, N.; Russo, M.; Piccirillo, R.; et al. Costameric integrin and sarcoglycan protein levels are altered in a Drosophila model for Limb-girdle muscular dystrophy type 2H. Mol. Biol. Cell 2021, 32, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Ball, E.; Ball, S.P.; Sparrow, J.C. A mutation affecting larval muscle development in Drosophila melanogaster. Dev. Genet. 1985, 6, 77–92. [Google Scholar] [CrossRef]
- Sandri, M. Protein breakdown in muscle wasting: Role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol. 2013, 45, 2121–2129. [Google Scholar] [CrossRef]
- Dastur, D.K.; Razzak, Z.A. Possible neurogenic factor in muscular dystrophy: Its similarity to denervation atrophy. J. Neurol. Neurosurg. Psychiatry 1973, 36, 399–410. [Google Scholar] [CrossRef]
- Sandri, M.; Coletto, L.; Grumati, P.; Bonaldo, P. Misregulation of autophagy and protein degradation systems in myopathies and muscular dystrophies. J. Cell Sci. 2013, 126, 5325–5333. [Google Scholar] [CrossRef]
- Yin, L.; Li, N.; Jia, W.; Wang, N.; Liang, M.; Yang, X.; Du, G. Skeletal muscle atrophy: From mechanisms to treatments. Pharmacol. Res. 2021, 172, 105807. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Lexell, J. Human aging, muscle mass, and fiber type composition. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 1995, 50, 11–16. [Google Scholar] [CrossRef]
- Snijders, T.; Verdijk, L.B.; van Loon, L.J. The impact of sarcopenia and exercise training on skeletal muscle satellite cells. Ageing Res. Rev. 2009, 8, 328–338. [Google Scholar] [CrossRef]
- Janssen, I.; Heymsfield, S.B.; Wang, Z.M.; Ross, R. Skeletal muscle mass and distribution in 468 men and women aged 18–88 yr. J. Appl. Physiol. 2000, 89, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Lecker, S.H.; Jagoe, R.T.; Gilbert, A.; Gomes, M.; Baracos, V.; Bailey, J.; Price, S.R.; Mitch, W.E.; Goldberg, A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004, 18, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Peris-Moreno, D.; Taillandier, D.; Polge, C. MuRF1/TRIM63, Master Regulator of Muscle Mass. Int. J. Mol. Sci. 2020, 21, 6663. [Google Scholar] [CrossRef]
- Cohen, S.; Brault, J.J.; Gygi, S.P.; Glass, D.J.; Valenzuela, D.M.; Gartner, C.; Latres, E.; Goldberg, A.L. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J. Cell Biol. 2009, 185, 1083–1095. [Google Scholar] [CrossRef]
- Tixier, V.; Bataille, L.; Etard, C.; Jagla, T.; Weger, M.; Daponte, J.P.; Strahle, U.; Dickmeis, T.; Jagla, K. Glycolysis supports embryonic muscle growth by promoting myoblast fusion. Proc. Natl. Acad. Sci. USA 2013, 110, 18982–18987. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Jiao, J.; Demontis, F. Skeletal muscle autophagy and its role in sarcopenia and organismal aging. Curr. Opin. Pharmacol. 2017, 34, 1–6. [Google Scholar] [CrossRef]
- Di Rienzo, M.; Piacentini, M.; Fimia, G.M. A TRIM32-AMBRA1-ULK1 complex initiates the autophagy response in atrophic muscle cells. Autophagy 2019, 15, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef]
- Itakura, E.; Mizushima, N. p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J. Cell Biol. 2011, 192, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Bischof, O.; Dejean, A. SUMO is growing senescent. Cell Cycle 2007, 6, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Schonkeren, S.L.; Massen, M.; van der Horst, R.; Koch, A.; Vaes, N.; Melotte, V. Nervous NDRGs: The N-myc downstream-regulated gene family in the central and peripheral nervous system. Neurogenetics 2019, 20, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Foletta, V.C.; Prior, M.J.; Stupka, N.; Carey, K.; Segal, D.H.; Jones, S.; Swinton, C.; Martin, S.; Cameron-Smith, D.; Walder, K.R. NDRG2, a novel regulator of myoblast proliferation, is regulated by anabolic and catabolic factors. J. Physiol. 2009, 587, 1619–1634. [Google Scholar] [CrossRef]
- Cohen, S.; Lee, D.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. Trim32 reduces PI3K-Akt-FoxO signaling in muscle atrophy by promoting plakoglobin-PI3K dissociation. J. Cell Biol. 2014, 204, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.J.; Weihl, C.C.; Spencer, M.J. Molecular and cellular basis of genetically inherited skeletal muscle disorders. Nat. Rev. Mol. Cell Biol. 2021, 22, 713–732. [Google Scholar] [CrossRef] [PubMed]
- Megarbane, A.; Bizzari, S.; Deepthi, A.; Sabbagh, S.; Mansour, H.; Chouery, E.; Hmaimess, G.; Jabbour, R.; Mehawej, C.; Alame, S.; et al. A 20-year Clinical and Genetic Neuromuscular Cohort Analysis in Lebanon: An International Effort. J. Neuromuscul. Dis. 2022, 9, 193–210. [Google Scholar] [CrossRef]
- Kottlors, M.; Kirschner, J. Elevated satellite cell number in Duchenne muscular dystrophy. Cell Tissue Res. 2010, 340, 541–548. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef]
- Jiang, C.; Wen, Y.; Kuroda, K.; Hannon, K.; Rudnicki, M.A.; Kuang, S. Notch signaling deficiency underlies age-dependent depletion of satellite cells in muscular dystrophy. Dis. Model. Mech. 2014, 7, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Boldrin, L.; Zammit, P.S.; Morgan, J.E. Satellite cells from dystrophic muscle retain regenerative capacity. Stem Cell Res. 2015, 14, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Megeney, L.A.; Kablar, B.; Garrett, K.; Anderson, J.E.; Rudnicki, M.A. MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes Dev. 1996, 10, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Sacco, A.; Mourkioti, F.; Tran, R.; Choi, J.; Llewellyn, M.; Kraft, P.; Shkreli, M.; Delp, S.; Pomerantz, J.H.; Artandi, S.E.; et al. Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell 2010, 143, 1059–1071. [Google Scholar] [CrossRef]
- Kuang, S.; Kuroda, K.; Le Grand, F.; Rudnicki, M.A. Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell 2007, 129, 999–1010. [Google Scholar] [CrossRef]
- Chang, N.C.; Chevalier, F.P.; Rudnicki, M.A. Satellite Cells in Muscular Dystrophy—Lost in Polarity. Trends Mol. Med. 2016, 22, 479–496. [Google Scholar] [CrossRef]
- De Palma, C.; Perrotta, C.; Pellegrino, P.; Clementi, E.; Cervia, D. Skeletal muscle homeostasis in duchenne muscular dystrophy: Modulating autophagy as a promising therapeutic strategy. Front. Aging Neurosci. 2014, 6, 188. [Google Scholar] [CrossRef]
- De Palma, C.; Morisi, F.; Cheli, S.; Pambianco, S.; Cappello, V.; Vezzoli, M.; Rovere-Querini, P.; Moggio, M.; Ripolone, M.; Francolini, M.; et al. Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis. 2012, 3, e418. [Google Scholar] [CrossRef]
- Benson, M.A.; Newey, S.E.; Martin-Rendon, E.; Hawkes, R.; Blake, D.J. Dysbindin, a novel coiled-coil-containing protein that interacts with the dystrobrevins in muscle and brain. J. Biol. Chem. 2001, 276, 24232–24241. [Google Scholar] [CrossRef]
- Wakayama, Y.; Matsuzaki, Y.; Yamashita, S.; Inoue, M.; Jimi, T.; Hara, H.; Unaki, A.; Iijima, S.; Masaki, H. Dysbindin, syncoilin, and beta-synemin mRNA levels in dystrophic muscles. Int. J. Neurosci. 2010, 120, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Assereto, S.; Piccirillo, R.; Baratto, S.; Scudieri, P.; Fiorillo, C.; Massacesi, M.; Traverso, M.; Galietta, L.J.; Bruno, C.; Minetti, C.; et al. The ubiquitin ligase tripartite-motif-protein 32 is induced in Duchenne muscular dystrophy. Lab. Investig. 2016, 96, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Harisseh, R.; Chatelier, A.; Magaud, C.; Deliot, N.; Constantin, B. Involvement of TRPV2 and SOCE in calcium influx disorder in DMD primary human myotubes with a specific contribution of alpha1-syntrophin and PLC/PKC in SOCE regulation. Am. J. Physiol. Cell Physiol. 2013, 304, C881–C894. [Google Scholar] [CrossRef]
- Zhao, X.; Moloughney, J.G.; Zhang, S.; Komazaki, S.; Weisleder, N. Orai1 mediates exacerbated Ca2+ entry in dystrophic skeletal muscle. PLoS ONE 2012, 7, e49862. [Google Scholar] [CrossRef]
- Zhu, J.W.; Zou, M.M.; Li, Y.F.; Chen, W.J.; Liu, J.C.; Chen, H.; Fang, L.P.; Zhang, Y.; Wang, Z.T.; Chen, J.B.; et al. Absence of TRIM32 Leads to Reduced GABAergic Interneuron Generation and Autism-like Behaviors in Mice via Suppressing mTOR Signaling. Cereb. Cortex 2020, 30, 3240–3258. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.Y.; Chen, W.J.; Huang, Z.P.; Yang, G.; Wu, M.L.; Xu, D.E.; Yang, W.L.; Luo, Y.C.; Xiao, Z.C.; Xu, R.X.; et al. TRIM32 Deficiency Impairs the Generation of Pyramidal Neurons in Developing Cerebral Cortex. Cells 2022, 11, 449. [Google Scholar] [CrossRef]
- Zhu, J.W.; Jia, W.Q.; Zhou, H.; Li, Y.F.; Zou, M.M.; Wang, Z.T.; Wu, B.S.; Xu, R.X. Deficiency of TRIM32 Impairs Motor Function and Purkinje Cells in Mid-Aged Mice. Front. Aging Neurosci. 2021, 13, 697494. [Google Scholar] [CrossRef]
- Yokota, T.; Mishra, M.; Akatsu, H.; Tani, Y.; Miyauchi, T.; Yamamoto, T.; Kosaka, K.; Nagai, Y.; Sawada, T.; Heese, K. Brain site-specific gene expression analysis in Alzheimer’s disease patients. Eur. J. Clin. Investig. 2006, 36, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, T.T.; Xie, G.G.; Zhu, X.Q.; Wang, Y. Ubiquitin ligase TRIM32 promotes dendrite arborization by mediating degradation of the epigenetic factor CDYL. FASEB J. 2022, 36, e22087. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Luo, W.; Zhang, Y.; Yang, R.; Li, X.; Guo, Y.; Zhang, C.; Yang, R.; Gao, W.Q. Trim32 suppresses cerebellar development and tumorigenesis by degrading Gli1/sonic hedgehog signaling. Cell Death Differ. 2020, 27, 1286–1299. [Google Scholar] [CrossRef] [PubMed]
- Nicklas, S.; Hillje, A.L.; Okawa, S.; Rudolph, I.M.; Collmann, F.M.; van Wuellen, T.; Del Sol, A.; Schwamborn, J.C. A complex of the ubiquitin ligase TRIM32 and the deubiquitinase USP7 balances the level of c-Myc ubiquitination and thereby determines neural stem cell fate specification. Cell Death Differ. 2019, 26, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Hillje, A.L.; Pavlou, M.A.; Beckmann, E.; Worlitzer, M.M.; Bahnassawy, L.; Lewejohann, L.; Palm, T.; Schwamborn, J.C. TRIM32-dependent transcription in adult neural progenitor cells regulates neuronal differentiation. Cell Death Dis. 2013, 4, e976. [Google Scholar] [CrossRef]
- Sato, T.; Okumura, F.; Kano, S.; Kondo, T.; Ariga, T.; Hatakeyama, S. TRIM32 promotes neural differentiation through retinoic acid receptor-mediated transcription. J. Cell Sci. 2011, 124, 3492–3502. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, J.; Ji, Y.; Zhang, X.; Wang, P.; Deng, K.; Jiang, X.; Ma, G.; Li, H. Tripartite motif 32 prevents pathological cardiac hypertrophy. Clin. Sci. 2016, 130, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Horn, E.J.; Albor, A.; Liu, Y.; El-Hizawi, S.; Vanderbeek, G.E.; Babcock, M.; Bowden, G.T.; Hennings, H.; Lozano, G.; Weinberg, W.C.; et al. RING protein Trim32 associated with skin carcinogenesis has anti-apoptotic and E3-ubiquitin ligase properties. Carcinogenesis 2004, 25, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Albor, A.; Kulesz-Martin, M. Novel initiation genes in squamous cell carcinomagenesis: A role for substrate-specific ubiquitylation in the control of cell survival. Mol. Carcinog. 2007, 46, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Zhou, X.; Jiao, Q.; Chen, X. The Functions of TRIM56 in Antiviral Innate Immunity and Tumorigenesis. Int. J. Mol. Sci. 2023, 24, 5046. [Google Scholar] [CrossRef]
- Aley, P.K.; Mikolajczyk, A.M.; Munz, B.; Churchill, G.C.; Galione, A.; Berger, F. Nicotinic acid adenine dinucleotide phosphate regulates skeletal muscle differentiation via action at two-pore channels. Proc. Natl. Acad. Sci. USA 2010, 107, 19927–19932. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Autosomal Dominant LGMDs | |||
|---|---|---|---|
| Refined Classification | Classical Classification | Gene Name | Protein Name (Reasons for the Exclusion) |
| None | LGMD1A | MYOT | Myotilin; excluded (myofibrillar myopathy, distal weakness) |
| None | LGMD1B | LMNA | Lamin A/C; excluded (Emery–Dreifuss muscular dystrophy, high risk of cardiac arrhythmia) |
| None | LGMD1C | CAV3 | Caveolin 3; excluded (rippling muscle disease and myalgia) |
| LGMDD1 | LGMD1D | DNAJB6 | DnaJ heat shock protein family (Hsp40) member B6 |
| None | LGMD1E | DES | Desmin; excluded (false linkage, distal weakness and cardiomyopathy) |
| LGMDD2 | LGMD1F | TNPO3 | Transportin 3 |
| LGMDD3 | LGMD1G | HNRNPDL | Heterogeneous nuclear ribonucleoprotein D-like protein |
| None | LGMD1H | unknown | Unknown; excluded (false linkage) |
| LGMDD4 | LGMD1I | CAPN3 | Calpain 3 |
| LGMDD5 | None | COL6A1, COL6A2, or COL6A3 | Collagen type VI α1, α2, or α3 chain |
| Autosomal Recessive LGMDs | |||
| Refined Classification | Classical Classification | Gene Name | Protein Name (Reasons for the Exclusion) |
| LGMDR1 | LGMD2A | CAPN3 | Calpain 3 |
| LGMDR2 | LGMD2B | DYSF | Dysferlin |
| LGMDR3 | LGMD2D | SGCA | α-Sarcoglycan |
| LGMDR4 | LGMD2E | SGCB | β-Sarcoglycan |
| LGMDR5 | LGMD2C | SGCG | γ-Sarcoglycan |
| LGMDR6 | LGMD2F | SGCD | δ-Sarcoglycan |
| LGMDR7 | LGMD2G | TCAP | Telethonin |
| LGMDR8 | LGMD2H | TRIM32 | Tripartite motif-containing 32 (TRIM32) |
| LGMDR9 | LGMD2I | FKRP | Fukutin-related protein |
| LGMDR10 | LGMD2J | TTN | Titin |
| LGMDR11 | LGMD2K | POMT1 | Protein O-mannosyl-transferase 1 |
| LGMDR12 | LGMD2L | ANO5 | Anoctamin 5 |
| LGMDR13 | LGMD2M | FKTN/FCMD | Fukutin |
| LGMDR14 | LGMD2N | POMT2 | Protein O-mannosyl-transferase 2 |
| LGMDR15 | LGMD2O | POMGnT1 | Protein O-linked mannose N-acetylglucosaminyl transferase 1 |
| LGMDR16 | LGMD2P | DAG1 | Dystroglycan 1 |
| LGMDR17 | LGMD2Q | PLEC | Plectin |
| None | LGMD2R | DES | Desmin; excluded (myofibrillar myopathy, distal weakness) |
| LGMDR18 | LGMD2S | TRAPPC11 | Trafficking protein particle complex 11 |
| LGMDR19 | LGMD2T | GMPPB | GDP-mannose pyrophosphorylase B |
| LGMDR20 | LGMD2U | CRPPA/ISPD | CDP-L-ribitol pyrophosphorylase A |
| None | LGMD2V | GAA | Acid α -glucosidase; excluded (Pompe disease) |
| None | LGMD2W | PINCH2 | Particularly interesting Cys-His-rich protein 2; excluded (reported in only one family) |
| None | LGMD2Y | TORIAIP1 | Torsin 1A interacting protein 1; excluded (reported in only one family) |
| LGMDR21 | LGMD2Z | POGLUT1 | Protein O-glucosyltransferase 1 |
| LGMDR22 | None | COL6A1, COL6A2, or COL6A3 | Collagen type VI α1, α2, or α3 chain |
| LGMDR23 | None | LAMA2 | Laminin subunit α2 |
| LGMDR24 | None | POMGNT2 | Protein O-linked mannose N-acetylglucosaminyl transferase 2 |
| LGMDR25 | LGMD2X | BVES | Blood vessel epicardial substance |
| LGMDR (number pending) | None | PYROXD1 | Pyridine nucleotide-disulfide oxidoreductase domain-containing protein 1 |
| Variation in DNA | Heredity | Variant Name (Protein) | Position of Mutation | Phenotype | Reference Article |
|---|---|---|---|---|---|
| 35dupA | Homozygous | D12EfsTer44 | Intervening region before the RING domain | LGMD2H | [110] |
| 115_116insT | Homozygous | C39LfsX17 | RING domain | LGMD2H | [87] |
| 650A>G, 1701_1703del | Heterozygous | N217S/F568del | Intervening region between coiled-coil region and NHL repeats, 4th NHL repeat | LGMD2H | [87] |
| 1180G>A | Homozygous | R394H | 2th NHL repeat | LGMD2H | [51] |
| 1459G>A | Homozygous | D487N | 3th NHL repeat | LGMD2H | [3] |
| 1559delC | Homozygous | T520TfsX13 | 5th NHL repeat | LGMD2H | [51] |
| 1560delC | Heterozygous | C521VfsX13 | 5th NHL repeat | LGMD2H | [111] |
| 1761_1763delGAT | Heterozygous | D588del | 5th NHL repeat | LGMD2H | [51] |
| 1753_1766dup14 | Homozygous | I590LfsX38 | 5th NHL repeat | LGMD2H | [112] |
| 1771G>A | Homozygous | V591M | 5th NHL repeat | LGMD2H | [87] |
| 1781G>A | Homozygous | S594N | 5th NHL repeat | LGMD2H | [113] |
| 1837C>T | Heterozygous | R613X | 6th NHL repeat | LGMD2H | [114] |
| 1855C>T | Homozygous | P619S | 6th NHL repeat | LGMD2H | [115] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, S.Y.; Choi, J.H.; Kim, J.; Woo, J.S.; Lee, E.H. Tripartite Motif-Containing Protein 32 (TRIM32): What Does It Do for Skeletal Muscle? Cells 2023, 12, 2104. https://doi.org/10.3390/cells12162104
Jeong SY, Choi JH, Kim J, Woo JS, Lee EH. Tripartite Motif-Containing Protein 32 (TRIM32): What Does It Do for Skeletal Muscle? Cells. 2023; 12(16):2104. https://doi.org/10.3390/cells12162104
Chicago/Turabian StyleJeong, Seung Yeon, Jun Hee Choi, Jooho Kim, Jin Seok Woo, and Eun Hui Lee. 2023. "Tripartite Motif-Containing Protein 32 (TRIM32): What Does It Do for Skeletal Muscle?" Cells 12, no. 16: 2104. https://doi.org/10.3390/cells12162104
APA StyleJeong, S. Y., Choi, J. H., Kim, J., Woo, J. S., & Lee, E. H. (2023). Tripartite Motif-Containing Protein 32 (TRIM32): What Does It Do for Skeletal Muscle? Cells, 12(16), 2104. https://doi.org/10.3390/cells12162104