
Dendritic Cell Vaccines: A Shift from Conventional Approach to New Generations

Abstract
1. Introduction
2. Classical DC Vaccine
3. Limitations of Classical DC Vaccine
4. Biomaterial-Based DC Vaccines
5. Combinatory Immunogenic Cell Death-Inducing DC Vaccines
6. mRNA-Pulsed DC Vaccines
7. DC-Derived Small Extracellular Vesicle (sEV) Vaccines
8. Tumor-Derived sEV Vaccines
9. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic Immunity in Cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.D.; Cabral, H.; Stylianopoulos, T.; Jain, R.K. Improving Cancer Immunotherapy Using Nanomedicines: Progress, Opportunities and Challenges. Nat. Rev. Clin. Oncol. 2020, 17, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Makkouk, A.; Weiner, G.J. Cancer Immunotherapy and Breaking Immune Tolerance: New Approaches to an Old Challenge. Cancer Res. 2015, 75, 5–10. [Google Scholar] [CrossRef]
- Phuengkham, H.; Ren, L.; Shin, I.W.; Lim, Y.T. Nanoengineered Immune Niches for Reprogramming the Immunosuppressive Tumor Microenvironment and Enhancing Cancer Immunotherapy. Adv. Mater. 2019, 31, 1803322. [Google Scholar] [CrossRef]
- Del Prete, A.; Salvi, V.; Soriani, A.; Laffranchi, M.; Sozio, F.; Bosisio, D.; Sozzani, S. Dendritic Cell Subsets in Cancer Immunity and Tumor Antigen Sensing. Cell Mol. Immunol. 2023, 20, 432–447. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic Cells in Cancer Immunology and Immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Saxena, M.; Bhardwaj, N. Re-Emergence of Dendritic Cell Vaccines for Cancer Treatment. Trends Cancer 2018, 4, 119–137. [Google Scholar] [CrossRef]
- Lin, M.J.; Svensson-Arvelund, J.; Lubitz, G.S.; Marabelle, A.; Melero, I.; Brown, B.D.; Brody, J.D. Cancer Vaccines: The next Immunotherapy Frontier. Nat. Cancer 2022, 3, 911–926. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T Cell Therapy: Current Limitations and Potential Strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Kartikasari, A.E.R.; Prakash, M.D.; Cox, M.; Wilson, K.; Boer, J.C.; Cauchi, J.A.; Plebanski, M. Therapeutic Cancer Vaccines—T Cell Responses and Epigenetic Modulation. Front. Immunol. 2019, 9, 3109. [Google Scholar] [CrossRef]
- Appleton, E.; Hassan, J.; Chan Wah Hak, C.; Sivamanoharan, N.; Wilkins, A.; Samson, A.; Ono, M.; Harrington, K.J.; Melcher, A.; Wennerberg, E. Kickstarting Immunity in Cold Tumours: Localised Tumour Therapy Combinations with Immune Checkpoint Blockade. Front. Immunol. 2021, 12, 754436. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic Cancer Vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ Cytotoxic T Lymphocytes in Cancer Immunotherapy: A Review. J. Cell Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer Vaccines as Promising Immuno-Therapeutics: Platforms and Current Progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Gutiérrez-Martínez, E.; Planès, R.; Anselmi, G.; Reynolds, M.; Menezes, S.; Adiko, A.C.; Saveanu, L.; Guermonprez, P. Cross-Presentation of Cell-Associated Antigens by MHC Class I in Dendritic Cell Subsets. Front. Immunol. 2015, 6, 363. [Google Scholar] [CrossRef]
- Kumbhari, A.; Kim, P.S.; Lee, P.P. Optimisation of Anti-Cancer Peptide Vaccines to Preferentially Elicit High-Avidity T Cells. J. Theor. Biol. 2020, 486, 110067. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J. Dendritic-Cell-Based Therapeutic Cancer Vaccines. Immunity 2013, 39, 38–48. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J. Cancer Immunotherapy via Dendritic Cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic Cell-Based Immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef]
- Mastelic-Gavillet, B.; Balint, K.; Boudousquie, C.; Gannon, P.O.; Kandalaft, L.E. Personalized Dendritic Cell Vaccines-Recent Breakthroughs and Encouraging Clinical Results. Front. Immunol. 2019, 10, 766. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic Cells and the Control of Immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Sprooten, J.; Ceusters, J.; Coosemans, A.; Agostinis, P.; De Vleeschouwer, S.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; Garg, A.D. Trial Watch: Dendritic Cell Vaccination for Cancer Immunotherapy. Oncoimmunology 2019, 8, e1638212. [Google Scholar] [CrossRef] [PubMed]
- Eisenbarth, S.C. Dendritic Cell Subsets in T Cell Programming: Location Dictates Function. Nat. Rev. Immunol. 2019, 19, 89–103. [Google Scholar] [CrossRef]
- Satpathy, A.T.; Wu, X.; Albring, J.C.; Murphy, K.M. Re(de)Fining the Dendritic Cell Lineage. Nat. Immunol. 2012, 13, 1145–1154. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Guilliams, M.; Merad, M. Expanding Dendritic Cell Nomenclature in the Single-Cell Era. Nat. Rev. Immunol. 2022, 22, 67–68. [Google Scholar] [CrossRef]
- Cytlak, U.; Resteu, A.; Pagan, S.; Green, K.; Milne, P.; Maisuria, S.; McDonald, D.; Hulme, G.; Filby, A.; Carpenter, B.; et al. Differential IRF8 Transcription Factor Requirement Defines Two Pathways of Dendritic Cell Development in Humans. Immunity 2020, 53, 353–370.e8. [Google Scholar] [CrossRef]
- Villani, A.-C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-Cell RNA-Seq Reveals New Types of Human Blood Dendritic Cells, Monocytes, and Progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef]
- Dutertre, C.-A.; Becht, E.; Irac, S.E.; Khalilnezhad, A.; Narang, V.; Khalilnezhad, S.; Ng, P.Y.; van den Hoogen, L.L.; Leong, J.Y.; Lee, B.; et al. Single-Cell Analysis of Human Mononuclear Phagocytes Reveals Subset-Defining Markers and Identifies Circulating Inflammatory Dendritic Cells. Immunity 2019, 51, 573–589.e8. [Google Scholar] [CrossRef]
- Brown, C.C.; Gudjonson, H.; Pritykin, Y.; Deep, D.; Lavallée, V.P.; Mendoza, A.; Fromme, R.; Mazutis, L.; Ariyan, C.; Leslie, C.; et al. Transcriptional Basis of Mouse and Human Dendritic Cell Heterogeneity. Cell 2019, 179, 846–863.e24. [Google Scholar] [CrossRef]
- Marmonti, E.; Oliva-Ramirez, J.; Haymaker, C. Dendritic Cells: The Long and Evolving Road towards Successful Targetability in Cancer. Cells 2022, 11, 3028. [Google Scholar] [CrossRef]
- Maier, B.; Leader, A.M.; Chen, S.T.; Tung, N.; Chang, C.; LeBerichel, J.; Chudnovskiy, A.; Maskey, S.; Walker, L.; Finnigan, J.P.; et al. A Conserved Dendritic-Cell Regulatory Program Limits Antitumour Immunity. Nature 2020, 580, 257–262. [Google Scholar] [CrossRef]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.-R.; Hildebrand, W.H.; Mardis, E.R.; et al. A Dendritic Cell Vaccine Increases the Breadth and Diversity of Melanoma Neoantigen-Specific T Cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 2019, 50, 1317–1334.e10. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M. Decisions About Dendritic Cells: Past, Present, and Future. Annu. Rev. Immunol. 2012, 30, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [PubMed]
- Gallo, P.M.; Gallucci, S. The Dendritic Cell Response to Classic, Emerging, and Homeostatic Danger Signals. Implications for Autoimmunity. Front. Immunol. 2013, 4, 138. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-like Receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Oliveira, M.M.S.; D’Aulerio, R.; Yong, T.; He, M.; Baptista, M.A.P.; Nylén, S.; Westerberg, L.S. Increased Cross-Presentation by Dendritic Cells and Enhanced Anti-Tumour Therapy Using the Arp2/3 Inhibitor CK666. Br. J. Cancer 2023, 128, 982–991. [Google Scholar] [CrossRef]
- Kurts, C.; Robinson, B.W.S.; Knolle, P.A. Cross-Priming in Health and Disease. Nat. Rev. Immunol. 2010, 10, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Cabeza-Cabrerizo, M.; Cardoso, A.; Minutti, C.M.; Pereira da Costa, M.; Reis e Sousa, C. Dendritic Cells Revisited. Annu. Rev. Immunol. 2021, 39, 131–166. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Wherry, E.J.; Ahmed, R. Effector and Memory T-Cell Differentiation: Implications for Vaccine Development. Nat. Rev. Immunol. 2002, 2, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Curtsinger, J.M.; Mescher, M.F. Inflammatory Cytokines as a Third Signal for T Cell Activation. Curr. Opin. Immunol. 2010, 22, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-Presentation by Dendritic Cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef]
- Fu, C.; Zhou, L.; Mi, Q.-S.; Jiang, A. DC-Based Vaccines for Cancer Immunotherapy. Vaccines 2020, 8, 706. [Google Scholar] [CrossRef]
- Calmeiro, J.; Carrascal, M.; Gomes, C.; Falcão, A.; Cruz, M.T.; Neves, B.M. Biomaterial-Based Platforms for in Situ Dendritic Cell Programming and Their Use in Antitumor Immunotherapy. J. Immunother. Cancer 2019, 7, 238. [Google Scholar] [CrossRef]
- Constantino, J.; Gomes, C.; Falcão, A.; Cruz, M.T.; Neves, B.M. Antitumor Dendritic Cell–Based Vaccines: Lessons from 20 Years of Clinical Trials and Future Perspectives. Transl. Res. 2016, 168, 74–95. [Google Scholar] [CrossRef]
- Sallusto, F.; Lanzavecchia, A. Efficient Presentation of Soluble Antigen by Cultured Human Dendritic Cells Is Maintained by Granulocyte/Macrophage Colony-Stimulating Factor plus Interleukin 4 and Downregulated by Tumor Necrosis Factor Alpha. J. Exp. Med. 1994, 179, 1109–1118. [Google Scholar] [CrossRef]
- Ghorbaninezhad, F.; Asadzadeh, Z.; Masoumi, J.; Mokhtarzadeh, A.; Kazemi, T.; Aghebati-Maleki, L.; Shotorbani, S.S.; Shadbad, M.A.; Baghbanzadeh, A.; Hemmat, N.; et al. Dendritic Cell-Based Cancer Immunotherapy in the Era of Immune Checkpoint Inhibitors: From Bench to Bedside. Life Sci. 2022, 297, 120466. [Google Scholar] [CrossRef]
- Massa, C.; Thomas, C.; Wang, E.; Marincola, F.; Seliger, B. Different Maturation Cocktails Provide Dendritic Cells with Different Chemoattractive Properties. J. Transl. Med. 2015, 13, 175. [Google Scholar] [CrossRef]
- Ratzinger, G.; Baggers, J.; de Cos, M.A.; Yuan, J.; Dao, T.; Reagan, J.L.; Münz, C.; Heller, G.; Young, J.W. Mature Human Langerhans Cells Derived from CD34+ Hematopoietic Progenitors Stimulate Greater Cytolytic T Lymphocyte Activity in the Absence of Bioactive IL-12p70, by Either Single Peptide Presentation or Cross-Priming, Than Do Dermal-Interstitial or Monocyte-Derived Dendritic Cells. J. Immunol. 2004, 173, 2780–2791. [Google Scholar] [CrossRef] [PubMed]
- Romano, E.; Rossi, M.; Ratzinger, G.; De Cos, M.A.; Chung, D.J.; Panageas, K.S.; Wolchock, J.D.; Houghton, A.N.; Chapman, P.B.; Heller, G.; et al. Peptide-Loaded Langerhans Cells, despite Increased IL15 Secretion and T-Cell Activation in Vitro, Elicit Antitumor T-Cell Responses Comparable to Peptide-Loaded Monocyte-Derived Dendritic Cells In Vivo. Clin. Cancer Res. 2011, 17, 1984–1997. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.J.; Carvajal, R.D.; Postow, M.A.; Sharma, S.; Pronschinske, K.B.; Shyer, J.A.; Singh-Kandah, S.; Dickson, M.A.; D’Angelo, S.P.; Wolchok, J.D.; et al. Langerhans-Type Dendritic Cells Electroporated with TRP-2 MRNA Stimulate Cellular Immunity against Melanoma: Results of a Phase I Vaccine Trial. Oncoimmunology 2018, 7, e1372081. [Google Scholar] [CrossRef] [PubMed]
- Higano, C.S.; Schellhammer, P.F.; Small, E.J.; Burch, P.A.; Nemunaitis, J.; Yuh, L.; Provost, N.; Frohlich, M.W. Integrated Data from 2 Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trials of Active Cellular Immunotherapy with Sipuleucel-T in Advanced Prostate Cancer. Cancer 2009, 115, 3670–3679. [Google Scholar] [CrossRef]
- Cheever, M.A.; Higano, C.S. PROVENGE (Sipuleucel-T) in Prostate Cancer: The First FDA-Approved Therapeutic Cancer Vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef]
- Scholz, M.; Yep, S.; Chancey, M.; Kelly, C.; Chau, K.; Turner, J.; Lam, R.; Drake, C. Phase I Clinical Trial of Sipuleucel-T Combined with Escalating Doses of Ipilimumab in Progressive Metastatic Castrate-Resistant Prostate Cancer. Immunotargets Ther. 2017, 6, 11–16. [Google Scholar] [CrossRef][Green Version]
- Sinha, M.; Zhang, L.; Subudhi, S.; Chen, B.; Marquez, J.; Liu, E.V.; Allaire, K.; Cheung, A.; Ng, S.; Nguyen, C.; et al. Pre-Existing Immune Status Associated with Response to Combination of Sipuleucel-T and Ipilimumab in Patients with Metastatic Castration-Resistant Prostate Cancer. J. Immunother. Cancer 2021, 9, e002254. [Google Scholar] [CrossRef]
- Pachynski, R.K.; Morishima, C.; Szmulewitz, R.; Harshman, L.; Appleman, L.; Monk, P.; Bitting, R.L.; Kucuk, O.; Millard, F.; Seigne, J.D.; et al. IL-7 Expands Lymphocyte Populations and Enhances Immune Responses to Sipuleucel-T in Patients with Metastatic Castration-Resistant Prostate Cancer (MCRPC). J. Immunother. Cancer 2021, 9, e002903. [Google Scholar] [CrossRef]
- Gilboa, E. DC-Based Cancer Vaccines. J. Clin. Investig. 2007, 117, 1195–1203. [Google Scholar] [CrossRef]
- Kastenmüller, W.; Kastenmüller, K.; Kurts, C.; Seder, R.A. Dendritic Cell-Targeted Vaccines—Hope or Hype? Nat. Rev. Immunol. 2014, 14, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Tel, J.; Benitez-Ribas, D.; Hoosemans, S.; Cambi, A.; Adema, G.J.; Figdor, C.G.; Tacken, P.J.; de Vries, I.J.M. DEC-205 Mediates Antigen Uptake and Presentation by Both Resting and Activated Human Plasmacytoid Dendritic Cells. Eur. J. Immunol. 2011, 41, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Lahoud, M.H.; Ahmet, F.; Kitsoulis, S.; Wan, S.S.; Vremec, D.; Lee, C.-N.; Phipson, B.; Shi, W.; Smyth, G.K.; Lew, A.M.; et al. Targeting Antigen to Mouse Dendritic Cells via Clec9A Induces Potent CD4 T Cell Responses Biased toward a Follicular Helper Phenotype. J. Immunol. 2011, 187, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Caminschi, I.; Maraskovsky, E.; Heath, W.R. Targeting Dendritic Cells in Vivo for Cancer Therapy. Front. Immunol. 2012, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; de Mingo Pulido, Á.; Ruffell, B. Dendritic Cells and Their Role in Immunotherapy. Front. Immunol. 2020, 11, 924. [Google Scholar] [CrossRef]
- Gardner, A.; Ruffell, B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef]
- Bonifaz, L.; Bonnyay, D.; Mahnke, K.; Rivera, M.; Nussenzweig, M.C.; Steinman, R.M. Efficient Targeting of Protein Antigen to the Dendritic Cell Receptor DEC-205 in the Steady State Leads to Antigen Presentation on Major Histocompatibility Complex Class I Products and Peripheral CD8+ T Cell Tolerance. J. Exp. Med. 2002, 196, 1627–1638. [Google Scholar] [CrossRef]
- Fu, C.; Ma, T.; Zhou, L.; Mi, Q.S.; Jiang, A. Dendritic Cell-Based Vaccines Against Cancer: Challenges, Advances and Future Opportunities. Immunol. Investig. 2022, 51, 2133–2158. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Belladonna, M.L.; Volpi, C.; Bianchi, R.; Vacca, C.; Orabona, C.; Pallotta, M.T.; Boon, L.; Gizzi, S.; Fioretti, M.C.; Grohmann, U.; et al. Cutting Edge: Autocrine TGF-β Sustains Default Tolerogenesis by IDO-Competent Dendritic Cells. J. Immunol. 2008, 181, 5194–5198. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of Vascular Endothelial Growth Factor by Human Tumors Inhibits the Functional Maturation of Dendritic Cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The Immunosuppressive Tumour Network: Myeloid-Derived Suppressor Cells, Regulatory T Cells and Natural Killer T Cells. Immunology 2013, 138, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T Cell Exclusion, Immune Privilege, and the Tumor Microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Regulatory T Cells, Tumour Immunity and Immunotherapy. Nat. Rev. Immunol. 2006, 6, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Balan, S.; Ollion, V.; Colletti, N.; Chelbi, R.; Montanana-Sanchis, F.; Liu, H.; Vu Manh, T.-P.; Sanchez, C.; Savoret, J.; Perrot, I.; et al. Human XCR1+ Dendritic Cells Derived In Vitro from CD34+ Progenitors Closely Resemble Blood Dendritic Cells, Including Their Adjuvant Responsiveness, Contrary to Monocyte-Derived Dendritic Cells. J. Immunol. 2014, 193, 1622–1635. [Google Scholar] [CrossRef]
- Laoui, D.; Keirsse, J.; Morias, Y.; Van Overmeire, E.; Geeraerts, X.; Elkrim, Y.; Kiss, M.; Bolli, E.; Lahmar, Q.; Sichien, D.; et al. The Tumour Microenvironment Harbours Ontogenically Distinct Dendritic Cell Populations with Opposing Effects on Tumour Immunity. Nat. Commun. 2016, 7, 13720. [Google Scholar] [CrossRef]
- Kleindienst, P.; Brocker, T. Endogenous Dendritic Cells Are Required for Amplification of T Cell Responses Induced by Dendritic Cell Vaccines In Vivo. J. Immunol. 2003, 170, 2817–2823. [Google Scholar] [CrossRef]
- Martín-Fontecha, A.; Sebastiani, S.; Höpken, U.E.; Uguccioni, M.; Lipp, M.; Lanzavecchia, A.; Sallusto, F. Regulation of Dendritic Cell Migration to the Draining Lymph Node: Impact on T Lymphocyte Traffic and Priming. J. Exp. Med. 2003, 198, 615–621. [Google Scholar] [CrossRef]
- Verdijk, P.; Aarntzen, E.H.J.G.; Lesterhuis, W.J.; Boullart, A.C.I.; Kok, E.; Van Rossum, M.M.; Strijk, S.; Eijckeler, F.; Bonenkamp, J.J.; Jacobs, J.F.M.; et al. Limited Amounts of Dendritic Cells Migrate into Thet-Cell Area of Lymph Nodes but Have High Immune Activating Potential in Melanoma Patients. Clin. Cancer Res. 2009, 15, 2531–2540. [Google Scholar] [CrossRef] [PubMed]
- Adema, G.J.; De Vries, I.J.M.; Punt, C.J.A.; Figdor, C.G. Migration of Dendritic Cell Based Cancer Vaccines: In Vivo Veritas? Curr. Opin. Immunol. 2005, 17, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Noubade, R.; Majri-Morrison, S.; Tarbell, K.V. Beyond CDC1: Emerging Roles of DC Crosstalk in Cancer Immunity. Front. Immunol. 2019, 10, 1014. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Banchereau, J. Taking Dendritic Cells into Medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef]
- Cai, L.; Xu, J.; Yang, Z.; Tong, R.; Dong, Z.; Wang, C.; Leong, K.W. Engineered Biomaterials for Cancer Immunotherapy. MedComm 2020, 1, 35–46. [Google Scholar] [CrossRef]
- Bachmann, M.F.; Jennings, G.T. Vaccine Delivery: A Matter of Size, Geometry, Kinetics and Molecular Patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef]
- Johansen, P.; Storni, T.; Rettig, L.; Qiu, Z.; Der-Sarkissian, A.; Smith, K.A.; Manolova, V.; Lang, K.S.; Senti, G.; Müllhaupt, B.; et al. Antigen Kinetics Determines Immune Reactivity. Proc. Natl. Acad. Sci. USA 2008, 105, 5189–5194. [Google Scholar] [CrossRef]
- Hawiger, D.; Inaba, K.; Dorsett, Y.; Guo, M.; Mahnke, K.; Rivera, M.; Ravetch, J.V.; Steinman, R.M.; Nussenzweig, M.C. Dendritic Cells Induce Peripheral T Cell Unresponsiveness under Steady State Conditions In Vivo. J. Exp. Med. 2001, 194, 769–780. [Google Scholar] [CrossRef]
- Ali, O.A.; Huebsch, N.; Cao, L.; Dranoff, G.; Mooney, D.J. Infection-Mimicking Materials to Program Dendritic Cells in Situ. Nat. Mater. 2009, 8, 151–158. [Google Scholar] [CrossRef]
- Gu, L.; Mooney, D.J. Biomaterials and Emerging Anticancer Therapeutics: Engineering the Microenvironment. Nat. Rev. Cancer 2016, 16, 56–66. [Google Scholar] [CrossRef]
- Gentile, P.; Chiono, V.; Carmagnola, I.; Hatton, P.V. An Overview of Poly(Lactic-Co-Glycolic) Acid (PLGA)-Based Biomaterials for Bone Tissue Engineering. Int. J. Mol. Sci. 2014, 15, 3640–3659. [Google Scholar] [CrossRef] [PubMed]
- van de Laar, L.; Coffer, P.J.; Woltman, A.M. Regulation of Dendritic Cell Development by GM-CSF: Molecular Control and Implications for Immune Homeostasis and Therapy. Blood 2012, 119, 3383–3393. [Google Scholar] [CrossRef] [PubMed]
- Ali, O.A.; Doherty, E.; Mooney, D.J.; Emerich, D. Relationship of Vaccine Efficacy to the Kinetics of DC and T-Cell Responses Induced by PLG-Based Cancer Vaccines. Biomatter 2011, 1, 66–75. [Google Scholar] [CrossRef]
- Ali, O.A.; Emerich, D.; Dranoff, G.; Mooney, D.J. In Situ Regulation of DC Subsets and T Cells Mediates Tumor Regression in Mice. Sci. Transl. Med. 2009, 1, 8ra19. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.; Murphy, K.M. Dendritic Cell Regulation of TH1-TH2 Development. Nat. Immunol. 2000, 1, 199–205. [Google Scholar] [CrossRef]
- den Haan, J.M.M.; Lehar, S.M.; Bevan, M.J. CD8+ but Not CD8− Dendritic Cells Cross-Prime Cytotoxic T Cells in Vivo. J. Exp. Med. 2000, 192, 1685–1696. [Google Scholar] [CrossRef]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Diamond, M.S.; et al. Batf3 Deficiency Reveals a Critical Role for CD8α+ Dendritic Cells in Cytotoxic T Cell Immunity. Science 2008, 322, 1097–1100. [Google Scholar] [CrossRef]
- Ali, O.A.; Verbeke, C.; Johnson, C.; Sands, R.W.; Lewin, S.A.; White, D.; Doherty, E.; Dranoff, G.; Mooney, D.J. Identification of Immune Factors Regulating Antitumor Immunity Using Polymeric Vaccines with Multiple Adjuvants. Cancer Res. 2014, 74, 1670–1681. [Google Scholar] [CrossRef]
- Shortman, K.; Heath, W.R. The CD8+ Dendritic Cell Subset. Immunol. Rev. 2010, 234, 18–31. [Google Scholar] [CrossRef]
- Quezada, S.A.; Peggs, K.S.; Curran, M.A.; Allison, J.P. CTLA4 Blockade and GM-CSF Combination Immunotherapy Alters the Intratumor Balance of Effector and Regulatory T Cells. J. Clin. Investig. 2006, 116, 1935–1945. [Google Scholar] [CrossRef]
- Ali, O.A.; Lewin, S.A.; Dranoff, G.; Mooney, D.J. Vaccines Combined with Immune Checkpoint Antibodies Promote Cytotoxic T-Cell Activity and Tumor Eradication. Cancer Immunol. Res. 2016, 4, 95–100. [Google Scholar] [CrossRef]
- Bencherif, S.A.; Sands, R.W.; Bhatta, D.; Arany, P.; Verbeke, C.S.; Edwards, D.A.; Mooney, D.J. Injectable Preformed Scaffolds with Shape-Memory Properties. Proc. Natl. Acad. Sci. USA 2012, 109, 19590–19595. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Qin, H.; Fernandez, I.; Wei, J.; Lin, J.; Kwak, L.W.; Roy, K. An Injectable Synthetic Immune-Priming Center Mediates Efficient T-Cell Class Switching and T-Helper 1 Response against B Cell Lymphoma. J. Control. Release 2011, 155, 184–192. [Google Scholar] [CrossRef]
- Shah, N.J.; Najibi, A.J.; Shih, T.Y.; Mao, A.S.; Sharda, A.; Scadden, D.T.; Mooney, D.J. A Biomaterial-Based Vaccine Eliciting Durable Tumour-Specific Responses against Acute Myeloid Leukaemia. Nat. Biomed. Eng. 2020, 4, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Henderson, T.M.A.; Ladewig, K.; Haylock, D.N.; McLean, K.M.; O’Connor, A.J. Cryogels for Biomedical Applications. J. Mater. Chem. B 2013, 1, 2682. [Google Scholar] [CrossRef]
- Hwang, Y.; Zhang, C.; Varghese, S. Poly(Ethylene Glycol) Cryogels as Potential Cell Scaffolds: Effect of Polymerization Conditions on Cryogel Microstructure and Properties. J. Mater. Chem. 2010, 20, 345–351. [Google Scholar] [CrossRef]
- Koshy, S.T.; Ferrante, T.C.; Lewin, S.A.; Mooney, D.J. Injectable, Porous, and Cell-Responsive Gelatin Cryogels. Biomaterials 2014, 35, 2477–2487. [Google Scholar] [CrossRef] [PubMed]
- Shih, T.Y.; Blacklow, S.O.; Li, A.W.; Freedman, B.R.; Bencherif, S.; Koshy, S.T.; Darnell, M.C.; Mooney, D.J. Injectable, Tough Alginate Cryogels as Cancer Vaccines. Adv. Healthc. Mater. 2018, 7, e1701469. [Google Scholar] [CrossRef] [PubMed]
- Augst, A.D.; Kong, H.J.; Mooney, D.J. Alginate Hydrogels as Biomaterials. Macromol. Biosci. 2006, 6, 623–633. [Google Scholar] [CrossRef]
- Najibi, A.J.; Shih, T.-Y.; Mooney, D.J. Cryogel Vaccines Effectively Induce Immune Responses Independent of Proximity to the Draining Lymph Nodes. Biomaterials 2022, 281, 121329. [Google Scholar] [CrossRef]
- Dellacherie, M.O.; Seo, B.R.; Mooney, D.J. Macroscale Biomaterials Strategies for Local Immunomodulation. Nat. Rev. Mater. 2019, 4, 379–397. [Google Scholar] [CrossRef]
- Adu-Berchie, K.; Mooney, D.J. Biomaterials as Local Niches for Immunomodulation. Acc. Chem. Res. 2020, 53, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Kukutsch, N.A.; Roßner, S.; Austyn, J.M.; Schuler, G.; Lutz, M.B. Formation and Kinetics of MHC Class I-Ovalbumin Peptide Complexes on Immature and Mature Murine Dendritic Cells. J. Investig. Dermatol. 2000, 115, 449–453. [Google Scholar] [CrossRef]
- Hori, Y.; Winans, A.M.; Huang, C.C.; Horrigan, E.M.; Irvine, D.J. Injectable Dendritic Cell-Carrying Alginate Gels for Immunization and Immunotherapy. Biomaterials 2008, 29, 3671–3682. [Google Scholar] [CrossRef] [PubMed]
- Bencherif, S.A.; Sands, R.W.; Ali, O.A.; Li, W.A.; Lewin, S.A.; Braschler, T.M.; Shih, T.Y.; Verbeke, C.S.; Bhatta, D.; Dranoff, G.; et al. Injectable Cryogel-Based Whole-Cell Cancer Vaccines. Nat. Commun. 2015, 6, 7556. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vacchelli, E.; Eggermont, A.; Fridman, W.H.; Galon, J.; Sautès-Fridman, C.; Tartour, E.; Zitvogel, L.; Kroemer, G. Trial Watch: Dendritic Cell-Based Interventions for Cancer Therapy. Oncoimmunology 2012, 1, 1111–1134. [Google Scholar] [CrossRef]
- Wang, H.; Mooney, D.J. Biomaterial-Assisted Targeted Modulation of Immune Cells in Cancer Treatment. Nat. Mater. 2018, 17, 761–772. [Google Scholar] [CrossRef]
- Wang, C.; Ye, Y.; Hu, Q.; Bellotti, A.; Gu, Z. Tailoring Biomaterials for Cancer Immunotherapy: Emerging Trends and Future Outlook. Adv. Mater. 2017, 29, 1606036. [Google Scholar] [CrossRef]
- Hu, Q.; Li, H.; Archibong, E.; Chen, Q.; Ruan, H.; Ahn, S.; Dukhovlinova, E.; Kang, Y.; Wen, D.; Dotti, G.; et al. Inhibition of Post-Surgery Tumour Recurrence via a Hydrogel Releasing CAR-T Cells and Anti-PDL1-Conjugated Platelets. Nat. Biomed. Eng. 2021, 5, 1038–1047. [Google Scholar] [CrossRef]
- Guo, Y.; Xie, Y.-Q.; Gao, M.; Zhao, Y.; Franco, F.; Wenes, M.; Siddiqui, I.; Bevilacqua, A.; Wang, H.; Yang, H.; et al. Metabolic Reprogramming of Terminally Exhausted CD8+ T Cells by IL-10 Enhances Anti-Tumor Immunity. Nat. Immunol. 2021, 22, 746–756. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3-Dioxygenase and Tumor-Induced Tolerance. J. Clin. Investig. 2007, 117, 1147–1154. [Google Scholar] [CrossRef]
- Weber, J.S.; Mulé, J.J. Cancer Immunotherapy Meets Biomaterials. Nat. Biotechnol. 2015, 33, 44–45. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Bhatta, R.; Liu, Y.; Bo, Y.; Wang, H. In Situ Dendritic Cell Recruitment and T Cell Activation for Cancer Immunotherapy. Front. Pharmacol. 2022, 13, 954955. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus Pembrolizumab versus Placebo plus Pembrolizumab in Patients with Unresectable or Metastatic Melanoma (ECHO-301/KEYNOTE-252): A Phase 3, Randomised, Double-Blind Study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef] [PubMed]
- Van den Eynde, B.J.; van Baren, N.; Baurain, J.-F. Is There a Clinical Future for IDO1 Inhibitors After the Failure of Epacadostat in Melanoma? Annu. Rev. Cancer Biol. 2020, 4, 241–256. [Google Scholar] [CrossRef]
- Campesato, L.F.; Budhu, S.; Tchaicha, J.; Weng, C.-H.; Gigoux, M.; Cohen, I.J.; Redmond, D.; Mangarin, L.; Pourpe, S.; Liu, C.; et al. Blockade of the AHR Restricts a Treg-Macrophage Suppressive Axis Induced by L-Kynurenine. Nat. Commun. 2020, 11, 4011. [Google Scholar] [CrossRef]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef]
- Wang, B.; Chen, J.; Caserto, J.S.; Wang, X.; Ma, M. An In Situ Hydrogel-Mediated Chemo-Immunometabolic Cancer Therapy. Nat. Commun. 2022, 13, 3821. [Google Scholar] [CrossRef]
- Zeng, Z.; Zhang, C.; Li, J.; Cui, D.; Jiang, Y.; Pu, K. Activatable Polymer Nanoenzymes for Photodynamic Immunometabolic Cancer Therapy. Adv. Mater. 2021, 33, 2007247. [Google Scholar] [CrossRef]
- Triplett, T.A.; Garrison, K.C.; Marshall, N.; Donkor, M.; Blazeck, J.; Lamb, C.; Qerqez, A.; Dekker, J.D.; Tanno, Y.; Lu, W.-C.; et al. Reversal of Indoleamine 2,3-Dioxygenase–Mediated Cancer Immune Suppression by Systemic Kynurenine Depletion with a Therapeutic Enzyme. Nat. Biotechnol. 2018, 36, 758–764. [Google Scholar] [CrossRef]
- Wang, H.; Najibi, A.J.; Sobral, M.C.; Seo, B.R.; Lee, J.Y.; Wu, D.; Li, A.W.; Verbeke, C.S.; Mooney, D.J. Biomaterial-Based Scaffold for in Situ Chemo-Immunotherapy to Treat Poorly Immunogenic Tumors. Nat. Commun. 2020, 11, 5696. [Google Scholar] [CrossRef] [PubMed]
- Li, A.W.; Sobral, M.C.; Badrinath, S.; Choi, Y.; Graveline, A.; Stafford, A.G.; Weaver, J.C.; Dellacherie, M.O.; Shih, T.-Y.; Ali, O.A.; et al. A Facile Approach to Enhance Antigen Response for Personalized Cancer Vaccination. Nat. Mater. 2018, 17, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Dellacherie, M.O.; Li, A.; Lu, B.Y.; Verbeke, C.S.; Gu, L.; Stafford, A.G.; Doherty, E.J.; Mooney, D.J. Single-Shot Mesoporous Silica Rods Scaffold for Induction of Humoral Responses Against Small Antigens. Adv. Funct. Mater. 2020, 30, 2002448. [Google Scholar] [CrossRef]
- Kang, B.H.; Lee, H.K. Dendritic Cell-Based Immunotherapy in Hot and Cold Tumors. Int. J. Mol. Sci. 2022, 23, 7325. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to Treat Immune Hot, Altered and Cold Tumours with Combination Immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef]
- Whiteside, T.L.; Demaria, S.; Rodriguez-Ruiz, M.E.; Zarour, H.M.; Melero, I. Emerging Opportunities and Challenges in Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 1845–1855. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Hett, E.; Kroemer, G.; Marincola, F.M. Immunogenic Cell Death in Cancer: Concept and Therapeutic Implications. J. Transl. Med. 2023, 21, 162. [Google Scholar] [CrossRef]
- Jin, M.Z.; Wang, X.P. Immunogenic Cell Death-Based Cancer Vaccines. Front. Immunol. 2021, 12, 697964. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus Guidelines for the Definition, Detection and Interpretation of Immunogenic Cell Death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of Immunogenic Cell Death and Its Relevance for Cancer Therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zhu, G.; Wang, S.; Yu, G.; Yang, Z.; Lin, L.; Zhou, Z.; Liu, Y.; Dai, Y.; Zhang, F.; et al. In Situ Dendritic Cell Vaccine for Effective Cancer Immunotherapy. ACS Nano 2019, 13, 3083–3094. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic Cell Death in Cancer and Infectious Disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Chargari, C.; Galluzzi, L.; Kroemer, G. Optimising Efficacy and Reducing Toxicity of Anticancer Radioimmunotherapy. Lancet Oncol. 2019, 20, e452–e463. [Google Scholar] [CrossRef] [PubMed]
- Nomura, S.; Morimoto, Y.; Tsujimoto, H.; Arake, M.; Harada, M.; Saitoh, D.; Hara, I.; Ozeki, E.; Satoh, A.; Takayama, E.; et al. Highly Reliable, Targeted Photothermal Cancer Therapy Combined with Thermal Dosimetry Using a near-Infrared Absorbent. Sci. Rep. 2020, 10, 9765. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, X.; Wang, X.; Guan, X.; Zhang, W.; Ma, J. Recent Advances in Selective Photothermal Therapy of Tumor. J. Nanobiotechnol. 2021, 19, 335. [Google Scholar] [CrossRef]
- Bloy, N.; Garcia, P.; Laumont, C.M.; Pitt, J.M.; Sistigu, A.; Stoll, G.; Yamazaki, T.; Bonneil, E.; Buqué, A.; Humeau, J.; et al. Immunogenic Stress and Death of Cancer Cells: Contribution of Antigenicity vs Adjuvanticity to Immunosurveillance. Immunol. Rev. 2017, 280, 165–174. [Google Scholar] [CrossRef]
- Zitvogel, L.; Kepp, O.; Kroemer, G. Decoding Cell Death Signals in Inflammation and Immunity. Cell 2010, 140, 798–804. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Alzeibak, R.; Mishchenko, T.A.; Shilyagina, N.Y.; Balalaeva, I.V.; Vedunova, M.V.; Krysko, D.V. Targeting Immunogenic Cancer Cell Death by Photodynamic Therapy: Past, Present and Future. J. Immunother. Cancer 2021, 9, e001926. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Vitale, I.; Sistigu, A.; Manic, G.; Rudqvist, N.-P.; Trajanoski, Z.; Galluzzi, L. Mutational and Antigenic Landscape in Tumor Progression and Cancer Immunotherapy. Trends Cell Biol. 2019, 29, 396–416. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic Cell Death and DAMPs in Cancer Therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Yatim, N.; Cullen, S.; Albert, M.L. Dying Cells Actively Regulate Adaptive Immune Responses. Nat. Rev. Immunol. 2017, 17, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Moserova, I.; Urbanova, L.; Bezu, L.; Kepp, O.; Cremer, I.; Salek, C.; Strnad, P.; Kroemer, G.; Galluzzi, L.; et al. Prognostic and Predictive Value of DAMPs and DAMP-Associated Processes in Cancer. Front. Immunol. 2015, 6, 402. [Google Scholar] [CrossRef]
- Hayashi, K.; Nikolos, F.; Lee, Y.C.; Jain, A.; Tsouko, E.; Gao, H.; Kasabyan, A.; Leung, H.E.; Osipov, A.; Jung, S.Y.; et al. Tipping the Immunostimulatory and Inhibitory DAMP Balance to Harness Immunogenic Cell Death. Nat. Commun. 2020, 11, 6299. [Google Scholar] [CrossRef]
- Garg, A.D.; Galluzzi, L.; Apetoh, L.; Baert, T.; Birge, R.B.; Bravo-San Pedro, J.M.; Breckpot, K.; Brough, D.; Chaurio, R.; Cirone, M.; et al. Molecular and Translational Classifications of DAMPs in Immunogenic Cell Death. Front. Immunol. 2015, 6, 588. [Google Scholar] [CrossRef]
- Matzinger, P. The Danger Model: A Renewed Sense of Self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef]
- Vedunova, M.; Turubanova, V.; Vershinina, O.; Savyuk, M.; Efimova, I.; Mishchenko, T.; Raedt, R.; Vral, A.; Vanhove, C.; Korsakova, D.; et al. DC Vaccines Loaded with Glioma Cells Killed by Photodynamic Therapy Induce Th17 Anti-Tumor Immunity and Provide a Four-Gene Signature for Glioma Prognosis. Cell Death Dis. 2022, 13, 1062. [Google Scholar] [CrossRef]
- Wang, X.; Ji, J.; Zhang, H.; Fan, Z.; Zhang, L.; Shi, L.; Zhou, F.; Chen, W.R.; Wang, H.; Wang, X. Stimulation of Dendritic Cells by DAMPs in ALA-PDT Treated SCC Tumor Cells. Oncotarget 2015, 6, 44688–44702. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galassi, C.; Zitvogel, L.; Galluzzi, L. Immunogenic Cell Stress and Death. Nat. Immunol. 2022, 23, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin Exposure Dictates the Immunogenicity of Cancer Cell Death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Fosco, D.; Kline, D.E.; Kline, J. Calreticulin Promotes Immunity and Type I Interferon-Dependent Survival in Mice with Acute Myeloid Leukemia. Oncoimmunology 2017, 6, e1278332. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Wang, Y.; Michaud, M.; Ma, Y.; Sukkurwala, A.Q.; Shen, S.; Kepp, O.; Métivier, D.; Galluzzi, L.; Perfettini, J.-L.; et al. Molecular Mechanisms of ATP Secretion during Immunogenic Cell Death. Cell Death Differ. 2014, 21, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Portela Catani, J.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer Chemotherapy-Induced Intratumoral Recruitment and Differentiation of Antigen-Presenting Cells. Immunity 2013, 38, 729–741. [Google Scholar] [CrossRef]
- Schmitt, E.; Gehrmann, M.; Brunet, M.; Multhoff, G.; Garrido, C. Intracellular and Extracellular Functions of Heat Shock Proteins: Repercussions in Cancer Therapy. J. Leukoc. Biol. 2007, 81, 15–27. [Google Scholar] [CrossRef]
- Fucikova, J.; Kralikova, P.; Fialova, A.; Brtnicky, T.; Rob, L.; Bartunkova, J.; Špíšek, R. Human Tumor Cells Killed by Anthracyclines Induce a Tumor-Specific Immune Response. Cancer Res. 2011, 71, 4821–4833. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like Receptor 4-Dependent Contribution of the Immune System to Anticancer Chemotherapy and Radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Saenz, R.; Futalan, D.; Leutenez, L.; Eekhout, F.; Fecteau, J.F.; Sundelius, S.; Sundqvist, S.; Larsson, M.; Hayashi, T.; Minev, B.; et al. TLR4-Dependent Activation of Dendritic Cells by an HMGB1-Derived Peptide Adjuvant. J. Transl. Med. 2014, 12, 211. [Google Scholar] [CrossRef]
- Green, D.R.; Ferguson, T.; Zitvogel, L.; Kroemer, G. Immunogenic and Tolerogenic Cell Death. Nat. Rev. Immunol. 2009, 9, 353–363. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef]
- Reynders, K.; Illidge, T.; Siva, S.; Chang, J.Y.; De Ruysscher, D. The Abscopal Effect of Local Radiotherapy: Using Immunotherapy to Make a Rare Event Clinically Relevant. Cancer Treat. Rev. 2015, 41, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Systemic Effects of Local Radiotherapy. Lancet Oncol. 2009, 10, 718–726. [Google Scholar] [CrossRef]
- Xu, J.; Escamilla, J.; Mok, S.; David, J.; Priceman, S.; West, B.; Bollag, G.; McBride, W.; Wu, L. CSF1R Signaling Blockade Stanches Tumor-Infiltrating Myeloid Cells and Improves the Efficacy of Radiotherapy in Prostate Cancer. Cancer Res. 2013, 73, 2782–2794. [Google Scholar] [CrossRef] [PubMed]
- Bos, P.D.; Plitas, G.; Rudra, D.; Lee, S.Y.; Rudensky, A.Y. Transient Regulatory T Cell Ablation Deters Oncogene-Driven Breast Cancer and Enhances Radiotherapy. J. Exp. Med. 2013, 210, 2435–2466. [Google Scholar] [CrossRef]
- Demaria, S.; Golden, E.B.; Formenti, S.C. Role of Local Radiation Therapy in Cancer Immunotherapy. JAMA Oncol. 2015, 1, 1325–1332. [Google Scholar] [CrossRef]
- Haroun, R.; Naasri, S.; Oweida, A.J. Toll-Like Receptors and the Response to Radiotherapy in Solid Tumors: Challenges and Opportunities. Vaccines 2023, 11, 818. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Marron, T.U.; Upadhyay, R.; Svensson-Arvelund, J.; Dhainaut, M.; Hussein, S.; Zhan, Y.; Ostrowski, D.; Yellin, M.; Marsh, H.; et al. Systemic Clinical Tumor Regressions and Potentiation of PD1 Blockade with in Situ Vaccination. Nat. Med. 2019, 25, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Oba, T.; Kajihara, R.; Yokoi, T.; Repasky, E.A.; Ito, F. Neoadjuvant in Situ Immunomodulation Enhances Systemic Antitumor Immunity against Highly Metastatic Tumors. Cancer Res. 2021, 81, 6183–6195. [Google Scholar] [CrossRef]
- Marron, T.; Fasano, J.; Doroshow, D.; Ostrowski, D.; Sorich, J.; Van-Voorthuysen, M.; Coffey, J.; Moshier, E.; Salazar, A.; Yellin, M.; et al. Flt3L-Primed in Situ Vaccination and Pembrolizumab Induce Systemic Tumor Regressions of Bulky Tumors in Patients with Lymphomas and ER/PR+ Breast Cancer. In Regular and Young Investigator Award Abstracts; BMJ Publishing Group Ltd.: London, UK, 2022; Volume 25, p. A622. [Google Scholar]
- Frank, M.J.; Reagan, P.M.; Bartlett, N.L.; Gordon, L.I.; Friedberg, J.W.; Czerwinski, D.K.; Long, S.R.; Hoppe, R.T.; Janssen, R.; Candia, A.F.; et al. In Situ Vaccination with a TLR9 Agonist and Local Low-Dose Radiation Induces Systemic Responses in Untreated Indolent Lymphoma. Cancer Discov. 2018, 8, 1258–1269. [Google Scholar] [CrossRef] [PubMed]
- Brody, J.D.; Ai, W.Z.; Czerwinski, D.K.; Torchia, J.A.; Levy, M.; Advani, R.H.; Kim, Y.H.; Hoppe, R.T.; Knox, S.J.; Shin, L.K.; et al. In Situ Vaccination with a TLR9 Agonist Induces Systemic Lymphoma Regression: A Phase I/II Study. J. Clin. Oncol. 2010, 28, 4324–4332. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; van den Eertwegh, A.J.M.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H.; et al. Ipilimumab versus Placebo after Radiotherapy in Patients with Metastatic Castration-Resistant Prostate Cancer That Had Progressed after Docetaxel Chemotherapy (CA184-043): A Multicentre, Randomised, Double-Blind, Phase 3 Trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef]
- Slovin, S.F.; Higano, C.S.; Hamid, O.; Tejwani, S.; Harzstark, A.; Alumkal, J.J.; Scher, H.I.; Chin, K.; Gagnier, P.; McHenry, M.B.; et al. Ipilimumab Alone or in Combination with Radiotherapy in Metastatic Castration-Resistant Prostate Cancer: Results from an Open-Label, Multicenter Phase I/II Study. Ann. Oncol. 2013, 24, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The Future of Immune Checkpoint Therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Formenti, S.C.; Rudqvist, N.-P.; Golden, E.; Cooper, B.; Wennerberg, E.; Lhuillier, C.; Vanpouille-Box, C.; Friedman, K.; Ferrari de Andrade, L.; Wucherpfennig, K.W.; et al. Radiotherapy Induces Responses of Lung Cancer to CTLA-4 Blockade. Nat. Med. 2018, 24, 1845–1851. [Google Scholar] [CrossRef]
- Dolmans, D.E.J.G.J.; Fukumura, D.; Jain, R.K. Photodynamic Therapy for Cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic Therapy of Cancer: An Update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Niedre, M.J.; Yu, C.S.; Patterson, M.S.; Wilson, B.C. Singlet Oxygen Luminescence as an in Vivo Photodynamic Therapy Dose Metric: Validation in Normal Mouse Skin with Topical Amino-Levulinic Acid. Br. J. Cancer 2005, 92, 298–304. [Google Scholar] [CrossRef]
- Turubanova, V.D.; Balalaeva, I.V.; Mishchenko, T.A.; Catanzaro, E.; Alzeibak, R.; Peskova, N.N.; Efimova, I.; Bachert, C.; Mitroshina, E.V.; Krysko, O.; et al. Immunogenic Cell Death Induced by a New Photodynamic Therapy Based on Photosens and Photodithazine. J. Immunother. Cancer 2019, 7, 350. [Google Scholar] [CrossRef]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The Two Faces of IL-6 in the Tumor Microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-Tumor Immunity. Cell Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-Dependent Immunogenicity of Doxorubicin-Induced Tumor Cell Death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Shen, G.; Gao, W.; Huang, Z.; Huang, C.; Fu, L. Neoantigens: Promising Targets for Cancer Therapy. Signal Transduct. Target. Ther. 2023, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, S.R.; Nave, H.; Korangy, F.; Schlote, K.; Pabst, R.; Jaffee, E.M.; Manns, M.P.; Greten, T.F. Apoptotic, but Not Necrotic, Tumor Cell Vaccines Induce a Potent Immune Response In Vivo. Int. J. Cancer 2003, 103, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Gamrekelashvili, J.; Krüger, C.; von Wasielewski, R.; Hoffmann, M.; Huster, K.M.; Busch, D.H.; Manns, M.P.; Korangy, F.; Greten, T.F. Necrotic Tumor Cell Death In Vivo Impairs Tumor-Specific Immune Responses. J. Immunol. 2007, 178, 1573–1580. [Google Scholar] [CrossRef]
- Bartholomae, W.C.; Rininsland, F.H.; Eisenberg, J.C.; Boehm, B.O.; Lehmann, P.V.; Tary-Lehmann, M. T Cell Immunity Induced by Live, Necrotic, and Apoptotic Tumor Cells 1. J. Immunol. 2004, 173, 1012–1022. [Google Scholar] [CrossRef]
- Gamrekelashvili, J.; Greten, T.F.; Korangy, F. Immunogenicity of Necrotic Cell Death. Cell. Mol. Life Sci. 2015, 72, 273–283. [Google Scholar] [CrossRef]
- Harari, A.; Graciotti, M.; Bassani-Sternberg, M.; Kandalaft, L.E. Antitumour Dendritic Cell Vaccination in a Priming and Boosting Approach. Nat. Rev. Drug Discov. 2020, 19, 635–652. [Google Scholar] [CrossRef]
- Galluzzi, L.; Humeau, J.; Buqué, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with Chemotherapy in the Era of Immune Checkpoint Inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef]
- Luo, M.; Wang, H.; Wang, Z.; Cai, H.; Lu, Z.; Li, Y.; Du, M.; Huang, G.; Wang, C.; Chen, X.; et al. A STING-Activating Nanovaccine for Cancer Immunotherapy. Nat. Nanotechnol. 2017, 12, 648–654. [Google Scholar] [CrossRef]
- Tugues, S.; Burkhard, S.H.; Ohs, I.; Vrohlings, M.; Nussbaum, K.; vom Berg, J.; Kulig, P.; Becher, B. New Insights into IL-12-Mediated Tumor Suppression. Cell Death Differ. 2015, 22, 237–246. [Google Scholar] [CrossRef]
- Zakharova, M.; Ziegler, H.K. Paradoxical Anti-Inflammatory Actions of TNF-α: Inhibition of IL-12 and IL-23 via TNF Receptor 1 in Macrophages and Dendritic Cells. J. Immunol. 2005, 175, 5024–5033. [Google Scholar] [CrossRef] [PubMed]
- Han, H.S.; Choi, K.Y. Advances in Nanomaterial-mediated Photothermal Cancer Therapies: Toward Clinical Applications. Biomedicines 2021, 9, 305. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wang, C.; Cui, W.; Gong, H.; Liang, C.; Shi, X.; Li, Z.; Sun, B.; Liu, Z. Combined Photothermal and Photodynamic Therapy Delivered by PEGylated MoS 2 Nanosheets. Nanoscale 2014, 6, 11219–11225. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Xiao, Y.; Li, W.; Yang, Q.; Tan, L.; Jia, Y.; Qu, Y.; Qian, Z. Photosensitizer Micelles Together with IDO Inhibitor Enhance Cancer Photothermal Therapy and Immunotherapy. Adv. Sci. 2018, 5, 1700891. [Google Scholar] [CrossRef]
- Huang, Q.; Zhang, S.; Zhang, H.; Han, Y.; Liu, H.; Ren, F.; Sun, Q.; Li, Z.; Gao, M. Boosting the Radiosensitizing and Photothermal Performance of Cu 2– x Se Nanocrystals for Synergetic Radiophotothermal Therapy of Orthotopic Breast Cancer. ACS Nano 2019, 13, 1342–1353. [Google Scholar] [CrossRef]
- Nam, J.; Son, S.; Ochyl, L.J.; Kuai, R.; Schwendeman, A.; Moon, J.J. Chemo-Photothermal Therapy Combination Elicits Anti-Tumor Immunity against Advanced Metastatic Cancer. Nat. Commun. 2018, 9, 1074. [Google Scholar] [CrossRef]
- Ng, C.W.; Li, J.; Pu, K. Recent Progresses in Phototherapy-Synergized Cancer Immunotherapy. Adv. Funct. Mater. 2018, 28, 1804688. [Google Scholar] [CrossRef]
- Wang, C.; Xu, L.; Liang, C.; Xiang, J.; Peng, R.; Liu, Z. Immunological Responses Triggered by Photothermal Therapy with Carbon Nanotubes in Combination with Anti-CTLA-4 Therapy to Inhibit Cancer Metastasis. Adv. Mater. 2014, 26, 8154–8162. [Google Scholar] [CrossRef]
- Zhou, F.; Yang, J.; Zhang, Y.; Liu, M.; Lang, M.L.; Li, M.; Chen, W.R. Local Phototherapy Synergizes with Immunoadjuvant for Treatment of Pancreatic Cancer through Induced Immunogenic Tumor Vaccine. Clin. Cancer Res. 2018, 24, 5335–5346. [Google Scholar] [CrossRef]
- Chen, Q.; Xu, L.; Liang, C.; Wang, C.; Peng, R.; Liu, Z. Photothermal Therapy with Immune-Adjuvant Nanoparticles Together with Checkpoint Blockade for Effective Cancer Immunotherapy. Nat. Commun. 2016, 7, 13193. [Google Scholar] [CrossRef]
- Li, X.; Lovell, J.F.; Yoon, J.; Chen, X. Clinical Development and Potential of Photothermal and Photodynamic Therapies for Cancer. Nat. Rev. Clin. Oncol. 2020, 17, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Z.; Zhao, X.; Song, X.R. Ex Vivo Pulsed Dendritic Cell Vaccination against Cancer. Acta Pharmacol. Sin. 2020, 41, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Bol, K.F.; Schreibelt, G.; Rabold, K.; Wculek, S.K.; Schwarze, J.K.; Dzionek, A.; Teijeira, A.; Kandalaft, L.E.; Romero, P.; Coukos, G.; et al. The Clinical Application of Cancer Immunotherapy Based on Naturally Circulating Dendritic Cells. J. Immunother. Cancer 2019, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Erdmann, M.; Haendle, I.; Voland, S.; Berger, T.; Schultz, E.; Strasser, E.; Dankerl, P.; Janka, R.; Schliep, S.; et al. Twelve-Year Survival and Immune Correlates in Dendritic Cell–Vaccinated Melanoma Patients. JCI Insight 2017, 2, e91438. [Google Scholar] [CrossRef]
- Benteyn, D.; Heirman, C.; Bonehill, A.; Thielemans, K.; Breckpot, K. MRNA-Based Dendritic Cell Vaccines. Expert. Rev. Vaccines 2014, 14, 161–176. [Google Scholar] [CrossRef]
- Aarntzen, E.H.J.G.; Schreibelt, G.; Bol, K.; Lesterhuis, W.J.; Croockewit, A.J.; De Wilt, J.H.W.; Van Rossum, M.M.; Blokx, W.A.M.; Jacobs, J.F.M.; Duiveman-De Boer, T.; et al. Vaccination with MRNA-Electroporated Dendritic Cells Induces Robust Tumor Antigen-Specific CD4+ and CD8+ T Cells Responses in Stage III and IV Melanoma Patients. Clin. Cancer Res. 2012, 18, 5460–5470. [Google Scholar] [CrossRef]
- Beck, J.D.; Reidenbach, D.; Salomon, N.; Sahin, U.; Türeci, Ö.; Vormehr, M.; Kranz, L.M. MRNA Therapeutics in Cancer Immunotherapy. Mol. Cancer 2021, 20, 69. [Google Scholar] [CrossRef]
- Van Lint, S.; Heirman, C.; Thielemans, K.; Breckpot, K. mRNA: From a Chemical Blueprint for Protein Production to an Off-the-Shelf Therapeutic. Hum. Vaccines Immunother. 2013, 9, 265–274. [Google Scholar] [CrossRef]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of Double-Stranded RNA and Activation of NF-ΚB by Toll-like Receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Pastor, F.; Berraondo, P.; Etxeberria, I.; Frederick, J.; Sahin, U.; Gilboa, E.; Melero, I. An RNA Toolbox for Cancer Immunotherapy. Nat. Rev. Drug Discov. 2018, 17, 751–767. [Google Scholar] [CrossRef] [PubMed]
- Boczkowski, D.; Nair, S.K.; Snyder, D.; Gilboa, E. Dendritic Cells Pulsed with RNA Are Potent Antigen-Presenting Cells in Vitro and in Vivo. J. Exp. Med. 1996, 184, 465–472. [Google Scholar] [CrossRef]
- Müller, M.R.; Tsakou, G.; Grünebach, F.; Schmidt, S.M.; Brossart, P. Induction of Chronic Lymphocytic Leukemia (CLL)-Specific CD4- and CD8-Mediated T-Cell Responses Using RNA-Transfected Dendritic Cells. Blood 2004, 103, 1763–1769. [Google Scholar] [CrossRef]
- Milazzo, C.; Reichardt, V.L.; Müller, M.R.; Grünebach, F.; Brossart, P. Induction of Myeloma-Specific Cytotoxic T Cells Using Dendritic Cells Transfected with Tumor-Derived RNA. Blood 2003, 101, 977–982. [Google Scholar] [CrossRef][Green Version]
- Nencioni, A.; Müller, M.R.; Grünebach, F.; Garuti, A.; Mingari, M.C.; Patrone, F.; Ballestrero, A.; Brossart, P. Dendritic Cells Transfected with Tumor RNA for the Induction of Antitumor CTL in Colorectal Cancer. Cancer Gene Ther. 2003, 10, 209–214. [Google Scholar] [CrossRef][Green Version]
- Nair, S.K.; Morse, M.; Boczkowski, D.; Ian Cumming, R.; Vasovic, L.; Gilboa, E.; Kim Lyerly, H. Induction of Tumor-Specific Cytotoxic T Lymphocytes in Cancer Patients by Autologous Tumor RNA-Transfected Dendritic Cells. Ann. Surg. 2002, 235, 540–549. [Google Scholar] [CrossRef]
- Gilboa, E.; Vieweg, J. Cancer Immunotherapy with MRNA-Transfected Dendritic Cells. Immunol. Rev. 2004, 199, 251–263. [Google Scholar] [CrossRef]
- Van Tendeloo, V.F.I.; Ponsaerts, P.; Lardon, F.; Nijs, G.; Lenjou, M.; Van Broeckhoven, C.; Van Bockstaele, D.R.; Berneman, Z.N. Highly Efficient Gene Delivery by MRNA Electroporation in Human Hematopoietic Cells: Superiority to Lipofection and Passive Pulsing of MRNA and to Electroporation of Plasmid CDNA for Tumor Antigen Loading of Dendritic Cells. Blood 2001, 98, 49–56. [Google Scholar] [CrossRef]
- Van Nuffel, A.M.; Corthals, J.; Neyns, B.; Heirman, C.; Thielemans, K.; Bonehill, A. Immunotherapy of Cancer with Dendritic Cells Loaded with Tumor Antigens and Activated through MRNA Electroporation. Methods Mol. Biol. 2010, 629, 405–452. [Google Scholar] [CrossRef] [PubMed]
- Gerer, K.F.; Hoyer, S.; Dörrie, J.; Schaft, N. Electroporation of Mrna as Universal Technology Platform to Transfect a Variety of Primary Cells with Antigens and Functional Proteins. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1499, pp. 165–178. [Google Scholar]
- Markovic, S.N.; Dietz, A.B.; Greiner, C.W.; Maas, M.L.; Butler, G.W.; Padley, D.J.; Bulur, P.A.; Allred, J.B.; Creagan, E.T.; Ingle, J.N.; et al. Preparing Clinical-Grade Myeloid Dendritic Cells by Electroporation-Mediated Transfection of in Vitro Amplified Tumor-Derived MRNA and Safety Testing in Stage IV Malignant Melanoma. J. Transl. Med. 2006, 4, 35. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kyte, J.A.; Mu, L.; Aamdal, S.; Kvalheim, G.; Dueland, S.; Hauser, M.; Gullestad, H.P.; Ryder, T.; Lislerud, K.; Hammerstad, H.; et al. Phase I/II Trial of Melanoma Therapy with Dendritic Cells Transfected with Autologous Tumor-MRNA. Cancer Gene Ther. 2006, 13, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Dannull, J.; Heiser, A.; Yancey, D.; Pruitt, S.; Madden, J.; Coleman, D.; Niedzwiecki, D.; Gilboa, E.; Vieweg, J. Immunological and Clinical Responses in Metastatic Renal Cancer Patients Vaccinated with Tumor RNA-Transfected Dendritic Cells. Cancer Res. 2003, 63, 2127–2133. [Google Scholar]
- Kyte, J.A.; Aamdal, S.; Dueland, S.; Sæbøe-Larsen, S.; Inderberg, E.M.; Madsbu, U.E.; Skovlund, E.; Gaudernack, G.; Kvalheim, G. Immune Response and Long-Term Clinical Outcome in Advanced Melanoma Patients Vaccinated with Tumor-MRNA-Transfected Dendritic Cells. Oncoimmunology 2016, 5, e1232237. [Google Scholar] [CrossRef] [PubMed]
- Javorovic, M.; Pohla, H.; Frankenberger, B.; Wölfel, T.; Schendel, D.J. RNA Transfer by Electroporation into Mature Dendritic Cells Leading to Reactivation of Effector-Memory Cytotoxic T Lymphocytes: A Quantitative Analysis. Mol. Ther. 2005, 12, 734–743. [Google Scholar] [CrossRef]
- Anguille, S.; Van de Velde, A.L.; Smits, E.L.; Van Tendeloo, V.F.; Juliusson, G.; Cools, N.; Nijs, G.; Stein, B.; Lion, E.; Van Driessche, A.; et al. Dendritic Cell Vaccination as Postremission Treatment to Prevent or Delay Relapse in Acute Myeloid Leukemia. Blood 2017, 130, 1713–1721. [Google Scholar] [CrossRef]
- Kongsted, P.; Borch, T.H.; Ellebaek, E.; Iversen, T.Z.; Andersen, R.; Met, Ö.; Hansen, M.; Lindberg, H.; Sengeløv, L.; Svane, I.M. Dendritic Cell Vaccination in Combination with Docetaxel for Patients with Metastatic Castration-Resistant Prostate Cancer: A Randomized Phase II Study. Cytotherapy 2017, 19, 500–513. [Google Scholar] [CrossRef]
- Bol, K.F.; Aarntzen, E.H.J.G.; in ’t Hout, F.E.M.; Schreibelt, G.; Creemers, J.H.A.; Lesterhuis, W.J.; Gerritsen, W.R.; Grunhagen, D.J.; Verhoef, C.; Punt, C.J.A.; et al. Favorable Overall Survival in Stage III Melanoma Patients after Adjuvant Dendritic Cell Vaccination. Oncoimmunology 2016, 5, e1057673. [Google Scholar] [CrossRef]
- Boudewijns, S.; Bloemendal, M.; de Haas, N.; Westdorp, H.; Bol, K.F.; Schreibelt, G.; Aarntzen, E.H.J.G.; Lesterhuis, W.J.; Gorris, M.A.J.; Croockewit, A.; et al. Autologous Monocyte-Derived DC Vaccination Combined with Cisplatin in Stage III and IV Melanoma Patients: A Prospective, Randomized Phase 2 Trial. Cancer Immunol. Immunother. 2020, 69, 477–488. [Google Scholar] [CrossRef]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, Ö.; Sahin, U. Modification of Antigen-Encoding RNA Increases Stability, Translational Efficacy, and T-Cell Stimulatory Capacity of Dendritic Cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef]
- Galaine, J.; Borg, C.; Godet, Y.; Adotévi, O. Interest of Tumor-Specific CD4 T Helper 1 Cells for Therapeutic Anticancer Vaccine. Vaccines 2015, 3, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, B.J.; Craft, J.E.; Kaech, S.M. The Multifaceted Role of CD4+ T Cells in CD8+ T Cell Memory. Nat. Rev. Immunol. 2016, 16, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Ahrends, T.; Busselaar, J.; Severson, T.M.; Bąbała, N.; de Vries, E.; Bovens, A.; Wessels, L.; van Leeuwen, F.; Borst, J. CD4+ T Cell Help Creates Memory CD8+ T Cells with Innate and Help-Independent Recall Capacities. Nat. Commun. 2019, 10, 5531. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Vieweg, J.; Weizer, A.Z.; Dahm, P.; Yancey, D.; Turaga, V.; Higgins, J.; Boczkowski, D.; Gilboa, E.; Dannull, J. Enhanced Induction of Telomerase-Specific CD4+ T Cells Using Dendritic Cells Transfected with RNA Encoding a Chimeric Gene Product. Cancer Res. 2002, 62, 5041–5048. [Google Scholar]
- Benteyn, D.; Anguille, S.; Van Lint, S.; Heirman, C.; Van Nuffel, A.M.T.; Corthals, J.; Ochsenreither, S.; Waelput, W.; Van Beneden, K.; Breckpot, K.; et al. Design of an Optimized Wilms’ Tumor 1 (WT1) MRNA Construct for Enhanced WT1 Expression and Improved Immunogenicity in Vitro and in Vivo. Mol. Ther. Nucleic Acids 2013, 2, e134. [Google Scholar] [CrossRef]
- Kreiter, S.; Selmi, A.; Diken, M.; Sebastian, M.; Osterloh, P.; Schild, H.; Huber, C.; Türeci, Ö.; Sahin, U. Increased Antigen Presentation Efficiency by Coupling Antigens to MHC Class I Trafficking Signals. J. Immunol. 2008, 180, 309–318. [Google Scholar] [CrossRef]
- Bonehill, A.; Heirman, C.; Tuyaerts, S.; Michiels, A.; Breckpot, K.; Brasseur, F.; Zhang, Y.; van der Bruggen, P.; Thielemans, K. Messenger RNA-Electroporated Dendritic Cells Presenting MAGE-A3 Simultaneously in HLA Class I and Class II Molecules. J. Immunol. 2004, 172, 6649–6657. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, Y.; Huang, L. MRNA Vaccine for Cancer Immunotherapy. Mol. Cancer 2021, 20, 41. [Google Scholar] [CrossRef]
- Batich, K.A.; Mitchell, D.A.; Healy, P.; Herndon, J.E.; Sampson, J.H. Once, Twice, Three Times a Finding: Reproducibility of Dendritic Cell Vaccine Trials Targeting Cytomegalovirus in Glioblastoma. Clin. Cancer Res. 2020, 26, 5297–5303. [Google Scholar] [CrossRef]
- Dannull, J.; Nair, S.; Su, Z.; Boczkowski, D.; DeBeck, C.; Yang, B.; Gilboa, E.; Vieweg, J. Enhancing the Immunostimulatory Function of Dendritic Cells by Transfection with MRNA Encoding OX40 Ligand. Blood 2005, 105, 3206–3213. [Google Scholar] [CrossRef][Green Version]
- Tcherepanova, I.Y.; Adams, M.D.; Feng, X.; Hinohara, A.; Horvatinovich, J.; Calderhead, D.; Healey, D.; Nicolette, C.A. Ectopic Expression of a Truncated CD40L Protein from Synthetic Post-Transcriptionally Capped RNA in Dendritic Cells Induces High Levels of IL-12 Secretion. BMC Mol. Biol. 2008, 9, 90. [Google Scholar] [CrossRef]
- Tuyaerts, S.; Van Meirvenne, S.; Bonehill, A.; Heirman, C.; Corthals, J.; Waldmann, H.; Breckpot, K.; Thielemans, K.; Aerts, J.L. Expression of Human GITRL on Myeloid Dendritic Cells Enhances Their Immunostimulatory Function but Does Not Abrogate the Suppressive Effect of CD4+CD25+ Regulatory T Cells. J. Leukoc. Biol. 2007, 82, 93–105. [Google Scholar] [CrossRef]
- Grünebach, F.; Kayser, K.; Weck, M.M.; Müller, M.R.; Appel, S.; Brossart, P. Cotransfection of Dendritic Cells with RNA Coding for HER-2/Neu and 4-1BBL Increases the Induction of Tumor Antigen Specific Cytotoxic T Lymphocytes. Cancer Gene Ther. 2005, 12, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Aerts-Toegaert, C.; Heirman, C.; Tuyaerts, S.; Corthals, J.; Aerts, J.L.; Bonehill, A.; Thielemans, K.; Breckpot, K.; Breckpot, K. CD83 Expression on Dendritic Cells and T Cells: Correlation with Effective Immune Responses. Eur. J. Immunol. 2007, 37, 686–695. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, B.; Heirman, C.; Corthals, J.; Empsen, C.; van Grunsven, L.A.; Allard, S.D.; Pen, J.; Lacor, P.; Thielemans, K.; Aerts, J.L. The Combination of 4-1BBL and CD40L Strongly Enhances the Capacity of Dendritic Cells to Stimulate HIV-Specific T Cell Responses. J. Leukoc. Biol. 2011, 89, 989–999. [Google Scholar] [CrossRef]
- Amin, A.; Dudek, A.Z.; Logan, T.F.; Lance, R.S.; Holzbeierlein, J.M.; Knox, J.J.; Master, V.A.; Pal, S.K.; Miller, W.H.; Karsh, L.I.; et al. Survival with AGS-003, an Autologous Dendritic Cell-Based Immunotherapy, in Combination with Sunitinib in Unfavorable Risk Patients with Advanced Renal Cell Carcinoma (RCC): Phase 2 Study Results. J. Immunother. Cancer 2015, 3, 14. [Google Scholar] [CrossRef]
- Figlin, R.A.; Tannir, N.M.; Uzzo, R.G.; Tykodi, S.S.; Chen, D.Y.T.; Master, V.; Kapoor, A.; Vaena, D.; Lowrance, W.; Bratslavsky, G.; et al. Results of the ADAPT Phase 3 Study of Rocapuldencel-T in Combination with Sunitinib as First-Line Therapy in Patients with Metastatic Renal Cell Carcinoma. Clin. Cancer Res. 2020, 26, 2327–2335. [Google Scholar] [CrossRef] [PubMed]
- Bonehill, A.; Tuyaerts, S.; Van Nuffel, A.M.; Heirman, C.; Bos, T.J.; Fostier, K.; Neyns, B.; Thielemans, K. Enhancing the T-Cell Stimulatory Capacity of Human Dendritic Cells by Co-Electroporation with CD40L, CD70 and Constitutively Active TLR4 Encoding MRNA. Mol. Ther. 2008, 16, 1170–1180. [Google Scholar] [CrossRef]
- Bonehill, A.; Van Nuffel, A.M.T.; Corthals, J.; Tuyaerts, S.; Heirman, C.; François, V.; Colau, D.; Van Der Bruggen, P.; Neyns, B.; Thielemans, K. Single-Step Antigen Loading and Activation of Dendritic Cells by MRNA Electroporation for the Purpose of Therapeutic Vaccination in Melanoma Patients. Clin. Cancer Res. 2009, 15, 3366–3375. [Google Scholar] [CrossRef]
- Pen, J.J.; De Keersmaecker, B.; Maenhout, S.K.; Van Nuffel, A.M.T.; Heirman, C.; Corthals, J.; Escors, D.; Bonehill, A.; Thielemans, K.; Breckpot, K.; et al. Modulation of Regulatory T Cell Function by Monocyte-Derived Dendritic Cells Matured through Electroporation with MRNA Encoding CD40 Ligand, Constitutively Active TLR4, and CD70. J. Immunol. 2013, 191, 1976–1983. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, S.; Wilgenhof, S.; Heirman, C.; Corthals, J.; Breckpot, K.; Bonehill, A.; Neyns, B.; Thielemans, K. Optimized Dendritic Cell-Based Immunotherapy for Melanoma: The TriMix-Formula. Cancer Immunol. Immunother. 2014, 63, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; Van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II Study of Autologous Monocyte-Derived MRNA Electroporated Dendritic Cells (TriMixDC-MEL) plus Ipilimumab in Patientswith Pretreated Advanced Melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and Tumor Antigen MRNA Electroporated Dendritic Cell Vaccination plus Ipilimumab: Link between T-Cell Activation and Clinical Responses in Advanced Melanoma. J. Immunother. Cancer 2020, 8, e000329. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.-A.; Reed, K.; et al. Nivolumab plus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer Immunoediting and Resistance to T Cell-Based Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus Ipilimumab or Nivolumab Alone versus Ipilimumab Alone in Advanced Melanoma (CheckMate 067): 4-Year Outcomes of a Multicentre, Randomised, Phase 3 Trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Van den Bergh, J.; Willemen, Y.; Lion, E.; Van Acker, H.; De Reu, H.; Anguille, S.; Goossens, H.; Berneman, Z.; Van Tendeloo, V.; Smits, E. Transpresentation of Interleukin-15 by IL-15/IL-15Rα MRNA-Engineered Human Dendritic Cells Boosts Antitumoral Natural Killer Cell Activity. Oncotarget 2015, 6, 44123–44133. [Google Scholar] [CrossRef]
- Minkis, K.; Kavanagh, D.G.; Alter, G.; Bogunovic, D.; O’Neill, D.; Adams, S.; Pavlick, A.; Walker, B.D.; Brockman, M.A.; Gandhi, R.T.; et al. Type 2 Bias of T Cells Expanded from the Blood of Melanoma Patients Switched to Type 1 by IL-12p70 MRNA-Transfected Dendritic Cells. Cancer Res. 2008, 68, 9441–9450. [Google Scholar] [CrossRef]
- Bontkes, H.J.; Kramer, D.; Ruizendaal, J.J.; Kueter, E.W.M.; van Tendeloo, V.F.I.; Meijer, C.J.L.M.; Hooijberg, E. Dendritic Cells Transfected with Interleukin-12 and Tumor-Associated Antigen Messenger RNA Induce High Avidity Cytotoxic T Cells. Gene Ther. 2007, 14, 366–375. [Google Scholar] [CrossRef]
- Naka, T.; Iwahashi, M.; Nakamura, M.; Ojima, T.; Nakamori, M.; Ueda, K.; Katsuda, M.; Miyazawa, M.; Ishida, K.; Yamaue, H. Tumor Vaccine Therapy against Recrudescent Tumor Using Dendritic Cells Simultaneously Transfected with Tumor RNA and Granulocyte Macrophage Colony-Stimulating Factor RNA. Cancer Sci. 2008, 99, 407–413. [Google Scholar] [CrossRef]
- de Mey, W.; Locy, H.; De Ridder, K.; De Schrijver, P.; Autaers, D.; Lakdimi, A.; Esprit, A.; Franceschini, L.; Thielemans, K.; Verdonck, M.; et al. An MRNA Mix Redirects Dendritic Cells towards an Antiviral Program, Inducing Anticancer Cytotoxic Stem Cell and Central Memory CD8+ T Cells. Front. Immunol. 2023, 14, 1111523. [Google Scholar] [CrossRef] [PubMed]
- Heufler, C.; Koch, F.; Stanzl, U.; Topar, G.; Wysocka, M.; Trinchieri, G.; Enk, A.; Steinman, R.M.; Romani, N.; Schuler, G. Interleukin-12 Is Produced by Dendritic Cells and Mediates T Helper 1 Development as Well as Interferon-γ Production by T Helper 1 Cells. Eur. J. Immunol. 1996, 26, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Gately, M.K.; Renzetti, L.M.; Magram, J.; Stern, A.S.; Adorini, L.; Gubler, U.; Presky, D.H. The Interleukin-12/ Interleukin-12-Receptor System: Role in Normal and Pathologic Immune Responses. Annu. Rev. Immunol. 1998, 16, 495–521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, Y.; Wang, Q.T.; Sun, S.N.; Li, S.Y.; Shang, H.; He, Y.W. Enhanced Human T Lymphocyte Antigen Priming by Cytokine-Matured Dendritic Cells Overexpressing Bcl-2 and IL-12. Front. Cell Dev. Biol. 2020, 8, 205. [Google Scholar] [CrossRef] [PubMed]
- Nopora, A.; Brocker, T. Bcl-2 Controls Dendritic Cell Longevity In Vivo. J. Immunol. 2002, 169, 3006–3014. [Google Scholar] [CrossRef]
- Granucci, F.; Vizzardelli, C.; Pavelka, N.; Feau, S.; Persico, M.; Virzi, E.; Rescigno, M.; Moro, G.; Ricciardi-Castagnoli, P. Inducible IL-2 Production by Dendritic Cells Revealed by Global Gene Expression Analysis. Nat. Immunol. 2001, 2, 882–888. [Google Scholar] [CrossRef]
- Castañón, E.; Zamarin, D.; Carneiro, B.A.; Marron, T.; Patel, S.P.; Subbiah, V.; Mehmi, I.; Oberoi, H.K.; El-Khoueiry, A.; Ridgway, B.; et al. Abstract CT004: Intratumoral (IT) MEDI1191 + Durvalumab (D): Update on the First-in-Human Study in Advanced Solid Tumors. Cancer Res. 2023, 83, CT004. [Google Scholar] [CrossRef]
- Carneiro, B.A.; Zamarin, D.; Marron, T.; Mehmi, I.; Patel, S.P.; Subbiah, V.; El-Khoueiry, A.; Grand, D.; Garcia-Reyes, K.; Goel, S.; et al. Abstract CT183: First-in-Human Study of MEDI1191 (MRNA Encoding IL-12) plus Durvalumab in Patients (Pts) with Advanced Solid Tumors. Cancer Res. 2022, 82, CT183. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. MRNA Vaccines-a New Era in Vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Van Lint, S.; Goyvaerts, C.; Maenhout, S.; Goethals, L.; Disy, A.; Benteyn, D.; Pen, J.; Bonehill, A.; Heirman, C.; Breckpot, K.; et al. Preclinical Evaluation of TriMix and Antigen MRNA-Based Antitumor Therapy. Cancer Res. 2012, 72, 1661–1671. [Google Scholar] [CrossRef]
- Van Lint, S.; Renmans, D.; Broos, K.; Goethals, L.; Maenhout, S.; Benteyn, D.; Goyvaerts, C.; Du Four, S.; Van Der Jeught, K.; Bialkowski, L.; et al. Intratumoral Delivery of TriMix MRNA Results in T-Cell Activation by Cross-Presenting Dendritic Cells. Cancer Immunol Res. 2016, 4, 146–156. [Google Scholar] [CrossRef]
- Van Lint, S.; Renmans, D.; Broos, K.; Dewitte, H.; Lentacker, I.; Heirman, C.; Breckpot, K.; Thielemans, K. The ReNAissanCe of MRNA-Based Cancer Therapy. Expert. Rev. Vaccines 2014, 14, 235–251. [Google Scholar] [CrossRef]
- Kreiter, S.; Selmi, A.; Diken, M.; Koslowski, M.; Britten, C.M.; Huber, C.; Türeci, Ö.; Sahin, U. Intranodal Vaccination with Naked Antigen-Encoding RNA Elicits Potent Prophylactic and Therapeutic Antitumoral Immunity. Cancer Res. 2010, 70, 9031–9040. [Google Scholar] [CrossRef]
- Liang, X.; Li, D.; Leng, S.; Zhu, X. RNA-Based Pharmacotherapy for Tumors: From Bench to Clinic and Back. Biomed. Pharmacother. 2020, 125, 109997. [Google Scholar] [CrossRef]
- Arance Fernandez, A.M.; Baurain, J.-F.; Vulsteke, C.; Rutten, A.; Soria, A.; Carrasco, J.; Neyns, B.; De Keersmaecker, B.; Van Assche, T.; Lindmark, B. A Phase I Study (E011-MEL) of a TriMix-Based MRNA Immunotherapy (ECI-006) in Resected Melanoma Patients: Analysis of Safety and Immunogenicity. J. Clin. Oncol. 2019, 37, 2641. [Google Scholar] [CrossRef]
- Clausen, B.E.; Stoitzner, P. Functional Specialization of Skin Dendritic Cell Subsets in Regulating T Cell Responses. Front. Immunol. 2015, 6, 534. [Google Scholar] [CrossRef]
- Weide, B.; Pascolo, S.; Scheel, B.; Derhovanessian, E.; Pflugfelder, A.; Eigentler, T.K.; Pawelec, G.; Hoerr, I.; Rammensee, H.-G.; Garbe, C. Direct Injection of Protamine-Protected MRNA: Results of a Phase 1/2 Vaccination Trial in Metastatic Melanoma Patients. J. Immunother. 2009, 32, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Kübler, H.; Scheel, B.; Gnad-Vogt, U.; Miller, K.; Schultze-Seemann, W.; Dorp, F.; Parmiani, G.; Hampel, C.; Wedel, S.; Trojan, L.; et al. Self-Adjuvanted MRNA Vaccination in Advanced Prostate Cancer Patients: A First-in-Man Phase I/IIa Study. J. Immunother. Cancer 2015, 3, 26. [Google Scholar] [CrossRef]
- Fotin-Mleczek, M.; Duchardt, K.M.; Lorenz, C.; Pfeiffer, R.; Ojkić-Zrna, S.; Probst, J.; Kallen, K.-J. Messenger RNA-Based Vaccines with Dual Activity Induce Balanced TLR-7 Dependent Adaptive Immune Responses and Provide Antitumor Activity. J. Immunother. 2011, 34, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA Delivery to Dendritic Cells Exploits Antiviral Defence for Cancer Immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid Nanoparticles for MRNA Delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA Vaccine Drives Immunity in Checkpoint-Inhibitor-Treated Melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef]
- Schmidt, M.; Vogler, I.; Derhovanessian, E.; Omokoko, T.; Godehardt, E.; Attig, S.; Cortini, A.; Newrzela, S.; Grützner, J.; Bolte, S.; et al. 88MO T-Cell Responses Induced by an Individualized Neoantigen Specific Immune Therapy in Post (Neo)Adjuvant Patients with Triple Negative Breast Cancer. Ann. Oncol. 2020, 31, S276. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; Neyns, B.; Thielemans, K. Clinical Trials with MRNA Electroporated Dendritic Cells for Stage III/IV Melanoma Patients. J. Immunother. Cancer 2015, 3, P211. [Google Scholar] [CrossRef][Green Version]
- Wilgenhof, S.; Van Nuffel, A.M.T.; Benteyn, D.; Corthals, J.; Aerts, C.; Heirman, C.; Van Riet, I.; Bonehill, A.; Thielemans, K.; Neyns, B. A Phase IB Study on Intravenous Synthetic MRNA Electroporated Dendritic Cell Immunotherapy in Pretreated Advanced Melanoma Patients. Ann. Oncol. 2013, 24, 2686–2693. [Google Scholar] [CrossRef]
- Jansen, Y.; Kruse, V.; Corthals, J.; Schats, K.; van Dam, P.J.; Seremet, T.; Heirman, C.; Brochez, L.; Kockx, M.; Thielemans, K.; et al. A Randomized Controlled Phase II Clinical Trial on MRNA Electroporated Autologous Monocyte-Derived Dendritic Cells (TriMixDC-MEL) as Adjuvant Treatment for Stage III/IV Melanoma Patients Who Are Disease-Free Following the Resection of Macrometastases. Cancer Immunol. Immunother. 2020, 69, 2589–2598. [Google Scholar] [CrossRef]
- Kalluri, R.; McAndrews, K.M. The Role of Extracellular Vesicles in Cancer. Cell 2023, 186, 1610–1626. [Google Scholar] [CrossRef]
- Pitt, J.M.; André, F.; Amigorena, S.; Soria, J.C.; Eggermont, A.; Kroemer, G.; Zitvogel, L. Dendritic Cell-Derived Exosomes for Cancer Therapy. J. Clin. Investig. 2016, 126, 1224–1232. [Google Scholar] [CrossRef]
- Couch, Y.; Buzàs, E.I.; Di Vizio, D.; Gho, Y.S.; Harrison, P.; Hill, A.F.; Lötvall, J.; Raposo, G.; Stahl, P.D.; Théry, C.; et al. A Brief History of Nearly Everything—The Rise and Rise of Extracellular Vesicles. J. Extracell. Vesicles 2021, 10, e12144. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular Vesicles in Cancer—Implications for Future Improvements in Cancer Care. Nat. Rev. Clin. Oncol. 2018, 15, 617–638. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; LeBleu, V.S. The Biology, Function, and Biomedical Applications of Exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Zuo, B.; Zhang, Y.; Zhao, K.; Wu, L.; Qi, H.; Yang, R.; Gao, X.; Geng, M.; Wu, Y.; Jing, R.; et al. Universal Immunotherapeutic Strategy for Hepatocellular Carcinoma with Exosome Vaccines That Engage Adaptive and Innate Immune Responses. J. Hematol. Oncol. 2022, 15, 46. [Google Scholar] [CrossRef]
- Chen, X.; Feng, J.; Chen, W.; Shao, S.; Chen, L.; Wan, H. Small Extracellular Vesicles: From Promoting Pre-Metastatic Niche Formation to Therapeutic Strategies in Breast Cancer. Cell Commun. Signal. 2022, 20, 141. [Google Scholar] [CrossRef]
- Zhang, B.; Yin, Y.; Lai, R.C.; Lim, S.K. Immunotherapeutic Potential of Extracellular Vesicles. Front. Immunol. 2014, 5, 518. [Google Scholar] [CrossRef] [PubMed]
- Besse, B.; Charrier, M.; Lapierre, V.; Dansin, E.; Lantz, O.; Planchard, D.; Le Chevalier, T.; Livartoski, A.; Barlesi, F.; Laplanche, A.; et al. Dendritic Cell-Derived Exosomes as Maintenance Immunotherapy after First Line Chemotherapy in NSCLC. Oncoimmunology 2016, 5, e1071008. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Dorval, T.; Chaput, N.; André, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of Metastatic Melanoma Patients with Autologous Dendritic Cell (DC) Derived-Exosomes: Results of the First Phase 1 Clinical Trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A Phase I Study of Dexosome Immunotherapy in Patients with Advanced Non-Small Cell Lung Cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Viaud, S.; Ploix, S.; Lapierre, V.; Théry, C.; Commere, P.-H.; Tramalloni, D.; Gorrichon, K.; Virault-Rocroy, P.; Tursz, T.; Lantz, O.; et al. Updated Technology to Produce Highly Immunogenic Dendritic Cell-Derived Exosomes of Clinical Grade. J. Immunother. 2011, 34, 65–75. [Google Scholar] [CrossRef]
- Qian, K.; Fu, W.; Li, T.; Zhao, J.; Lei, C.; Hu, S. The Roles of Small Extracellular Vesicles in Cancer and Immune Regulation and Translational Potential in Cancer Therapy. J. Exp. Clin. Cancer Res. 2022, 41, 286. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Nicco, C.; Lombard, B.; Véron, P.; Raposo, G.; Batteux, F.; Amigorena, S.; Théry, C. ICAM-1 on Exosomes from Mature Dendritic Cells Is Critical for Efficient Naive T-Cell Priming. Blood 2005, 106, 216–223. [Google Scholar] [CrossRef]
- Pang, X.-L.; Wang, Z.-G.; Liu, L.; Feng, Y.-H.; Wang, J.-X.; Xie, H.-C.; Yang, X.-L.; Li, J.-F.; Feng, G.-W. Immature Dendritic Cells Derived Exosomes Promotes Immune Tolerance by Regulating T Cell Differentiation in Renal Transplantation. Aging 2019, 11, 8911–8924. [Google Scholar] [CrossRef]
- Fu, C.; Zhou, L.; Mi, Q.-S.; Jiang, A. Plasmacytoid Dendritic Cells and Cancer Immunotherapy. Cells 2022, 11, 222. [Google Scholar] [CrossRef]
- Tel, J.; Erik, H.J.G.A.; Baba, T.; Schreibelt, G.; Schulte, B.M.; Benitez-Ribas, D.; Boerman, O.C.; Croockewit, S.; Wim, J.G.O.; Van Rossum, M.; et al. Natural Human Plasmacytoid Dendritic Cells Induce Antigen-Specific T-Cell Responses in Melanoma Patients. Cancer Res. 2013, 73, 1063–1075. [Google Scholar] [CrossRef]
- Fu, C.; Peng, P.; Loschko, J.; Feng, L.; Pham, P.; Cui, W.; Lee, K.P.; Krug, A.B.; Jiang, A. Plasmacytoid Dendritic Cells Cross-Prime Naive CD8 T Cells by Transferring Antigen to Conventional Dendritic Cells through Exosomes. Proc. Natl. Acad. Sci. USA 2020, 117, 23730–23741. [Google Scholar] [CrossRef]
- Greening, D.W.; Xu, R.; Ale, A.; Hagemeyer, C.E.; Chen, W. Extracellular Vesicles as next Generation Immunotherapeutics. Semin. Cancer Biol. 2023, 90, 73–100. [Google Scholar] [CrossRef]
- Lu, Z.; Zuo, B.; Jing, R.; Gao, X.; Rao, Q.; Liu, Z.; Qi, H.; Guo, H.; Yin, H.F. Dendritic Cell-Derived Exosomes Elicit Tumor Regression in Autochthonous Hepatocellular Carcinoma Mouse Models. J. Hepatol. 2017, 67, 739–748. [Google Scholar] [CrossRef]
- Jarnicki, A.G.; Lysaght, J.; Todryk, S.; Mills, K.H.G. Suppression of Antitumor Immunity by IL-10 and TGF-Producing T Cells Infiltrating the Growing Tumor: Influence of Tumor Environment on the Induction of CD4 and CD8 Regulatory T Cells 1. J. Immunol. 2006, 177, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Miao, Y.; Wang, X.; Huang, X.; Dai, J. Recent Progress of Dendritic Cell-Derived Exosomes (Dex) as an Anti-Cancer Nanovaccine. Biomed. Pharmacother. 2022, 152, 113250. [Google Scholar] [CrossRef] [PubMed]
- De Waele, J.; Verhezen, T.; van der Heijden, S.; Berneman, Z.N.; Peeters, M.; Lardon, F.; Wouters, A.; Smits, E.L.J.M. A Systematic Review on Poly(I:C) and Poly-ICLC in Glioblastoma: Adjuvants Coordinating the Unlocking of Immunotherapy. J. Exp. Clin. Cancer Res. 2021, 40, 213. [Google Scholar] [CrossRef] [PubMed]
- Phung, C.D.; Pham, T.T.; Nguyen, H.T.; Nguyen, T.T.; Ou, W.; Jeong, J.H.; Choi, H.G.; Ku, S.K.; Yong, C.S.; Kim, J.O. Anti-CTLA-4 Antibody-Functionalized Dendritic Cell-Derived Exosomes Targeting Tumor-Draining Lymph Nodes for Effective Induction of Antitumor T-Cell Responses. Acta Biomater. 2020, 115, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.K.; Redecke, V.; Prilliman, K.R.; Takabayashi, K.; Corr, M.; Tallant, T.; DiDonato, J.; Dziarski, R.; Akira, S.; Schoenberger, S.P.; et al. A Subset of Toll-Like Receptor Ligands Induces Cross-Presentation by Bone Marrow-Derived Dendritic Cells. J. Immunol. 2003, 170, 4102–4110. [Google Scholar] [CrossRef]
- Tran, T.H.; Tran, T.T.P.; Truong, D.H.; Nguyen, H.T.; Pham, T.T.; Yong, C.S.; Kim, J.O. Toll-like Receptor-Targeted Particles: A Paradigm to Manipulate the Tumor Microenvironment for Cancer Immunotherapy. Acta Biomater. 2019, 94, 82–96. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Pedicord, V.A.; Montalvo, W.; Leiner, I.M.; Allison, J.P. Single Dose of Anti-CTLA-4 Enhances CD8+ T-Cell Memory Formation, Function, and Maintenance. Proc. Natl. Acad. Sci. USA 2011, 108, 266–271. [Google Scholar] [CrossRef]
- Liu, C.; Liu, X.; Xiang, X.; Pang, X.; Chen, S.; Zhang, Y.; Ren, E.; Zhang, L.; Liu, X.; Lv, P.; et al. A Nanovaccine for Antigen Self-Presentation and Immunosuppression Reversal as a Personalized Cancer Immunotherapy Strategy. Nat. Nanotechnol. 2022, 17, 531–540. [Google Scholar] [CrossRef]
- Perez, C.R.; De Palma, M. Engineering Dendritic Cell Vaccines to Improve Cancer Immunotherapy. Nat. Commun. 2019, 10, 5408. [Google Scholar] [CrossRef]
- Squadrito, M.L.; Cianciaruso, C.; Hansen, S.K.; De Palma, M. EVIR: Chimeric Receptors That Enhance Dendritic Cell Cross-Dressing with Tumor Antigens. Nat. Methods 2018, 15, 183–186. [Google Scholar] [CrossRef]
- Mohammadzadeh, Y.; De Palma, M. Boosting Dendritic Cell Nanovaccines. Nat. Nanotechnol. 2022, 17, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Naseri, M.; Bozorgmehr, M.; Zöller, M.; Ranaei Pirmardan, E.; Madjd, Z. Tumor-Derived Exosomes: The next Generation of Promising Cell-Free Vaccines in Cancer Immunotherapy. Oncoimmunology 2020, 9, 1779991. [Google Scholar] [CrossRef]
- Andre, F.; Schartz, N.E.; Movassagh, M.; Flament, C.; Pautier, P.; Morice, P.; Pomel, C.; Lhomme, C.; Escudier, B.; Le Chevalier, T.; et al. Malignant Effusions and Immunogenic Tumour-Derived Exosomes. Lancet 2002, 360, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Théry, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T.; et al. Tumor-Derived Exosomes Are a Source of Shared Tumor Rejection Antigens for CTL Cross-Priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Barros, F.M.; Carneiro, F.; Machado, J.C.; Melo, S.A. Exosomes and Immune Response in Cancer: Friends or Foes? Front. Immunol. 2018, 9, 730. [Google Scholar] [CrossRef]
- Hao, S.; Bai, O.; Li, F.; Yuan, J.; Laferte, S.; Xiang, J. Mature Dendritic Cells Pulsed with Exosomes Stimulate Efficient Cytotoxic T-Lymphocyte Responses and Antitumour Immunity. Immunology 2007, 120, 90–102. [Google Scholar] [CrossRef]
- Rappa, G.; Santos, M.F.; Green, T.M.; Karbanová, J.; Hassler, J.; Bai, Y.; Barsky, S.H.; Corbeil, D.; Lorico, A. Nuclear Transport of Cancer Extracellular Vesicle-Derived Biomaterials through Nuclear Envelope Invagination-Associated Late Endosomes. Oncotarget 2017, 8, 14443–14461. [Google Scholar] [CrossRef]
- Dorayappan, K.D.P.; Wanner, R.; Wallbillich, J.J.; Saini, U.; Zingarelli, R.; Suarez, A.A.; Cohn, D.E.; Selvendiran, K. Hypoxia-Induced Exosomes Contribute to a More Aggressive and Chemoresistant Ovarian Cancer Phenotype: A Novel Mechanism Linking STAT3/Rab Proteins. Oncogene 2018, 37, 3806–3821. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.L.L.; Coukos, G.; Kandalaft, L.E. Whole Tumor Antigen Vaccines: Where Are We? Vaccines 2015, 3, 344–372. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Erb, U.; Büchler, M.W.; Zöller, M. Improved Vaccine Efficacy of Tumor Exosome Compared to Tumor Lysate Loaded Dendritic Cells in Mice. Int. J. Cancer 2015, 136, E74–E84. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, J.; Shao, C.; Liu, S.; Yu, Y.; Wang, Q.; Cao, X. Efficient Induction of Antitumor T Cell Immunity by Exosomes Derived from Heat-Shocked Lymphoma Cells. Eur. J. Immunol. 2006, 36, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Bu, N.; Wu, H.; Sun, B.; Zhang, G.; Zhan, S.; Zhang, R.; Zhou, L. Exosome-Loaded Dendritic Cells Elicit Tumor-Specific CD8+ Cytotoxic T Cells in Patients with Glioma. J. Neurooncol. 2011, 104, 659–667. [Google Scholar] [CrossRef]
- Li, W.; Mu, D.; Tian, F.; Hu, Y.; Jiang, T.; Han, Y.; Chen, J.; Han, G.; Li, X. Exosomes Derived from Rab27a-Overexpressing Tumor Cells Elicit Efficient Induction of Antitumor Immunity. Mol. Med. Rep. 2013, 8, 1876–1882. [Google Scholar] [CrossRef]
- Czernek, L.; Düchler, M. Functions of Cancer-Derived Extracellular Vesicles in Immunosuppression. Arch. Immunol. Ther. Exp. 2017, 65, 311–323. [Google Scholar] [CrossRef]
- André, F.; Schartz, N.E.C.; Chaput, N.; Flament, C.; Raposo, G.; Amigorena, S.; Angevin, E.; Zitvogel, L. Tumor-Derived Exosomes: A New Source of Tumor Rejection Antigens. Vaccine 2002, 20, A28–A31. [Google Scholar] [CrossRef]
- Bu, N.; Li, Q.-L.; Feng, Q.; Sun, B.-Z. Immune Protection Effect of Exosomes against Attack of L1210 Tumor Cells. Leuk. Lymphoma. 2006, 47, 913–918. [Google Scholar] [CrossRef]
- Liu, H.; Chen, L.; Peng, Y.; Yu, S.; Liu, J.; Wu, L.; Zhang, L.; Wu, Q.; Chang, X.; Yu, X.; et al. Dendritic Cells Loaded with Tumor Derived Exosomes for Cancer Immunotherapy. Oncotarget 2018, 9, 2887–2894. [Google Scholar] [CrossRef]
- Jafari, A.; Babajani, A.; Abdollahpour-Alitappeh, M.; Ahmadi, N.; Rezaei-Tavirani, M. Exosomes and Cancer: From Molecular Mechanisms to Clinical Applications. Med. Oncol. 2021, 38, 45. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Liu, C.; Su, K.; Wang, J.; Liu, Y.; Zhang, L.; Li, C.; Cong, Y.; Kimberly, R.; Grizzle, W.E.; et al. Tumor Exosomes Inhibit Differentiation of Bone Marrow Dendritic Cells. J. Immunol. 2007, 178, 6867–6875. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Shen, K.; Wu, Q.; Sun, X.; Bai, Y.; Xie, Y.; Pan, J.; Qi, C. Tumor Exosomes Block Dendritic Cells Maturation to Decrease the T Cell Immune Response. Immunol. Lett. 2018, 199, 36–43. [Google Scholar] [CrossRef]
- Huang, S.; Li, Y.; Zhang, J.; Rong, J.; Ye, S. Epidermal Growth Factor Receptor-Containing Exosomes Induce Tumor-Specific Regulatory T Cells. Cancer Investig. 2013, 31, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Maia, J.; Caja, S.; Strano Moraes, M.C.; Couto, N.; Costa-Silva, B. Exosome-Based Cell-Cell Communication in the Tumor Microenvironment. Front. Cell Dev. Biol. 2018, 6, 18. [Google Scholar] [CrossRef]
- Liu, Y.; Gu, Y.; Cao, X. The Exosomes in Tumor Immunity. Oncoimmunology 2015, 4, e1027472. [Google Scholar] [CrossRef]
- Yang, C.; Kim, S.H.; Bianco, N.R.; Robbins, P.D. Tumor-Derived Exosomes Confer Antigen-Specific Immunosuppression in a Murine Delayed-Type Hypersensitivity Model. PLoS ONE 2011, 6, e22517. [Google Scholar] [CrossRef]
- Szajnik, M.; Czystowska, M.; Szczepanski, M.J.; Mandapathil, M.; Whiteside, T.L. Tumor-Derived Microvesicles Induce, Expand and Up-Regulate Biological Activities of Human Regulatory T Cells (Treg). PLoS ONE 2010, 5, e11469. [Google Scholar] [CrossRef]
- Wieckowski, E.U.; Visus, C.; Szajnik, M.; Szczepanski, M.J.; Storkus, W.J.; Whiteside, T.L. Tumor-Derived Microvesicles Promote Regulatory T Cell Expansion and Induce Apoptosis in Tumor-Reactive Activated CD8+ T Lymphocytes. J. Immunol. 2009, 183, 3720–3730. [Google Scholar] [CrossRef]
- Lee, E.-Y.; Park, K.-S.; Yoon, Y.J.; Lee, J.; Moon, H.-G.; Jang, S.C.; Choi, K.-H.; Kim, Y.-K.; Gho, Y.S. Therapeutic Effects of Autologous Tumor-Derived Nanovesicles on Melanoma Growth and Metastasis. PLoS ONE 2012, 7, e33330. [Google Scholar] [CrossRef]
- Ren, G.; Wang, Y.; Yuan, S.; Wang, B. Dendritic Cells Loaded with HeLa-Derived Exosomes Simulate an Antitumor Immune Response. Oncol. Lett. 2018, 15, 6636–6640. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; An, Y.; Jin, T.; Zhang, F.; He, P. Detection of MUC1 Protein on Tumor Cells and Their Derived Exosomes for Breast Cancer Surveillance with an Electrochemiluminescence Aptasensor. J. Electroanal. Chem. 2021, 882, 115011. [Google Scholar] [CrossRef]
- Kufe, D.W. Mucins in Cancer: Function, Prognosis and Therapy. Nat. Rev. Cancer 2009, 9, 874–885. [Google Scholar] [CrossRef]
- Lapointe, J.; Li, C.; Higgins, J.P.; van de Rijn, M.; Bair, E.; Montgomery, K.; Ferrari, M.; Egevad, L.; Rayford, W.; Bergerheim, U.; et al. Gene Expression Profiling Identifies Clinically Relevant Subtypes of Prostate Cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.F.; Sekine, H.; Ohno, T.; Abe, M.; Keefe, K.; Kufe, D.W. Use of a Murine Monoclonal Antibody for Detection of Circulating Plasma DF3 Antigen Levels in Breast Cancer Patients. J. Clin. Investig. 1985, 75, 1671–1678. [Google Scholar] [CrossRef]
- Cho, J.A.; Yeo, D.J.; Son, H.Y.; Kim, H.W.; Jung, D.S.; Ko, J.K.; Jason, S.K.; Kim, Y.N.; Kim, C.W. Exosomes: A New Delivery System for Tumor Antigens in Cancer Immunotherapy. Int. J. Cancer 2005, 114, 613–622. [Google Scholar] [CrossRef]
- Shi, X.; Sun, J.; Li, H.; Lin, H.; Xie, W.; Li, J.; Tan, W. Antitumor Efficacy of Interferon-γ-Modified Exosomal Vaccine in Prostate Cancer. Prostate 2020, 80, 811–823. [Google Scholar] [CrossRef]
- Yang, M.Q.; Du, Q.; Varley, P.R.; Goswami, J.; Liang, Z.; Wang, R.; Li, H.; Stolz, D.B.; Geller, D.A. Interferon Regulatory Factor 1 Priming of Tumour-Derived Exosomes Enhances the Antitumour Immune Response. Br. J. Cancer 2018, 118, 62–71. [Google Scholar] [CrossRef]
- Accolla, R.S.; Ramia, E.; Tedeschi, A.; Forlani, G. CIITA-Driven MHC Class II Expressing Tumor Cells as Antigen Presenting Cell Performers: Toward the Construction of an Optimal Anti-Tumor Vaccine. Front. Immunol. 2019, 10, 1806. [Google Scholar] [CrossRef]
- Daar, A.S.; Fuggle, S.V.; Fabre, J.W.; Ting, A.; Morris, P.J. The Detailed Distribution of MHC Class II Antigens in Normal Human Organs. Transplantation 1984, 38, 293–298. [Google Scholar] [CrossRef]
- Garcia-Lora, A.; Algarra, I.; Garrido, F. MHC Class I Antigens, Immune Surveillance, and Tumor Immune Escape. J. Cell. Physiol. 2003, 195, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Accolla, R.S.; Tosi, G. Optimal MHC-II-Restricted Tumor Antigen Presentation to CD4+ T Helper Cells: The Key Issue for Development of Anti-Tumor Vaccines. J. Transl. Med. 2012, 10, 154. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, S.H.; Cho, J.A.; Kim, C.W. Introduction of the CIITA Gene into Tumor Cells Produces Exosomes with Enhanced Anti-Tumor Effects. Exp. Mol. Med. 2011, 43, 281–290. [Google Scholar] [CrossRef]
- Morishita, M.; Takahashi, Y.; Matsumoto, A.; Nishikawa, M.; Takakura, Y. Exosome-Based Tumor Antigens–Adjuvant Co-Delivery Utilizing Genetically Engineered Tumor Cell-Derived Exosomes with Immunostimulatory CpG DNA. Biomaterials 2016, 111, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Nishikawa, M.; Takakura, Y. In Vivo Tracking of Extracellular Vesicles in Mice Using Fusion Protein Comprising Lactadherin and Gaussia Luciferase. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2017; Volume 1660, pp. 245–254. [Google Scholar]
- Green, N.M. The Use of [14C] Biotin for Kinetic Studies and for Assay. Biochem. J. 1963, 89, 585–591. [Google Scholar] [CrossRef]
- Yildirim, M.; Yildirim, T.C.; Turay, N.; Bildik, T.; Ibibik, B.; Evcili, I.; Ersan, P.G.; Tokat, U.M.; Sahin, O.; Gursel, I. TLR Ligand Loaded Exosome Mediated Immunotherapy of Established Mammary Tumor in Mice. Immunol. Lett. 2021, 239, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.L.; Kadima, A.N.; Cole, D.J.; Gillanders, W.E. Defining the Antigen-Specific T-Cell Response to Vaccination and Poly(I:C)/TLR3 Signaling. J. Immunother. 2005, 28, 220–228. [Google Scholar] [CrossRef]
- Zuo, B.; Qi, H.; Lu, Z.; Chen, L.; Sun, B.; Yang, R.; Zhang, Y.; Liu, Z.; Gao, X.; You, A.; et al. Alarmin-Painted Exosomes Elicit Persistent Antitumor Immunity in Large Established Tumors in Mice. Nat. Commun. 2020, 11, 1790. [Google Scholar] [CrossRef]
- Huang, L.; Rong, Y.; Tang, X.; Yi, K.; Qi, P.; Hou, J.; Liu, W.; He, Y.; Gao, X.; Yuan, C.; et al. Engineered Exosomes as an in Situ DC-Primed Vaccine to Boost Antitumor Immunity in Breast Cancer. Mol. Cancer 2022, 21, 45. [Google Scholar] [CrossRef]
- Hosseini, R.; Asef-Kabiri, L.; Yousefi, H.; Sarvnaz, H.; Salehi, M.; Akbari, M.E.; Eskandari, N. The Roles of Tumor-Derived Exosomes in Altered Differentiation, Maturation and Function of Dendritic Cells. Mol Cancer 2021, 20, 83. [Google Scholar] [CrossRef]
- Wang, Y.; Yi, J.; Chen, X.; Zhang, Y.; Xu, M.; Yang, Z. The Regulation of Cancer Cell Migration by Lung Cancer Cell-Derived Exosomes through TGF-β and IL-10. Oncol. Lett. 2016, 11, 1527–1530. [Google Scholar] [CrossRef]
- Lenart, M.; Rutkowska-Zapala, M.; Baj-Krzyworzeka, M.; Szatanek, R.; Węglarczyk, K.; Smallie, T.; Ziegler-Heitbrock, L.; Zembala, M.; Siedlar, M. Hyaluronan Carried by Tumor-Derived Microvesicles Induces IL-10 Production in Classical (CD14++ CD16−) Monocytes via PI3K/Akt/MTOR-Dependent Signalling Pathway. Immunobiology 2017, 222, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, L.; Li, D.; Yin, Y.; Zhang, C.-Y.; Li, J.; Zhang, Y. Microvesicle-Delivery MiR-150 Promotes Tumorigenesis by up-Regulating VEGF, and the Neutralization of MiR-150 Attenuate Tumor Development. Protein Cell 2013, 4, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.-P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-Associated Hsp72 from Tumor-Derived Exosomes Mediates STAT3-Dependent Immunosuppressive Function of Mouse and Human Myeloid-Derived Suppressor Cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Shen, H.; Li, Z.; Wang, T.; Wang, S. Tumor-Derived Exosomes, Myeloid-Derived Suppressor Cells, and Tumor Microenvironment. J. Hematol. Oncol. 2019, 12, 84. [Google Scholar] [CrossRef]
- Whiteside, T.L. Immune Modulation of T-Cell and NK (Natural Killer) Cell Activities by TEXs (Tumour-Derived Exosomes). Biochem. Soc. Trans. 2013, 41, 245–251. [Google Scholar] [CrossRef]
- Ji, B.; Wei, M.; Yang, B. Recent Advances in Nanomedicines for Photodynamic Therapy (PDT)-Driven Cancer Immunotherapy. Theranostics 2022, 12, 434–458. [Google Scholar] [CrossRef]
- Nam, J.; Son, S.; Park, K.S.; Moon, J.J. Photothermal Therapy Combined with Neoantigen Cancer Vaccination for Effective Immunotherapy against Large Established Tumors and Distant Metastasis. Adv. Ther. 2021, 4, 2100093. [Google Scholar] [CrossRef]
- Roth, G.A.; Picece, V.C.T.M.; Ou, B.S.; Luo, W.; Pulendran, B.; Appel, E.A. Designing Spatial and Temporal Control of Vaccine Responses. Nat. Rev. Mater. 2021, 7, 174–195. [Google Scholar] [CrossRef]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the Mutanome for Tumor Vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity against Cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards Personalized, Tumour-Specific, Therapeutic Vaccines for Cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.; Ibn-Salem, J.; Sorn, P.; Suchan, M.; Holtsträter, C.; Lahrmann, U.; Vogler, I.; Schmoldt, K.; Lang, F.; Schrörs, B.; et al. Accurate Detection of Tumor-Specific Gene Fusions Reveals Strongly Immunogenic Personal Neo-Antigens. Nat. Biotechnol. 2022, 40, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Song, H.; Qin, Y.; Huang, P.; Zhang, C.; Kong, D.; Wang, W. Engineering Dendritic-Cell-Based Vaccines and PD-1 Blockade in Self-Assembled Peptide Nanofibrous Hydrogel to Amplify Antitumor T-Cell Immunity. Nano Lett. 2018, 18, 4377–4385. [Google Scholar] [CrossRef]



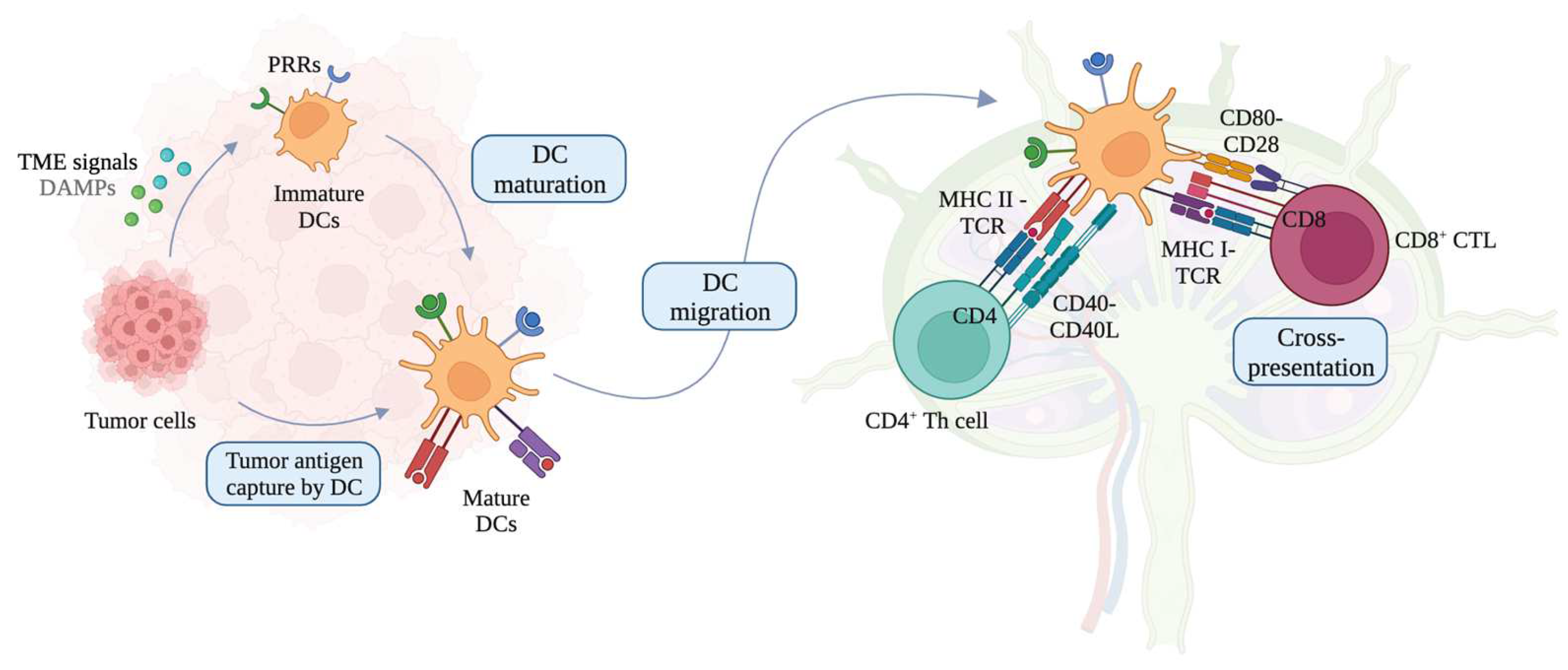{kind=link}
{kind=link}
| DC Vaccine Type | Advantages | Limitations | Reference |
|---|---|---|---|
| Conventional MoDC-based |
|
| NCT01832870 (Phase I); NCT01804465, NCT01881867 (Phase II); NCT00065442 (Phase III) [56,57,58,59] |
| Biomaterial-based |
|
| NCT01753089 (Phase I) [89,94,101,103,114,115,123,128,131,132,133,397] |
| Combinatory ICD-inducing |
|
| NCT01976585, NCT03789097, NCT00185965, NCT00323882, NCT02221739, (Phase I/II); NCT00880581 (Phase II); NCT00861614 (Phase III) [143,160,181,182,183,184,185,187,202,208,211,213] |
| mRNA-based |
|
| NCT00626483, NCT00639639, NCT03946800, NCT03788083, NCT03394937, NCT02410733, NCT02316457 (Phase I); NCT01066390 (Phase Ib); NCT02366728, NCT01302496, NCT01676779 (Phase II) [248,252,256,258,263,265,266,280,283,284,287,289,291,298,299,302] |
| DCsEV-based |
|
| NCT01159288 (Phase II) [309,312,313,314,315,318,321,327,333] |
| Tu-sEV-based |
|
| [368,369,370,376,379,381,382] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.-W.; Yam, J.W.P.; Mao, X. Dendritic Cell Vaccines: A Shift from Conventional Approach to New Generations. Cells 2023, 12, 2147. https://doi.org/10.3390/cells12172147
Lee K-W, Yam JWP, Mao X. Dendritic Cell Vaccines: A Shift from Conventional Approach to New Generations. Cells. 2023; 12(17):2147. https://doi.org/10.3390/cells12172147
Chicago/Turabian StyleLee, Kyu-Won, Judy Wai Ping Yam, and Xiaowen Mao. 2023. "Dendritic Cell Vaccines: A Shift from Conventional Approach to New Generations" Cells 12, no. 17: 2147. https://doi.org/10.3390/cells12172147
APA StyleLee, K.-W., Yam, J. W. P., & Mao, X. (2023). Dendritic Cell Vaccines: A Shift from Conventional Approach to New Generations. Cells, 12(17), 2147. https://doi.org/10.3390/cells12172147