
Foxo3 Knockdown Mediates Decline of Myod1 and Myog Reducing Myoblast Conversion to Myotubes

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Culture
2.2. Cell Line Selection
2.3. Comparison of Growth Speed in-between Selected Cell Lines
2.4. Quantitative Real Time PCR
2.5. Western Blot
2.6. Immunofluorescence Staining
2.7. Functional Contraction Analysis
2.8. Bulk RNA-Sequencing
2.9. Single Cell RNA-Sequencing
2.10. Statistics and Graphs
3. Results
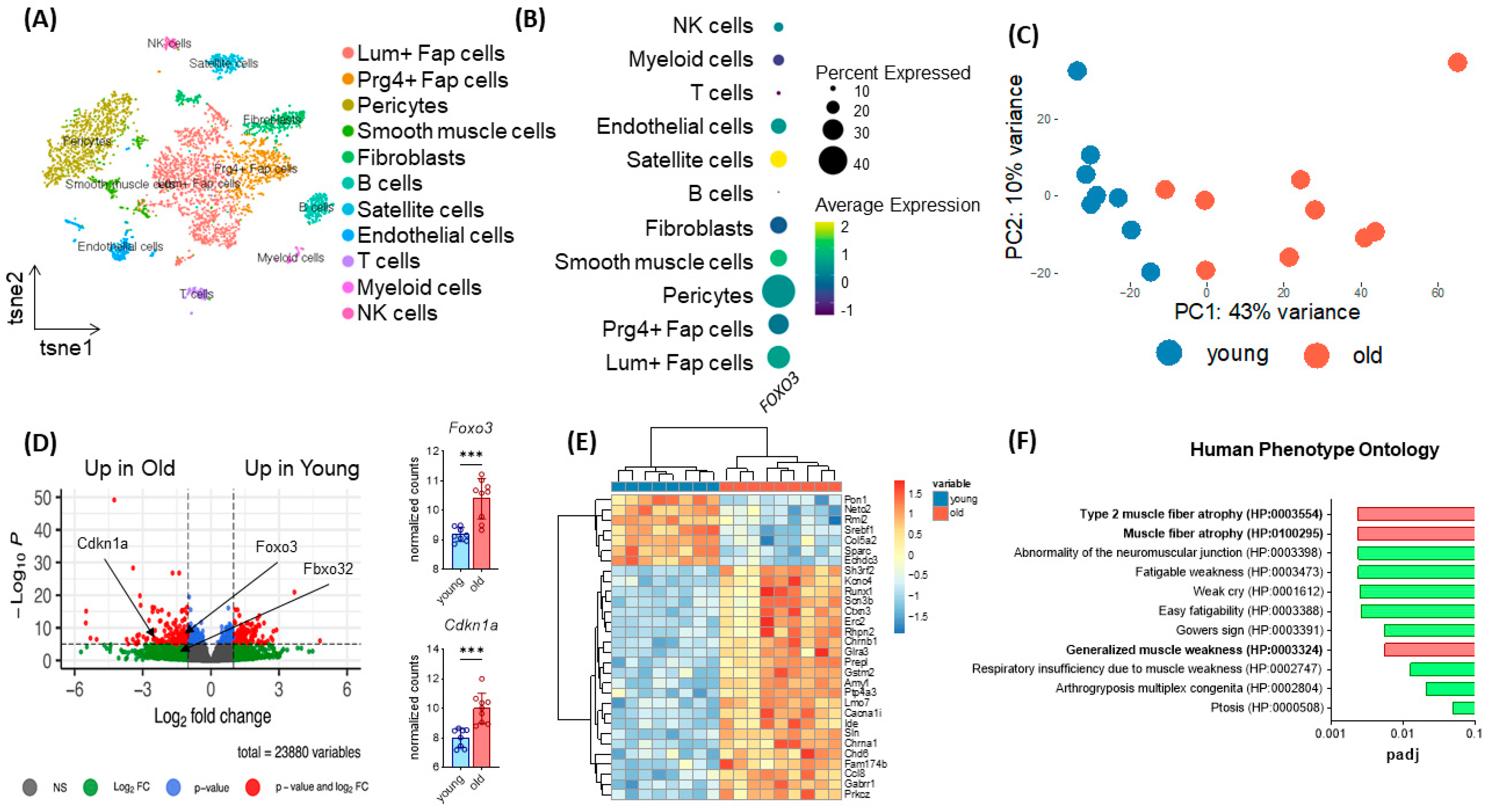
3.1. Increased Expression of Foxo3 in Satellite Cells Reflects Aging in Human and Murine Skeletal Muscle
3.2. AAV-Mediated Foxo3 Knockdown Leads to a Stable and Lasting Gene Knockdown in C2C12 Myoblasts
3.3. Foxo3 Knockdown Level and Selection of Groups with Equal Growth Rate
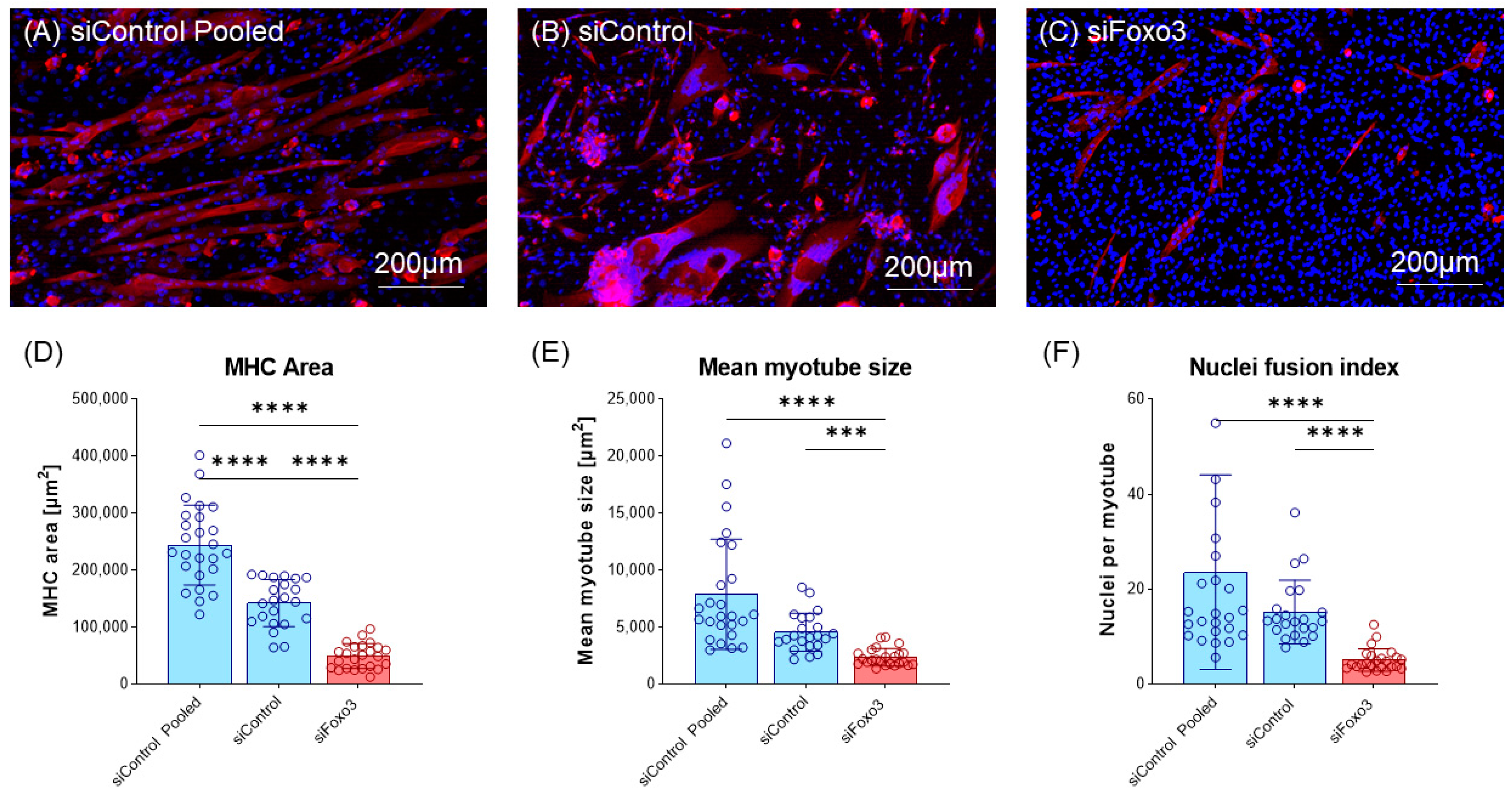
3.4. Foxo3 Knockdown Myotubes Display a Reduced Myotube Size and Lower Myoblast Fusion
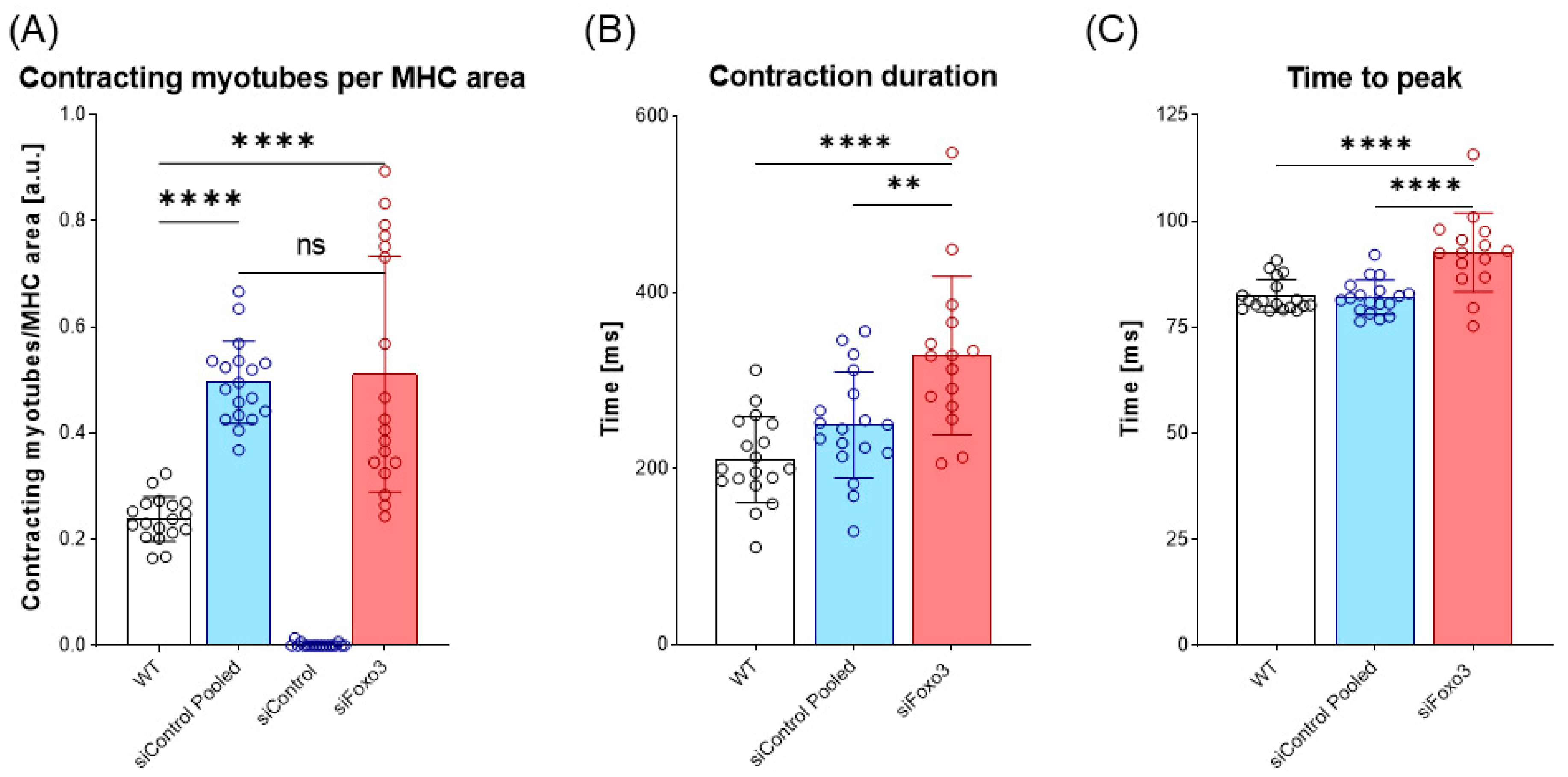
3.5. Foxo3 Knockdown Leads to Comparable Contracting Myotubes per MHC Area and Prolonged Contraction Duration and Time-to-Peak
3.6. Differentiation Marker and Genes upon Myogenic Differentiation
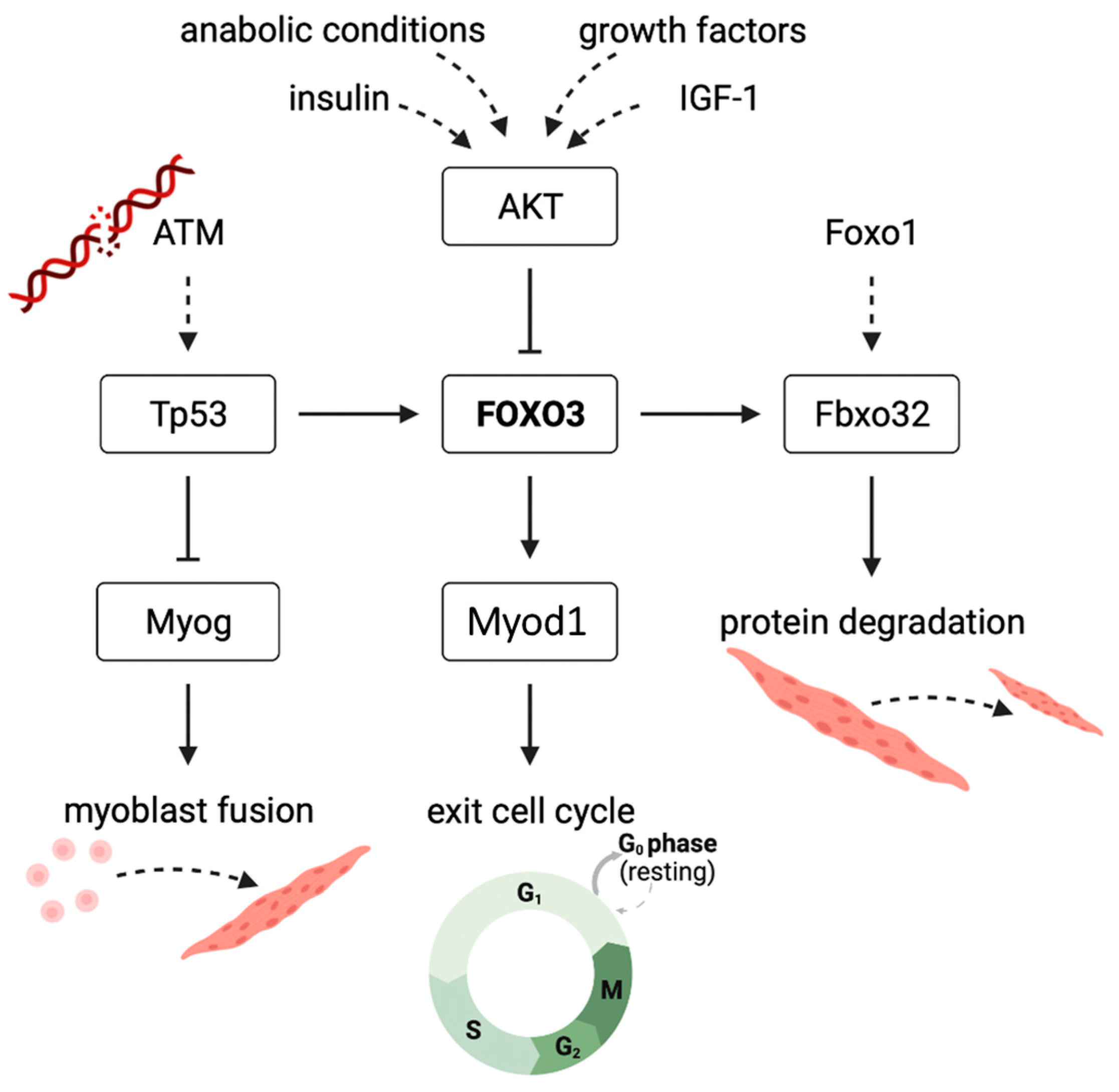
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European Consensus on Definition and Diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef]
- Martone, A.M.; Bianchi, L.; Abete, P.; Bellelli, G.; Bo, M.; Cherubini, A.; Corica, F.; Di Bari, M.; Maggio, M.; Manca, G.M.; et al. The Incidence of Sarcopenia among Hospitalized Older Patients: Results from the Glisten Study. J. Cachexia Sarcopenia Muscle 2017, 8, 907–914. [Google Scholar] [CrossRef]
- Bischoff-Ferrari, H.A.; Orav, J.E.; Kanis, J.A.; Rizzoli, R.; Schlögl, M.; Staehelin, H.B.; Willett, W.C.; Dawson-Hughes, B. Comparative Performance of Current Definitions of Sarcopenia against the Prospective Incidence of Falls among Community-Dwelling Seniors Age 65 and Older. Osteoporos Int. 2015, 26, 2793–2802. [Google Scholar] [CrossRef]
- Gariballa, S.; Alessa, A. Sarcopenia: Prevalence and Prognostic Significance in Hospitalized Patients. Clin. Nutr. 2013, 32, 772–776. [Google Scholar] [CrossRef]
- Beaudart, C.; Biver, E.; Reginster, J.; Rizzoli, R.; Rolland, Y.; Bautmans, I.; Petermans, J.; Gillain, S.; Buckinx, F.; Dardenne, N.; et al. Validation of the SarQoL®, a Specific Health-related Quality of Life Questionnaire for Sarcopenia. J. Cachexia Sarcopenia Muscle 2017, 8, 238–244. [Google Scholar] [CrossRef]
- Goates, S.; Du, K.; Arensberg, M.B.; Gaillard, T.; Guralnik, J.; Pereira, S.L. Economic Impact of Hospitalizations in US Adults with Sarcopenia. J. Frailty Aging 2019, 8, 93–99. [Google Scholar] [CrossRef]
- Pahor, M.; Guralnik, J.M.; Ambrosius, W.T.; Blair, S.; Bonds, D.E.; Church, T.S.; Espeland, M.A.; Fielding, R.A.; Gill, T.M.; Groessl, E.J.; et al. Effect of Structured Physical Activity on Prevention of Major Mobility Disability in Older Adults: The LIFE Study Randomized Clinical Trial. JAMA 2014, 311, 2387–2396. [Google Scholar] [CrossRef]
- Singh, N.A.; Quine, S.; Clemson, L.M.; Williams, E.J.; Williamson, D.A.; Stavrinos, T.M.; Grady, J.N.; Perry, T.J.; Lloyd, B.D.; Smith, E.U.R.; et al. Effects of High-Intensity Progressive Resistance Training and Targeted Multidisciplinary Treatment of Frailty on Mortality and Nursing Home Admissions after Hip Fracture: A Randomized Controlled Trial. J. Am. Med. Dir. Assoc. 2012, 13, 24–30. [Google Scholar] [CrossRef]
- Brown, C.J.; Redden, D.T.; Flood, K.L.; Allman, R.M. The Underrecognized Epidemic of Low Mobility During Hospitalization of Older Adults. J. Am. Geriatr. Soc. 2009, 57, 1660–1665. [Google Scholar] [CrossRef]
- Bauer, J.M.; Verlaan, S.; Bautmans, I.; Brandt, K.; Donini, L.M.; Maggio, M.; McMurdo, M.E.T.; Mets, T.; Seal, C.; Wijers, S.L.; et al. Effects of a Vitamin D and Leucine-Enriched Whey Protein Nutritional Supplement on Measures of Sarcopenia in Older Adults, the PROVIDE Study: A Randomized, Double-Blind, Placebo-Controlled Trial. J. Am. Med. Dir. Assoc. 2015, 16, 740–747. [Google Scholar] [CrossRef]
- Cantley, L.C. The Phosphoinositide 3-Kinase Pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Hannenhalli, S.; Kaestner, K.H. The Evolution of Fox Genes and Their Role in Development and Disease. Nat. Rev. Genet. 2009, 10, 233–240. [Google Scholar] [CrossRef]
- Latres, E.; Amini, A.R.; Amini, A.A.; Griffiths, J.; Martin, F.J.; Wei, Y.; Lin, H.C.; Yancopoulos, G.D.; Glass, D.J. Insulin-like Growth Factor-1 (IGF-1) Inversely Regulates Atrophy-Induced Genes via the Phosphatidylinositol 3-Kinase/Akt/Mammalian Target of Rapamycin (PI3K/Akt/mTOR) Pathway. J. Biol. Chem. 2005, 280, 2737–2744. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo Transcription Factors Induce the Atrophy-Related Ubiquitin Ligase Atrogin-1 and Cause Skeletal Muscle Atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Sacheck, J.M.; Ohtsuka, A.; McLary, S.C.; Goldberg, A.L. IGF-I Stimulates Muscle Growth by Suppressing Protein Breakdown and Expression of Atrophy-Related Ubiquitin Ligases, Atrogin-1 and MuRF1. Am. J. Physiol.-Endocrinol. Metab. 2004, 287, E591–E601. [Google Scholar] [CrossRef] [PubMed]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/PI3K/Akt Pathway Prevents Expression of Muscle Atrophy-Induced Ubiquitin Ligases by Inhibiting FOXO Transcription Factors. Mol. Cell 2004, 14, 395–403. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.-M.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of Ubiquitin Ligases Required for Skeletal Muscle Atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Tjørve, K.M.C.; Tjørve, E. The Use of Gompertz Models in Growth Analyses, and New Gompertz-Model Approach: An Addition to the Unified-Richards Family. PLoS ONE 2017, 12, e0178691. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Wagdi, A.; Malan, D.; Sathyanarayanan, U.; Beauchamp, J.S.; Vogt, M.; Zipf, D.; Beiert, T.; Mansuroglu, B.; Dusend, V.; Meininghaus, M.; et al. Selective Optogenetic Control of Gq Signaling Using Human Neuropsin. Nat. Commun. 2022, 13, 1765. [Google Scholar] [CrossRef]
- Börsch, A.; Ham, D.J.; Mittal, N.; Tintignac, L.A.; Migliavacca, E.; Feige, J.N.; Rüegg, M.A.; Zavolan, M. Molecular and Phenotypic Analysis of Rodent Models Reveals Conserved and Species-Specific Modulators of Human Sarcopenia. Commun. Biol. 2021, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Saul, D.; Kosinsky, R.L. Single-Cell Transcriptomics Reveals the Expression of Aging- and Senescence-Associated Genes in Distinct Cancer Cell Populations. Cells 2021, 10, 3126. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Rubenstein, A.B.; Smith, G.R.; Raue, U.; Begue, G.; Minchev, K.; Ruf-Zamojski, F.; Nair, V.D.; Wang, X.; Zhou, L.; Zaslavsky, E.; et al. Single-Cell Transcriptional Profiles in Human Skeletal Muscle. Sci. Rep. 2020, 10, 229. [Google Scholar] [CrossRef] [PubMed]
- Saul, D.; Kosinsky, R.L.; Atkinson, E.J.; Doolittle, M.L.; Zhang, X.; LeBrasseur, N.K.; Pignolo, R.J.; Robbins, P.D.; Niedernhofer, L.J.; Ikeno, Y.; et al. A New Gene Set Identifies Senescent Cells and Predicts Senescence-Associated Pathways across Tissues. Nat. Commun. 2022, 13, 4827. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, D.; Saxel, O. Serial Passaging and Differentiation of Myogenic Cells Isolated from Dystrophic Mouse Muscle. Nature 1977, 270, 725–727. [Google Scholar] [CrossRef]
- Choi, J.; Costa, M.L.; Mermelstein, C.S.; Chagas, C.; Holtzer, S.; Holtzer, H. MyoD Converts Primary Dermal Fibroblasts, Chondroblasts, Smooth Muscle, and Retinal Pigmented Epithelial Cells into Striated Mononucleated Myoblasts and Multinucleated Myotubes. Proc. Natl. Acad. Sci. USA 1990, 87, 7988–7992. [Google Scholar] [CrossRef]
- Ganassi, M.; Badodi, S.; Ortuste Quiroga, H.P.; Zammit, P.S.; Hinits, Y.; Hughes, S.M. Myogenin Promotes Myocyte Fusion to Balance Fibre Number and Size. Nat. Commun. 2018, 9, 4232. [Google Scholar] [CrossRef]
- Shiloh, Y.; Ziv, Y. The ATM Protein Kinase: Regulating the Cellular Response to Genotoxic Stress, and More. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.L.; Raue, U.; Slivka, D.R.; Trappe, S. Resistance Exercise, Skeletal Muscle FOXO3A, and 85-Year-Old Women. J. Gerontol. Ser. A 2010, 65A, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Parolo, S.; Marchetti, L.; Lauria, M.; Misselbeck, K.; Scott-Boyer, M.-P.; Caberlotto, L.; Priami, C. Combined Use of Protein Biomarkers and Network Analysis Unveils Deregulated Regulatory Circuits in Duchenne Muscular Dystrophy. PLoS ONE 2018, 13, e0194225. [Google Scholar] [CrossRef] [PubMed]
- Attaix, D.; Bechet, D. FoxO3 Controls Dangerous Proteolytic Liaisons. Cell Metab. 2007, 6, 425–427. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhu, M.; Lu, Z.; Zhang, Y.; Li, L.; Li, N.; Yin, L.; Wang, H.; Song, W.; Xu, H. L-Carnitine Ameliorates the Muscle Wasting of Cancer Cachexia through the AKT/FOXO3a/MaFbx Axis. Nutr. Metab. 2021, 18, 98. [Google Scholar] [CrossRef] [PubMed]
- Sawicka, A.K.; Hartmane, D.; Lipinska, P.; Wojtowicz, E.; Lysiak-Szydlowska, W.; Olek, R.A. L-Carnitine Supplementation in Older Women. A Pilot Study on Aging Skeletal Muscle Mass and Function. Nutrients 2018, 10, 255. [Google Scholar] [CrossRef] [PubMed]
- Judge, S.M.; Wu, C.-L.; Beharry, A.W.; Roberts, B.M.; Ferreira, L.F.; Kandarian, S.C.; Judge, A.R. Genome-Wide Identification of FoxO-Dependent Gene Networks in Skeletal Muscle during C26 Cancer Cachexia. BMC Cancer 2014, 14, 997. [Google Scholar] [CrossRef]
- Che, J.; Xu, C.; Wu, Y.; Jia, P.; Han, Q.; Ma, Y.; Wang, X.; Zheng, Y. MiR-1290 Promotes Myoblast Differentiation and Protects against Myotube Atrophy via Akt/P70/FoxO3 Pathway Regulation. Skelet. Muscle 2021, 11, 6. [Google Scholar] [CrossRef]
- Tang, E.D.; Nuñez, G.; Barr, F.G.; Guan, K.-L. Negative Regulation of the Forkhead Transcription Factor FKHR by Akt. J. Biol. Chem. 1999, 274, 16741–16746. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.F.; Yan, X.; Zhu, M.J.; Du, M. AMP-Activated Protein Kinase Enhances the Expression of Muscle-Specific Ubiquitin Ligases despite Its Activation of IGF-1/Akt Signaling in C2C12 Myotubes. J. Cell. Biochem. 2009, 108, 458–468. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Hoshino, Y.; Ito, T.; Nariai, T.; Mohri, T.; Obana, M.; Hayata, N.; Uozumi, Y.; Maeda, M.; Fujio, Y.; et al. Atrogin-1 Ubiquitin Ligase Is Upregulated by Doxorubicin via P38-MAP Kinase in Cardiac Myocytes. Cardiovasc. Res. 2008, 79, 89–96. [Google Scholar] [CrossRef]
- Hu, P.; Geles, K.G.; Paik, J.-H.; DePinho, R.A.; Tjian, R. Codependent Activators Direct Myoblast-Specific MyoD Transcription. Dev. Cell 2008, 15, 534–546. [Google Scholar] [CrossRef]
- Jeong, J.; Choi, K.-H.; Kim, S.-H.; Lee, D.-K.; Oh, J.-N.; Lee, M.; Choe, G.C.; Lee, C.-K. Combination of Cell Signaling Molecules Can Facilitate MYOD1-Mediated Myogenic Transdifferentiation of Pig Fibroblasts. J. Anim. Sci. Biotechnol. 2021, 12, 64. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Sato, S.; Chikazawa-Nohtomi, N.; Soma, A.; Kimura, H.; Wakabayashi, S.; Ko, S.B.H.; Ko, M.S.H. Efficient Differentiation of Human Pluripotent Stem Cells into Skeletal Muscle Cells by Combining RNA-Based MYOD1-Expression and POU5F1-Silencing. Sci. Rep. 2018, 8, 1189. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Lv, Y.; Shen, X.Q.; Xu, J.H.; Lu, H.; Fu, L.C.; Duan, T. Implantation of Muscle Satellite Cells Overexpressing Myogenin Improves Denervated Muscle Atrophy in Rats. Braz. J. Med. Biol. Res. 2016, 49, e5124. [Google Scholar] [CrossRef]
- Klagge, A.; Weidinger, C.; Krause, K.; Jessnitzer, B.; Gutknecht, M.; Fuhrer, D. The Role of FOXO3 in DNA Damage Response in Thyrocytes. Endocr. Relat. Cancer 2011, 18, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Kurinna, S.; Stratton, S.A.; Tsai, W.-W.; Akdemir, K.C.; Gu, W.; Singh, P.; Goode, T.; Darlington, G.J.; Barton, M.C. Direct Activation of Forkhead Box O3 by Tumor Suppressors P53 and P73 Is Disrupted during Liver Regeneration in Mice. Hepatology 2010, 52, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Brüggenwirth, I.M.A.; Martins, P.N. RNA Interference Therapeutics in Organ Transplantation: The Dawn of a New Era. Am. J. Transplant. 2020, 20, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.P.; Kenzelmann Broz, D.; Noderer, W.L.; Ferreira, J.P.; Overton, K.W.; Spencer, S.L.; Meyer, T.; Tapscott, S.J.; Attardi, L.D.; Wang, C.L. P53 Suppresses Muscle Differentiation at the Myogenin Step in Response to Genotoxic Stress. Cell Death Differ. 2015, 22, 560–573. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Kumar, R.M.; Penn, B.H.; Berkes, C.A.; Kooperberg, C.; Boyer, L.A.; Young, R.A.; Tapscott, S.J. Global and Gene-Specific Analyses Show Distinct Roles for Myod and Myog at a Common Set of Promoters. EMBO J. 2006, 25, 502–511. [Google Scholar] [CrossRef]
- Lee, H.-Y.; Youn, S.-W.; Kim, J.-Y.; Park, K.-W.; Hwang, C.-I.; Park, W.-Y.; Oh, B.-H.; Park, Y.-B.; Walsh, K.; Seo, J.-S.; et al. FOXO3a Turns the Tumor Necrosis Factor Receptor Signaling towards Apoptosis through Reciprocal Regulation of C-Jun N-Terminal Kinase and NF-kappaB. Arter. Thromb. Vasc. Biol. 2008, 28, 112–120. [Google Scholar] [CrossRef]
- Badu-Mensah, A.; Guo, X.; McAleer, C.W.; Rumsey, J.W.; Hickman, J.J. Functional Skeletal Muscle Model Derived from SOD1-Mutant ALS Patient iPSCs Recapitulates Hallmarks of Disease Progression. Sci. Rep. 2020, 10, 14302. [Google Scholar] [CrossRef]
- Takagi, S.; Nakamura, T.; Fujisato, T. Effect of Heat Stress on Contractility of Tissue-Engineered Artificial Skeletal Muscle. J. Artif. Organs 2018, 21, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral Vector Platforms within the Gene Therapy Landscape. Sig. Transduct. Target. Ther. 2021, 6, 1–24. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′→3′) | Reverse Primer (5′→3′) |
|---|---|---|
| Foxo3 | CGC TGT GTG CCC TAC TTC | CCC GTG CCT TCA TTC TGA |
| Fbxo32 | CAG CTT CGT GAG CGA CCT C | GGC AGT CGA GAA GTC CAG TC |
| Myod1 | GCC CGC GCT CCA ACT GCT CTG | CCT ACG GTG GTG CGC CCT CTG |
| Myog | CAT CCA GTA CAT TGA GCG CCT | GAG CAA ATG ATC TCC TGG GTT |
| Tp53 | CAC AGC ACA TGA CGG AGG TC | TCC TTC CAC CCG GAT AAG ATG |
| ATM | GAG TGA GAC GGG CTG TTA CC | CAT GCT GCC TCC TTC TTT TC |
| Foxo1 | GCG GGC TGG AAG AAT TCA AT | TCC AGT TCC TTC ATT CTG CA |
| Gapdh | CTC CCA CTC TTC CAC CCT CG | GCC TCT CTT GCT GAG TGT CC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gellhaus, B.; Böker, K.O.; Gsaenger, M.; Rodenwaldt, E.; Hüser, M.A.; Schilling, A.F.; Saul, D. Foxo3 Knockdown Mediates Decline of Myod1 and Myog Reducing Myoblast Conversion to Myotubes. Cells 2023, 12, 2167. https://doi.org/10.3390/cells12172167
Gellhaus B, Böker KO, Gsaenger M, Rodenwaldt E, Hüser MA, Schilling AF, Saul D. Foxo3 Knockdown Mediates Decline of Myod1 and Myog Reducing Myoblast Conversion to Myotubes. Cells. 2023; 12(17):2167. https://doi.org/10.3390/cells12172167
Chicago/Turabian StyleGellhaus, Benjamin, Kai O. Böker, Marlene Gsaenger, Eyck Rodenwaldt, Marc A. Hüser, Arndt F. Schilling, and Dominik Saul. 2023. "Foxo3 Knockdown Mediates Decline of Myod1 and Myog Reducing Myoblast Conversion to Myotubes" Cells 12, no. 17: 2167. https://doi.org/10.3390/cells12172167