
The Role and Therapeutic Targeting of CCR5 in Breast Cancer


 ,
, 
Abstract
:1. Introduction
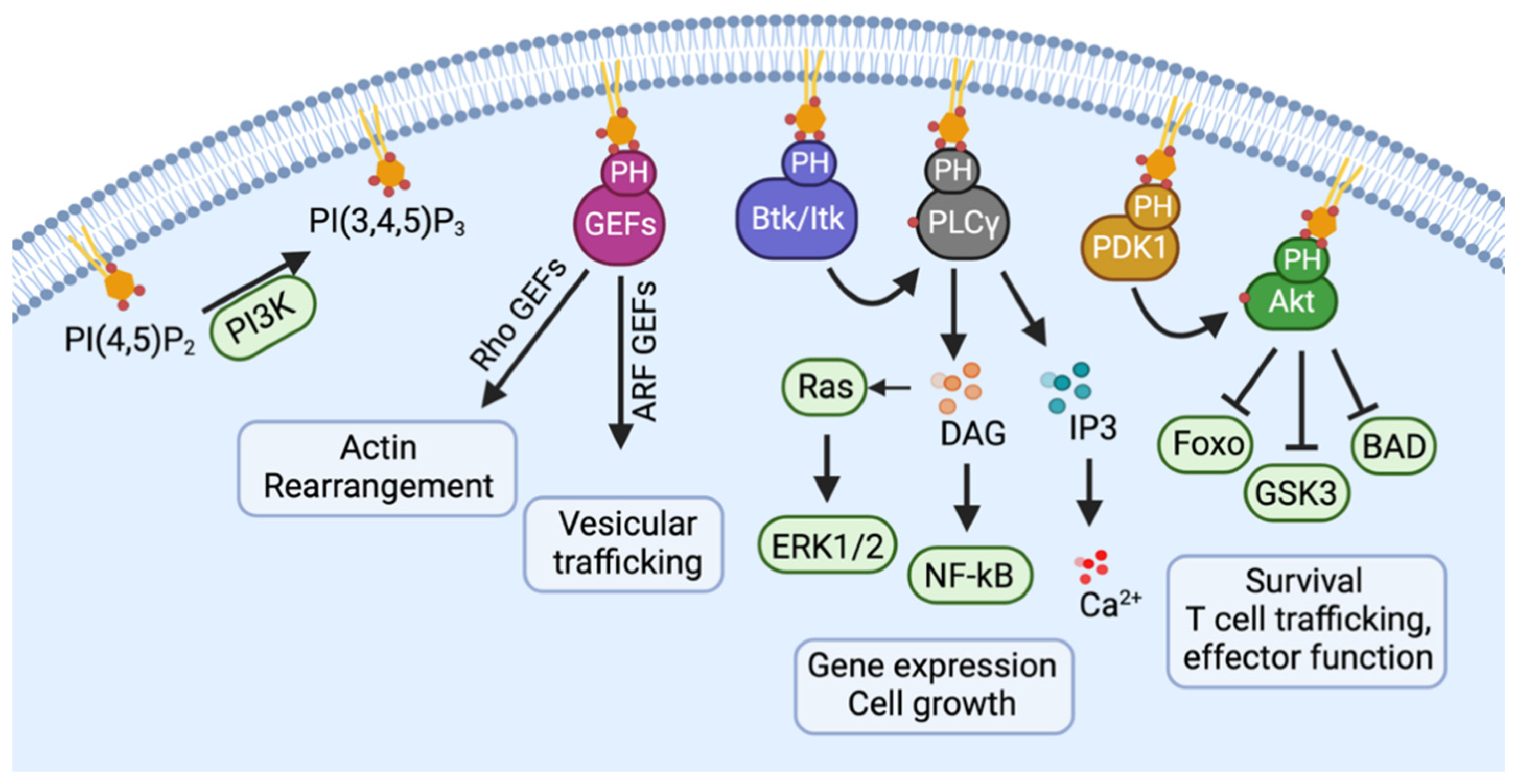
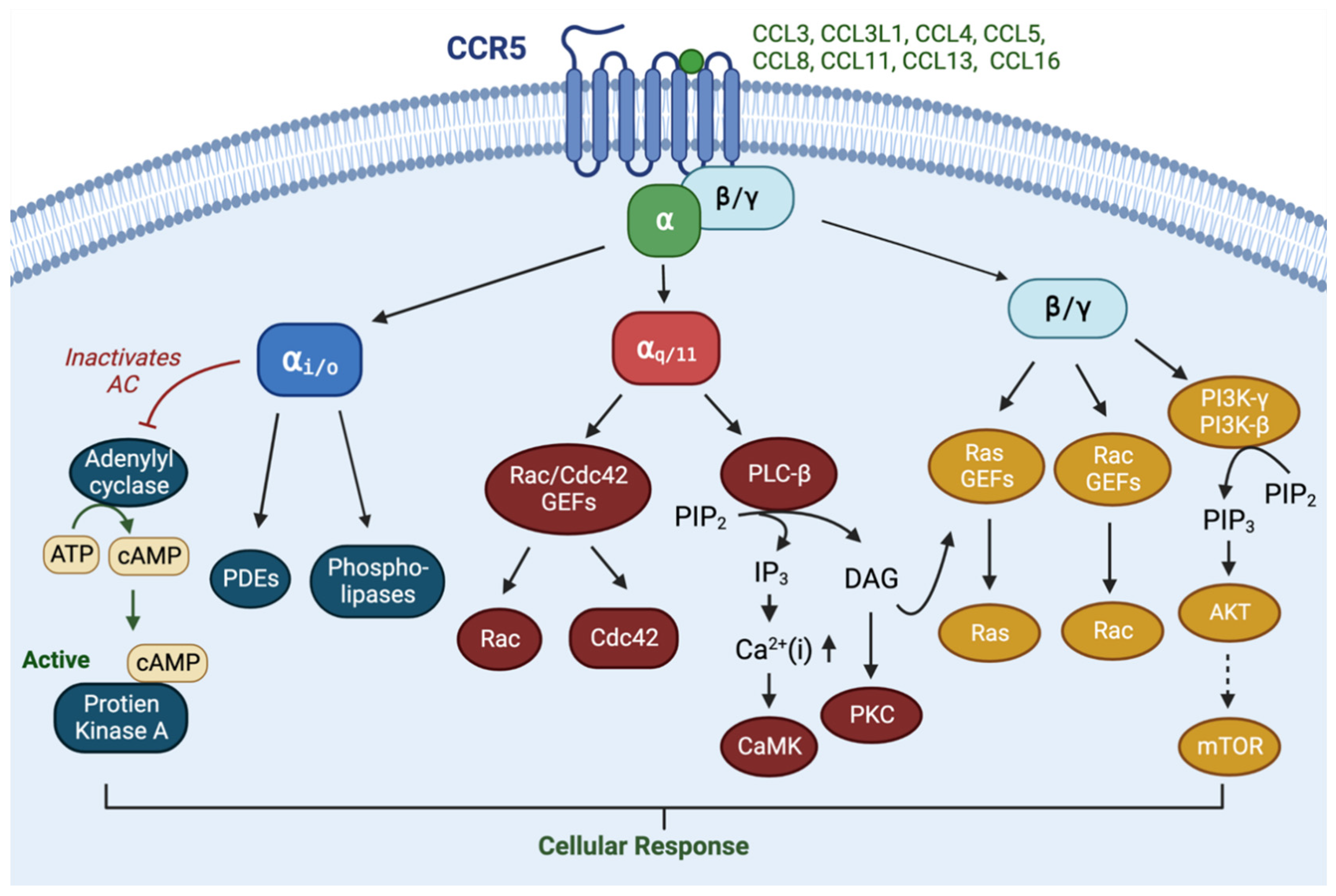
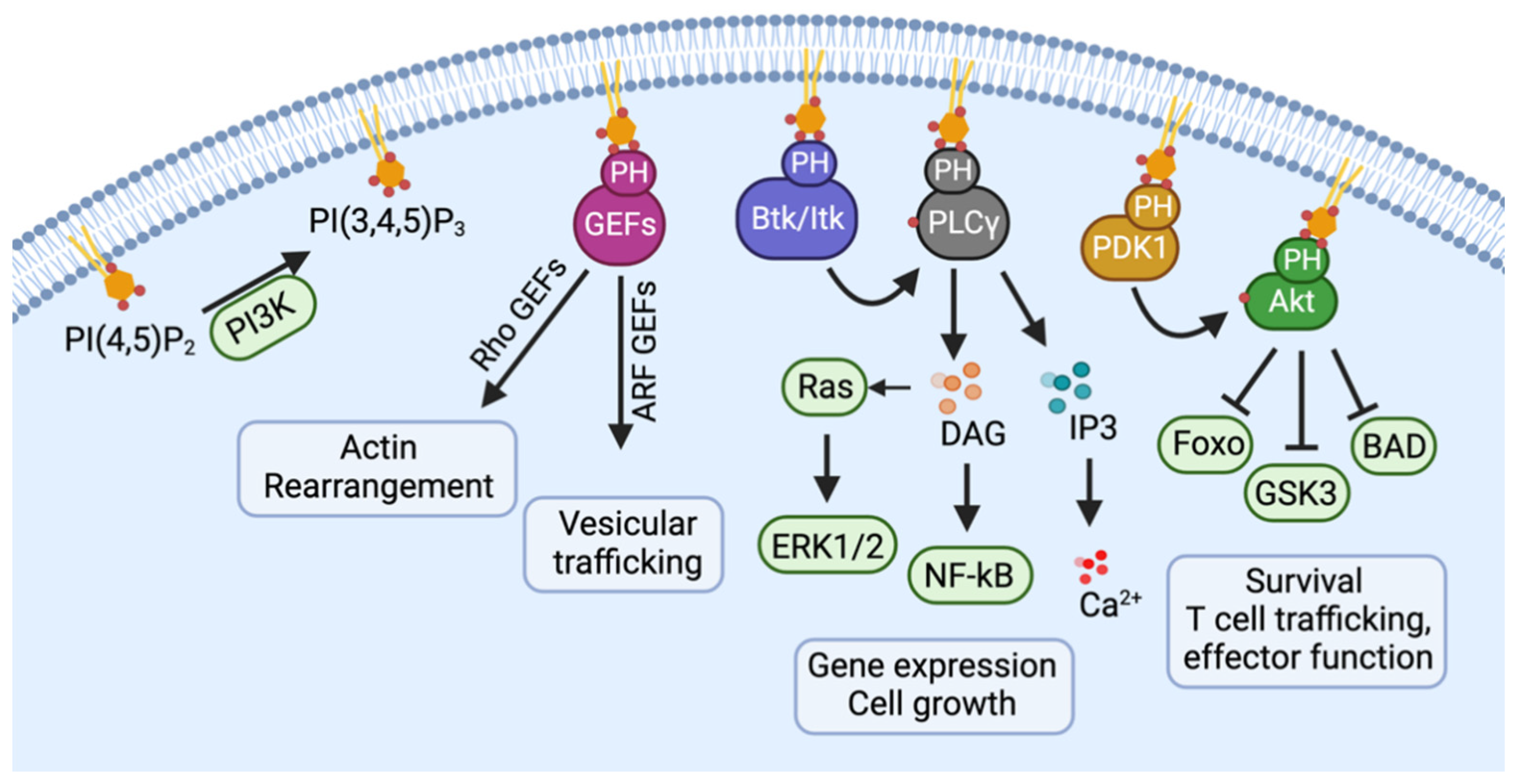
2. Normal Physiology of CCR5
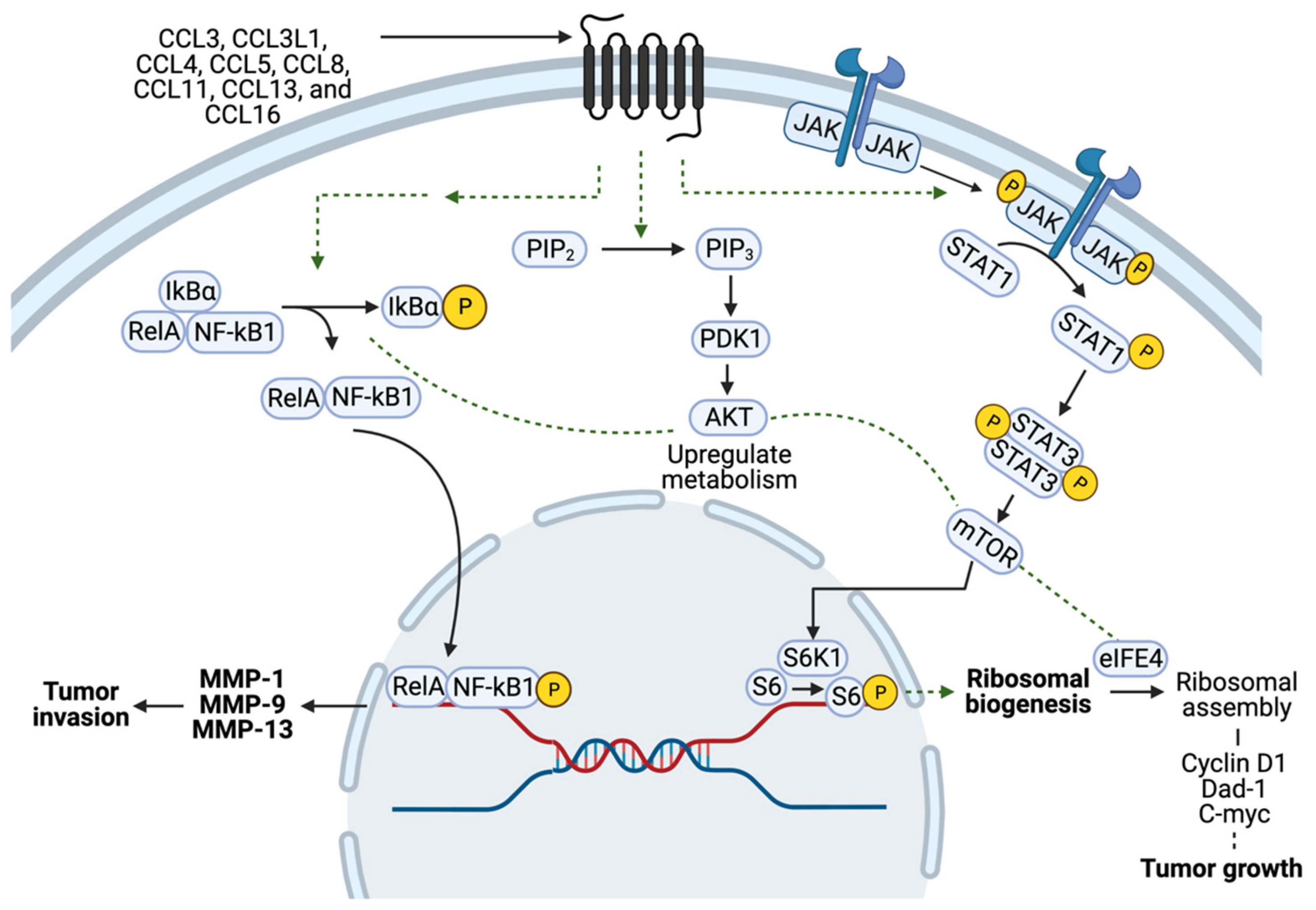
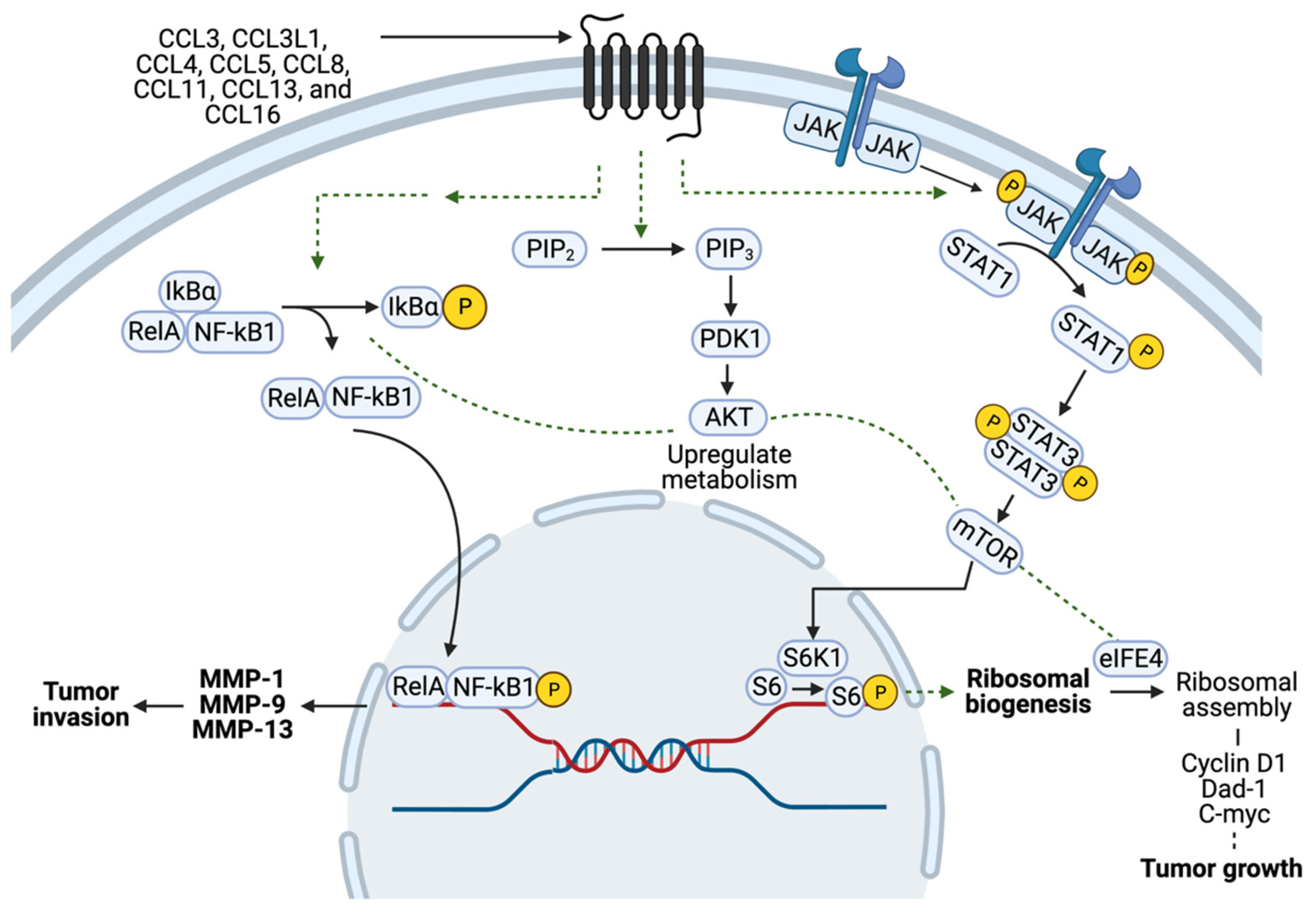
3. Pathophysiology of CCR5 in Cancers
4. The Role of CCR5 in Breast Cancer Cell Metabolism
5. The Role of CCR5 in Tumor Migration, Circulating Tumor Cells, and Tumor Metastasis
6. Involvement of CCR5 in Breast Tumor Angiogenesis
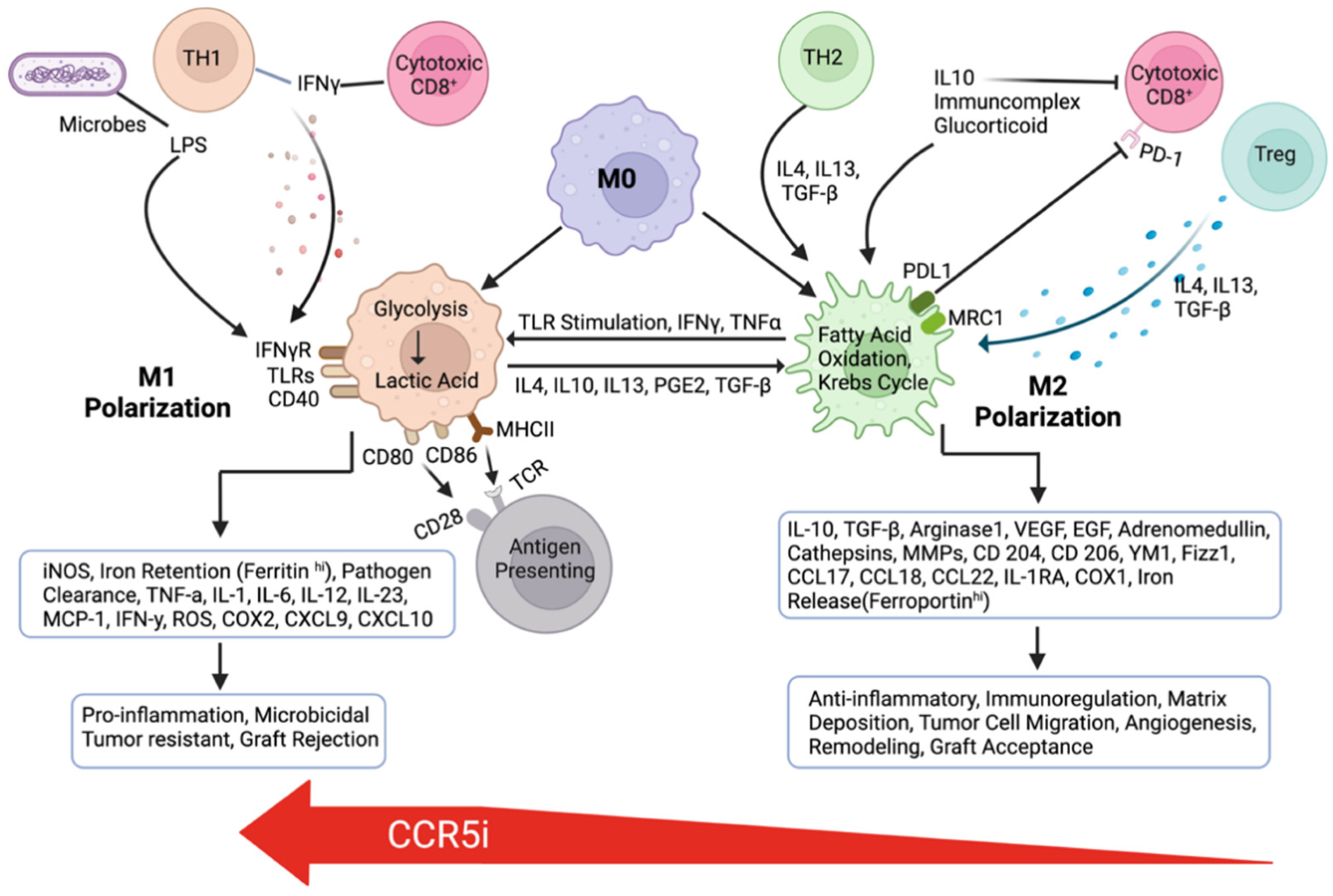
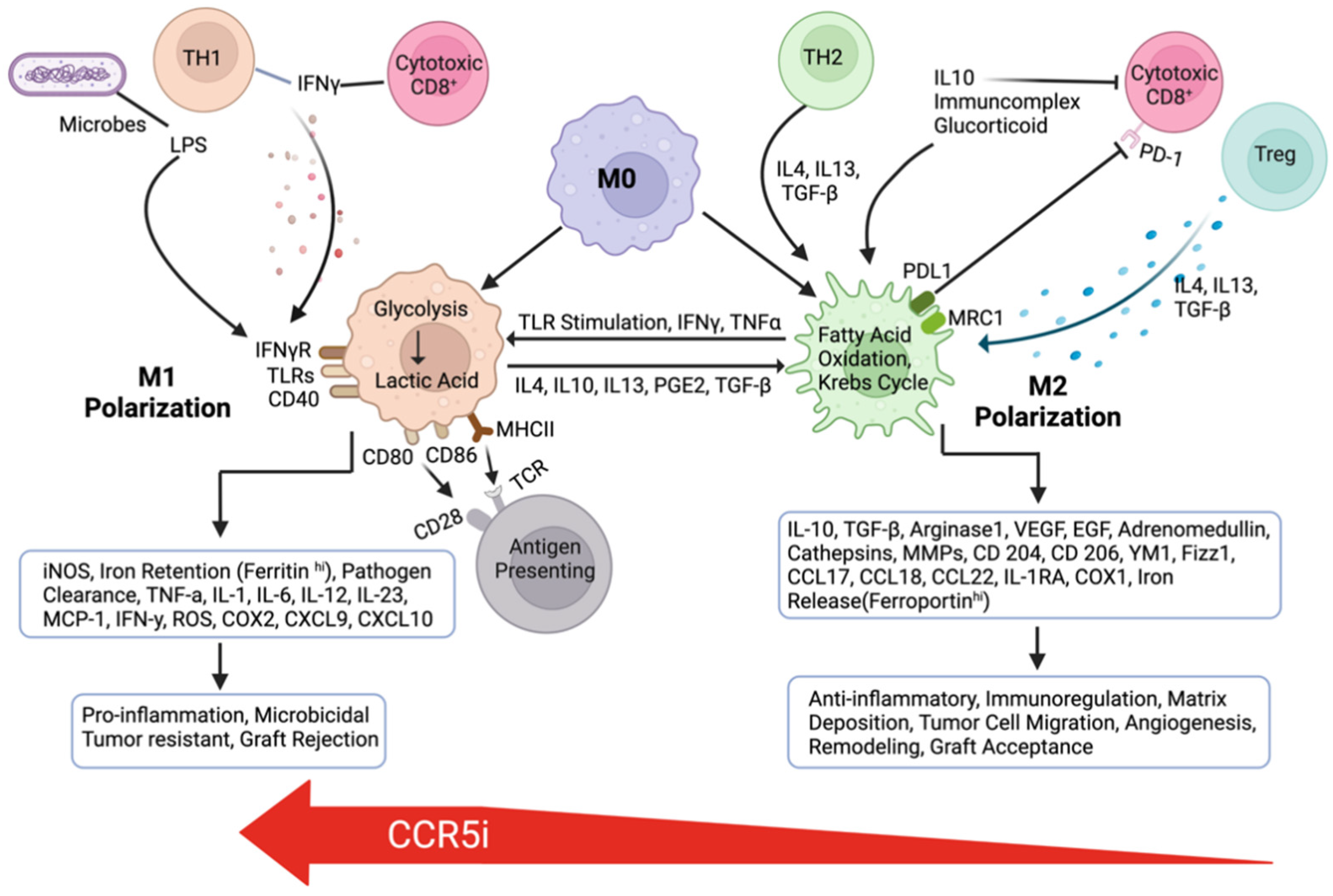
7. The Role of the CCR5-CCL5 Axis in Immune Evasion
8. The Role of CCR5 in Breast Cancer Stem Cell Expansion and Chemo-/Radio-Therapy Resistance
9. CCR5 Inhibitors for Treating Breast Cancer
10. Triple-Negative Breast Cancer Subtypes and CCR5 Inhibitor Therapy
11. Potential Role for CCR5 Inhibitors in Augmenting the Therapeutic Response to Current Breast Cancer Therapies
12. Checkpoint Inhibitors
13. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- SEER. Cancer Stat Facts: Female Breast Cancer. Available online: https://seer.cancer.gov/statfacts/html/breast.html (accessed on 5 August 2023).
- Chang, J.M.; McCullough, A.E.; Dueck, A.C.; Kosiorek, H.E.; Ocal, I.T.; Lidner, T.K.; Gray, R.J.; Wasif, N.; Northfelt, D.W.; Anderson, K.S.; et al. Back to Basics: Traditional Nottingham Grade Mitotic Counts Alone are Significant in Predicting Survival in Invasive Breast Carcinoma. Ann. Surg. Oncol. 2015, 22 (Suppl. S3), S509–S515. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Green, A.R. Molecular classification of breast cancer: What the pathologist needs to know. Pathology 2017, 49, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Makki, J. Diversity of Breast Carcinoma: Histological Subtypes and Clinical Relevance. Clin. Med. Insights Pathol. 2015, 8, 23–31. [Google Scholar] [CrossRef]
- Russnes, H.G.; Lingjaerde, O.C.; Borresen-Dale, A.L.; Caldas, C. Breast Cancer Molecular Stratification: From Intrinsic Subtypes to Integrative Clusters. Am. J. Pathol. 2017, 187, 2152–2162. [Google Scholar] [CrossRef]
- Wallden, B.; Storhoff, J.; Nielsen, T.; Dowidar, N.; Schaper, C.; Ferree, S.; Liu, S.; Leung, S.; Geiss, G.; Snider, J.; et al. Development and verification of the PAM50-based Prosigna breast cancer gene signature assay. BMC Med. Genom. 2015, 8, 54. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Velasco-Velazquez, M.; Jiao, X.; De La Fuente, M.; Pestell, T.G.; Ertel, A.; Lisanti, M.P.; Pestell, R.G. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. 2012, 72, 3839–3850. [Google Scholar] [CrossRef]
- Jiao, X.; Nawab, O.; Patel, T.; Kossenkov, A.V.; Halama, N.; Jaeger, D.; Pestell, R.G. Recent Advances Targeting CCR5 for Cancer and Its Role in Immuno-Oncology. Cancer Res. 2019, 79, 4801–4807. [Google Scholar] [CrossRef]
- Jiao, X.; Velasco-Velazquez, M.A.; Wang, M.; Li, Z.; Rui, H.; Peck, A.R.; Korkola, J.E.; Chen, X.; Xu, S.; DuHadaway, J.B.; et al. CCR5 Governs DNA Damage Repair and Breast Cancer Stem Cell Expansion. Cancer Res. 2018, 78, 1657–1671. [Google Scholar] [CrossRef]
- Span, P.N.; Pollakis, G.; Paxton, W.A.; Sweep, F.C.; Foekens, J.A.; Martens, J.W.; Sieuwerts, A.M.; van Laarhoven, H.W. Improved metastasis-free survival in nonadjuvantly treated postmenopausal breast cancer patients with chemokine receptor 5 del32 frameshift mutations. Int. J. Cancer 2015, 136, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Carmona, M.; Chaorentong, P.; Kather, J.N.; Rothenheber, R.; Ahmed, A.; Berthel, A.; Heinzelmann, A.; Moraleda, R.; Valous, N.A.; Kosaloglu, Z.; et al. CCR5 status and metastatic progression in colorectal cancer. Oncoimmunology 2019, 8, e1626193. [Google Scholar] [CrossRef] [PubMed]
- Wu, V.; Yeerna, H.; Nohata, N.; Chiou, J.; Harismendy, O.; Raimondi, F.; Inoue, A.; Russell, R.B.; Tamayo, P.; Gutkind, J.S. Illuminating the Onco-GPCRome: Novel G protein-coupled receptor-driven oncocrine networks and targets for cancer immunotherapy. J. Biol. Chem. 2019, 294, 11062–11086. [Google Scholar] [CrossRef] [PubMed]
- Gilman, A.G. G proteins: Transducers of receptor-generated signals. Annu. Rev. Biochem. 1987, 56, 615–649. [Google Scholar] [CrossRef]
- Lorenzen, E.; Ceraudo, E.; Berchiche, Y.A.; Rico, C.A.; Furstenberg, A.; Sakmar, T.P.; Huber, T. G protein subtype-specific signaling bias in a series of CCR5 chemokine analogs. Sci. Signal 2018, 11, eaao6152. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, M. Chemokine receptor CCR5: Insights into structure, function, and regulation. Cell Signal 2004, 16, 1201–1210. [Google Scholar] [CrossRef]
- Blanpain, C.; Migeotte, I.; Lee, B.; Vakili, J.; Doranz, B.J.; Govaerts, C.; Vassart, G.; Doms, R.W.; Parmentier, M. CCR5 binds multiple CC-chemokines: MCP-3 acts as a natural antagonist. Blood 1999, 94, 1899–1905. [Google Scholar] [CrossRef]
- Nomiyama, H.; Hieshima, K.; Nakayama, T.; Sakaguchi, T.; Fujisawa, R.; Tanase, S.; Nishiura, H.; Matsuno, K.; Takamori, H.; Tabira, Y.; et al. Human CC chemokine liver-expressed chemokine/CCL16 is a functional ligand for CCR1, CCR2 and CCR5, and constitutively expressed by hepatocytes. Int. Immunol. 2001, 13, 1021–1029. [Google Scholar] [CrossRef]
- O’Hayre, M.; Degese, M.S.; Gutkind, J.S. Novel insights into G protein and G protein-coupled receptor signaling in cancer. Curr. Opin. Cell Biol. 2014, 27, 126–135. [Google Scholar] [CrossRef]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef]
- Chen, X.; Wu, Q.; Depeille, P.; Chen, P.; Thornton, S.; Kalirai, H.; Coupland, S.E.; Roose, J.P.; Bastian, B.C. RasGRP3 Mediates MAPK Pathway Activation in GNAQ Mutant Uveal Melanoma. Cancer Cell 2017, 31, 685–696.e6. [Google Scholar] [CrossRef] [PubMed]
- Chikumi, H.; Vazquez-Prado, J.; Servitja, J.M.; Miyazaki, H.; Gutkind, J.S. Potent activation of RhoA by Galpha q and Gq-coupled receptors. J. Biol. Chem. 2002, 277, 27130–27134. [Google Scholar] [CrossRef] [PubMed]
- Vaque, J.P.; Dorsam, R.T.; Feng, X.; Iglesias-Bartolome, R.; Forsthoefel, D.J.; Chen, Q.; Debant, A.; Seeger, M.A.; Ksander, B.R.; Teramoto, H.; et al. A genome-wide RNAi screen reveals a Trio-regulated Rho GTPase circuitry transducing mitogenic signals initiated by G protein-coupled receptors. Mol. Cell 2013, 49, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hills, L.B.; Huang, Y.H. Lipid and Protein Co-Regulation of PI3K Effectors Akt and Itk in Lymphocytes. Front. Immunol. 2015, 6, 117. [Google Scholar] [CrossRef]
- Sicoli, D.; Jiao, X.; Ju, X.; Velasco-Velazquez, M.; Ertel, A.; Addya, S.; Li, Z.; Ando, S.; Fatatis, A.; Paudyal, B.; et al. CCR5 receptor antagonists block metastasis to bone of v-Src oncogene-transformed metastatic prostate cancer cell lines. Cancer Res. 2014, 74, 7103–7114. [Google Scholar] [CrossRef]
- Halama, N.; Zoernig, I.; Berthel, A.; Kahlert, C.; Klupp, F.; Suarez-Carmona, M.; Suetterlin, T.; Brand, K.; Krauss, J.; Lasitschka, F.; et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell 2016, 29, 587–601. [Google Scholar] [CrossRef]
- Pervaiz, A.; Ansari, S.; Berger, M.R.; Adwan, H. CCR5 blockage by maraviroc induces cytotoxic and apoptotic effects in colorectal cancer cells. Med. Oncol. 2015, 32, 158. [Google Scholar] [CrossRef]
- Liu, J.; Wang, C.; Ma, X.; Tian, Y.; Wang, C.; Fu, Y.; Luo, Y. High expression of CCR5 in melanoma enhances epithelial-mesenchymal transition and metastasis via TGFbeta1. J. Pathol. 2019, 247, 481–493. [Google Scholar] [CrossRef]
- Casagrande, N.; Borghese, C.; Visser, L.; Mongiat, M.; Colombatti, A.; Aldinucci, D. CCR5 antagonism by maraviroc inhibits Hodgkin lymphoma microenvironment interactions and xenograft growth. Haematologica 2019, 104, 564–575. [Google Scholar] [CrossRef]
- Gonzalez-Arriagada, W.A.; Lozano-Burgos, C.; Zuniga-Moreta, R.; Gonzalez-Diaz, P.; Coletta, R.D. Clinicopathological significance of chemokine receptor (CCR1, CCR3, CCR4, CCR5, CCR7 and CXCR4) expression in head and neck squamous cell carcinomas. J. Oral. Pathol. Med. 2018, 47, 755–763. [Google Scholar] [CrossRef]
- Aldinucci, D.; Casagrande, N. Inhibition of the CCL5/CCR5 Axis against the Progression of Gastric Cancer. Int. J. Mol. Sci. 2018, 19, 1477. [Google Scholar] [CrossRef]
- Wu, Y.C.; Shen, Y.C.; Chang, J.W.; Hsieh, J.J.; Chu, Y.; Wang, C.H. Autocrine CCL5 promotes tumor progression in esophageal squamous cell carcinoma in vitro. Cytokine 2018, 110, 94–103. [Google Scholar] [CrossRef]
- Zi, J.; Yuan, S.; Qiao, J.; Zhao, K.; Xu, L.; Qi, K.; Xu, K.; Zeng, L. Treatment with the C-C chemokine receptor type 5 (CCR5)-inhibitor maraviroc suppresses growth and induces apoptosis of acute lymphoblastic leukemia cells. Am. J. Cancer Res. 2017, 7, 869–880. [Google Scholar] [PubMed]
- Singh, S.K.; Mishra, M.K.; Eltoum, I.A.; Bae, S.; Lillard, J.W., Jr.; Singh, R. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci. Rep. 2018, 8, 1323. [Google Scholar] [CrossRef] [PubMed]
- Yaal-Hahoshen, N.; Shina, S.; Leider-Trejo, L.; Barnea, I.; Shabtai, E.L.; Azenshtein, E.; Greenberg, I.; Keydar, I.; Ben-Baruch, A. The chemokine CCL5 as a potential prognostic factor predicting disease progression in stage II breast cancer patients. Clin. Cancer Res. 2006, 12, 4474–4480. [Google Scholar] [CrossRef] [PubMed]
- Niwa, Y.; Akamatsu, H.; Niwa, H.; Sumi, H.; Ozaki, Y.; Abe, A. Correlation of tissue and plasma RANTES levels with disease course in patients with breast or cervical cancer. Clin. Cancer Res. 2001, 7, 285–289. [Google Scholar]
- Tsukishiro, S.; Suzumori, N.; Nishikawa, H.; Arakawa, A.; Suzumori, K. Elevated serum RANTES levels in patients with ovarian cancer correlate with the extent of the disorder. Gynecol. Oncol. 2006, 102, 542–545. [Google Scholar] [CrossRef]
- Sima, A.R.; Sima, H.R.; Rafatpanah, H.; Hosseinnezhad, H.; Ghaffarzadehgan, K.; Valizadeh, N.; Mehrabi Bahar, M.; Hakimi, H.R.; Masoom, A.; Noorbakhsh, A.; et al. Serum chemokine ligand 5 (CCL5/RANTES) level might be utilized as a predictive marker of tumor behavior and disease prognosis in patients with gastric adenocarcinoma. J. Gastrointest. Cancer 2014, 45, 476–480. [Google Scholar] [CrossRef]
- Sugasawa, H.; Ichikura, T.; Tsujimoto, H.; Kinoshita, M.; Morita, D.; Ono, S.; Chochi, K.; Tsuda, H.; Seki, S.; Mochizuki, H. Prognostic significance of expression of CCL5/RANTES receptors in patients with gastric cancer. J. Surg. Oncol. 2008, 97, 445–450. [Google Scholar] [CrossRef]
- Suenaga, M.; Mashima, T.; Kawata, N.; Wakatsuki, T.; Horiike, Y.; Matsusaka, S.; Dan, S.; Shinozaki, E.; Seimiya, H.; Mizunuma, N.; et al. Serum VEGF-A and CCL5 levels as candidate biomarkers for efficacy and toxicity of regorafenib in patients with metastatic colorectal cancer. Oncotarget 2016, 7, 34811–34823. [Google Scholar] [CrossRef]
- Wang, H.; Li, S.; Wang, Q.; Jin, Z.; Shao, W.; Gao, Y.; Li, L.; Lin, K.; Zhu, L.; Wang, H.; et al. Tumor immunological phenotype signature-based high-throughput screening for the discovery of combination immunotherapy compounds. Sci. Adv. 2021, 7, eabd7851. [Google Scholar] [CrossRef]
- Lin, S.; Wan, S.; Sun, L.; Hu, J.; Fang, D.; Zhao, R.; Yuan, S.; Zhang, L. Chemokine C-C motif receptor 5 and C-C motif ligand 5 promote cancer cell migration under hypoxia. Cancer Sci. 2012, 103, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Sun, L.; Lyu, X.; Ai, X.; Du, D.; Su, N.; Li, H.; Zhang, L.; Yu, J.; Yuan, S. Lactate-activated macrophages induced aerobic glycolysis and epithelial-mesenchymal transition in breast cancer by regulation of CCL5-CCR5 axis: A positive metabolic feedback loop. Oncotarget 2017, 8, 110426–110443. [Google Scholar] [CrossRef]
- Walens, A.; DiMarco, A.V.; Lupo, R.; Kroger, B.R.; Damrauer, J.S.; Alvarez, J.V. CCL5 promotes breast cancer recurrence through macrophage recruitment in residual tumors. eLife 2019, 8, e43653. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Huang, H.; Guo, M.; Chen, J.; Wu, W.; Li, W.; Xu, X.; Lin, X.; Fu, W.; Yao, Y.; et al. Breast Phyllodes Tumors Recruit and Repolarize Tumor-Associated Macrophages via Secreting CCL5 to Promote Malignant Progression, Which Can Be Inhibited by CCR5 Inhibition Therapy. Clin. Cancer Res. 2019, 25, 3873–3886. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Kim, S.S.; Lee, J. Cancer cell metabolism: Implications for therapeutic targets. Exp. Mol. Med. 2013, 45, e45. [Google Scholar] [CrossRef]
- Patra, S.; Elahi, N.; Armorer, A.; Arunachalam, S.; Omala, J.; Hamid, I.; Ashton, A.W.; Joyce, D.; Jiao, X.; Pestell, R.G. Mechanisms Governing Metabolic Heterogeneity in Breast Cancer and Other Tumors. Front. Oncol. 2021, 11, 700629. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef]
- Gao, D.; Rahbar, R.; Fish, E.N. CCL5 activation of CCR5 regulates cell metabolism to enhance proliferation of breast cancer cells. Open Biol. 2016, 6, 160122. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Aldinucci, D.; Borghese, C.; Casagrande, N. The CCL5/CCR5 Axis in Cancer Progression. Cancers 2020, 12, 1765. [Google Scholar] [CrossRef] [PubMed]
- Kocdor, M.A.; Kocdor, H.; Pereira, J.S.; Vanegas, J.E.; Russo, I.H.; Russo, J. Progressive increase of glucose transporter-3 (GLUT-3) expression in estrogen-induced breast carcinogenesis. Clin. Transl. Oncol. 2013, 15, 55–64. [Google Scholar] [CrossRef]
- Khalid, A.; Wolfram, J.; Ferrari, I.; Mu, C.; Mai, J.; Yang, Z.; Zhao, Y.; Ferrari, M.; Ma, X.; Shen, H. Recent Advances in Discovering the Role of CCL5 in Metastatic Breast Cancer. Mini-Rev. Med. Chem. 2015, 15, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, C.E.; Oda, J.M.; Losi Guembarovski, R.; de Oliveira, K.B.; Ariza, C.B.; Neto, J.S.; Banin Hirata, B.K.; Watanabe, M.A. CC chemokine receptor 5: The interface of host immunity and cancer. Dis. Markers 2014, 2014, 126954. [Google Scholar] [CrossRef]
- Lah Turnsek, T.; Jiao, X.; Novak, M.; Jammula, S.; Cicero, G.; Ashton, A.W.; Joyce, D.; Pestell, R.G. An Update on Glioblastoma Biology, Genetics, and Current Therapies: Novel Inhibitors of the G Protein-Coupled Receptor CCR5. Int. J. Mol. Sci. 2021, 22, 4464. [Google Scholar] [CrossRef]
- Novak, M.; Koprivnikar Krajnc, M.; Hrastar, B.; Breznik, B.; Majc, B.; Mlinar, M.; Rotter, A.; Porcnik, A.; Mlakar, J.; Stare, K.; et al. CCR5-Mediated Signaling Is Involved in Invasion of Glioblastoma Cells in Its Microenvironment. Int. J. Mol. Sci. 2020, 21, 4199. [Google Scholar] [CrossRef]
- Jiao, X.; Wang, M.; Zhang, Z.; Li, Z.; Ni, D.; Ashton, A.W.; Tang, H.Y.; Speicher, D.W.; Pestell, R.G. Leronlimab, a humanized monoclonal antibody to CCR5, blocks breast cancer cellular metastasis and enhances cell death induced by DNA damaging chemotherapy. Breast Cancer Res. 2021, 23, 11. [Google Scholar] [CrossRef] [PubMed]
- Upadhyaya, C.; Jiao, X.; Ashton, A.; Patel, K.; Kossenkov, A.V.; Pestell, R.G. The G protein coupled receptor CCR5 in cancer. Adv. Cancer Res. 2020, 145, 29–47. [Google Scholar] [CrossRef]
- Alix-Panabieres, C.; Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef]
- Velasco-Velazquez, M.; Pestell, R.G. The CCL5/CCR5 axis promotes metastasis in basal breast cancer. Oncoimmunology 2013, 2, e23660. [Google Scholar] [CrossRef]
- Velasco-Velazquez, M.; Xolalpa, W.; Pestell, R.G. The potential to target CCL5/CCR5 in breast cancer. Expert Opin. Ther. Targets 2014, 18, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Upadhyaya, C.; Zhang, Z.; Zhao, J.; Li, Z.; Patel, V.I.; Pestell, R.G. Assays for the Spectrum of Circulating Tumor Cells. Methods Mol. Biol. 2022, 2429, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Pestell, R.G.; Cristofanilli, M.; Jiao, X. Cancers Expressing CCR5 and Methods of Treatment of Same. WO/2016/209926, 6 August 2016. [Google Scholar]
- Raghavakaimal, A.; Cristofanilli, M.; Tang, C.M.; Alpaugh, R.K.; Gardner, K.P.; Chumsri, S.; Adams, D.L. CCR5 activation and endocytosis in circulating tumor-derived cells isolated from the blood of breast cancer patients provide information about clinical outcome. Breast Cancer Res. 2022, 24, 35. [Google Scholar] [CrossRef] [PubMed]
- Onstenk, W.; Sieuwerts, A.M.; Weekhout, M.; Mostert, B.; Reijm, E.A.; van Deurzen, C.H.; Bolt-de Vries, J.B.; Peeters, D.J.; Hamberg, P.; Seynaeve, C.; et al. Gene expression profiles of circulating tumor cells versus primary tumors in metastatic breast cancer. Cancer Lett. 2015, 362, 36–44. [Google Scholar] [CrossRef]
- D’Oronzo, S.; Lovero, D.; Palmirotta, R.; Stucci, L.S.; Tucci, M.; Felici, C.; Cascardi, E.; Giardina, C.; Cafforio, P.; Silvestris, F. Dissection of major cancer gene variants in subsets of circulating tumor cells in advanced breast cancer. Sci. Rep. 2019, 9, 17276. [Google Scholar] [CrossRef]
- Zhao, L.; Wu, X.; Li, T.; Luo, J.; Dong, D. ctcRbase: The gene expression database of circulating tumor cells and microemboli. Database 2020, 2020, baaa020. [Google Scholar] [CrossRef]
- Adams, D.L.; Raghavakaimal, A.; Tang, C.M.; Gardner, K.P.; Ray, N.G.; Pourhassan, N. Abstract CT156: Changes in circulating tumor associated cells predicts progression free and overall survival in metastatic TNBC patients after induction of the anti-CCR5 drug leronlimab. Cancer Res. 2022, 82, CT156. [Google Scholar] [CrossRef]
- Lianidou, E.S.; Markou, A. Circulating tumor cells in breast cancer: Detection systems, molecular characterization, and future challenges. Clin. Chem. 2011, 57, 1242–1255. [Google Scholar] [CrossRef]
- Plaks, V.; Koopman, C.D.; Werb, Z. Cancer. Circulating tumor cells. Science 2013, 341, 1186–1188. [Google Scholar] [CrossRef]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef]
- Wang, L.H.; Lin, C.Y.; Liu, S.C.; Liu, G.T.; Chen, Y.L.; Chen, J.J.; Chan, C.H.; Lin, T.Y.; Chen, C.K.; Xu, G.H.; et al. CCL5 promotes VEGF-C production and induces lymphangiogenesis by suppressing miR-507 in human chondrosarcoma cells. Oncotarget 2016, 7, 36896–36908. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.W.; Liu, S.C.; Sun, H.L.; Huang, T.Y.; Chan, C.H.; Yang, C.Y.; Yeh, H.I.; Huang, Y.L.; Chou, W.Y.; Lin, Y.M.; et al. CCL5/CCR5 axis induces vascular endothelial growth factor-mediated tumor angiogenesis in human osteosarcoma microenvironment. Carcinogenesis 2015, 36, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Sax, M.J.; Gasch, C.; Athota, V.R.; Freeman, R.; Rasighaemi, P.; Westcott, D.E.; Day, C.J.; Nikolic, I.; Elsworth, B.; Wei, M.; et al. Cancer cell CCL5 mediates bone marrow independent angiogenesis in breast cancer. Oncotarget 2016, 7, 85437–85449. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, J.; Fang, H.; Tang, L.; Chen, W.; Sun, Q.; Zhang, Q.; Yang, F.; Sun, Z.; Cao, L.; et al. Endothelial cells promote triple-negative breast cancer cell metastasis via PAI-1 and CCL5 signaling. FASEB J. 2018, 32, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.C.; Goedegebuure, P.S.; Belt, B.A.; Flaherty, B.; Sankpal, N.; Gillanders, W.E.; Eberlein, T.J.; Hsieh, C.S.; Linehan, D.C. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J. Immunol. 2009, 182, 1746–1755. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Xu, L.; Zeng, X.; Wu, H.; Liang, F.; Lv, Q.; Du, Z. CCL5 mediates breast cancer metastasis and prognosis through CCR5/Treg cells. Front. Oncol. 2022, 12, 972383. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Araujo, J.M.; Gomez, A.C.; Aguilar, A.; Salgado, R.; Balko, J.M.; Bravo, L.; Doimi, F.; Bretel, D.; Morante, Z.; Flores, C.; et al. Effect of CCL5 expression in the recruitment of immune cells in triple negative breast cancer. Sci. Rep. 2018, 8, 4899. [Google Scholar] [CrossRef]
- Zhao, X.; Qu, J.; Sun, Y.; Wang, J.; Liu, X.; Wang, F.; Zhang, H.; Wang, W.; Ma, X.; Gao, X.; et al. Prognostic significance of tumor-associated macrophages in breast cancer: A meta-analysis of the literature. Oncotarget 2017, 8, 30576–30586. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Du, Z.; Long, Q.; Lu, Q.; Li, H. Criteria derived from serum markers can precisely evaluate axillary status in breast cancer patients. J. Surg. Res. 2017, 208, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lv, D.; Kim, H.J.; Kurt, R.A.; Bu, W.; Li, Y.; Ma, X. A novel role of hematopoietic CCL5 in promoting triple-negative mammary tumor progression by regulating generation of myeloid-derived suppressor cells. Cell Res. 2013, 23, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, D.; Borghese, C.; Casagrande, N. Formation of the Immunosuppressive Microenvironment of Classic Hodgkin Lymphoma and Therapeutic Approaches to Counter It. Int. J. Mol. Sci. 2019, 20, 2416. [Google Scholar] [CrossRef] [PubMed]
- Zilio, S.; Bicciato, S.; Weed, D.; Serafini, P. CCR1 and CCR5 mediate cancer-induced myelopoiesis and differentiation of myeloid cells in the tumor. J. Immunother. Cancer 2022, 10, e003131. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Velazquez, M.A.; Popov, V.M.; Lisanti, M.P.; Pestell, R.G. The role of breast cancer stem cells in metastasis and therapeutic implications. Am. J. Pathol. 2011, 179, 2–11. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- De Francesco, E.M.; Sotgia, F.; Lisanti, M.P. Cancer stem cells (CSCs): Metabolic strategies for their identification and eradication. Biochem. J. 2018, 475, 1611–1634. [Google Scholar] [CrossRef]
- Peiris-Pages, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci. Rep. 2017, 7, 13856. [Google Scholar] [CrossRef]
- Dontu, G.; Al-Hajj, M.; Abdallah, W.M.; Clarke, M.F.; Wicha, M.S. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003, 36 (Suppl. S1), 59–72. [Google Scholar] [CrossRef] [PubMed]
- Ponti, D.; Costa, A.; Zaffaroni, N.; Pratesi, G.; Petrangolini, G.; Coradini, D.; Pilotti, S.; Pierotti, M.A.; Daidone, M.G. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005, 65, 5506–5511. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer stem cells. Int. J. Biochem. Cell Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Steed, A.; Co, M.; Chen, X. Cancer stem cells, epithelial-mesenchymal transition, ATP and their roles in drug resistance in cancer. Cancer Drug Resist. 2021, 4, 684–709. [Google Scholar] [CrossRef]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef]
- Zavadil, J.; Bottinger, E.P. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Lamouille, S.; Derynck, R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 2007, 178, 437–451. [Google Scholar] [CrossRef]
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. TGF-beta-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 2012, 125, 1259–1273. [Google Scholar] [CrossRef]
- Tyner, J.W.; Uchida, O.; Kajiwara, N.; Kim, E.Y.; Patel, A.C.; O’Sullivan, M.P.; Walter, M.J.; Schwendener, R.A.; Cook, D.N.; Danoff, T.M.; et al. CCL5-CCR5 interaction provides antiapoptotic signals for macrophage survival during viral infection. Nat. Med. 2005, 11, 1180–1187. [Google Scholar] [CrossRef]
- Wang, Y.C.; He, F.; Feng, F.; Liu, X.W.; Dong, G.Y.; Qin, H.Y.; Hu, X.B.; Zheng, M.H.; Liang, L.; Feng, L.; et al. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res. 2010, 70, 4840–4849. [Google Scholar] [CrossRef] [PubMed]
- Mirandola, L.; Chiriva-Internati, M.; Montagna, D.; Locatelli, F.; Zecca, M.; Ranzani, M.; Basile, A.; Locati, M.; Cobos, E.; Kast, W.M.; et al. Notch1 regulates chemotaxis and proliferation by controlling the CC-chemokine receptors 5 and 9 in T cell acute lymphoblastic leukaemia. J. Pathol. 2012, 226, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Alghibiwi, H.; Ansari, M.A.; Nadeem, A.; Algonaiah, M.A.; Attia, S.M.; Bakheet, S.A.; Albekairi, T.H.; Almudimeegh, S.; Alhamed, A.S.; Shahid, M.; et al. DAPTA, a C-C Chemokine Receptor 5 (CCR5), Leads to the Downregulation of Notch/NF-kappaB Signaling and Proinflammatory Mediators in CD40+ Cells in Experimental Autoimmune Encephalomyelitis Model in SJL/J Mice. Biomedicines 2023, 11, 1511. [Google Scholar] [CrossRef]
- Zhao, L.; Lei, J.; Gu, S.; Zhang, Y.; Jing, X.; Wang, L.; Zhang, L.; Ning, Q.; Luo, M.; Qi, Y.; et al. A yes-associated protein 1-Notch1 receptor positive feedback loop promotes breast cancer lung metastasis by attenuating the bone morphogenetic protein 4-SMAD family member 1/5 signaling. Carcinogenesis 2022, 43, 1162–1175. [Google Scholar] [CrossRef] [PubMed]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Y.; Hong, T.; Ye, J.; Chu, C.; Zuo, L.; Zhang, J.; Cui, X. Targeting a positive regulatory loop in the tumor-macrophage interaction impairs the progression of clear cell renal cell carcinoma. Cell Death Differ. 2021, 28, 932–951. [Google Scholar] [CrossRef]
- Yi, E.H.; Lee, C.S.; Lee, J.K.; Lee, Y.J.; Shin, M.K.; Cho, C.H.; Kang, K.W.; Lee, J.W.; Han, W.; Noh, D.Y.; et al. STAT3-RANTES autocrine signaling is essential for tamoxifen resistance in human breast cancer cells. Mol. Cancer Res. 2013, 11, 31–42. [Google Scholar] [CrossRef]
- Haag, G.M.; Springfeld, C.; Grun, B.; Apostolidis, L.; Zschabitz, S.; Dietrich, M.; Berger, A.K.; Weber, T.F.; Zoernig, I.; Schaaf, M.; et al. Pembrolizumab and maraviroc in refractory mismatch repair proficient/microsatellite-stable metastatic colorectal cancer—The PICCASSO phase I trial. Eur. J. Cancer 2022, 167, 112–122. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Chittoria, N.; Ehsani, S.; Rui, H.; Dolezal, M.; Stork-Sloots, L.; de Snoo, F.; Recknor, C.; Abramson, V. Abstract P5-17-08: A phase Ib/II study of leronlimab combined with carboplatin in patients with CCR5+ metastatic triple-negative breast cancer (mTNBC). Cancer Res. 2022, 82, P5-17-08. [Google Scholar] [CrossRef]
- Adams, D.; Raghavakaimal, A.; Tang, C.-M.; Gardner, K.P.; Kelly, S.; Pourhassan, N.; Ray, N. Safety, efficacy, and clinical outcomes of the anti-CCR5 inhibitor (Leronlimab): A pooled analysis of three clinical trials in patients with mTNBC. J. Clin. Oncol. 2022, 40, e13062. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Xing, X.; Zhuang, J.; Wang, J.; Wang, C.; Zhang, L.; Liu, L.; Feng, F.; Li, H.; et al. Immunogenomic landscape analyses of immune molecule signature-based risk panel for patients with triple-negative breast cancer. Mol. Ther. Nucleic Acids 2022, 28, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.Z.; Ma, D.; Suo, C.; Shi, J.; Xue, M.; Hu, X.; Xiao, Y.; Yu, K.D.; Liu, Y.R.; Yu, Y.; et al. Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and Treatment Strategies. Cancer Cell 2019, 35, 428–440.e5. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2019, 51, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Brett, E.; Duscher, D.; Pagani, A.; Daigeler, A.; Kolbenschlag, J.; Hahn, M. Naming the Barriers between Anti-CCR5 Therapy, Breast Cancer and Its Microenvironment. Int. J. Mol. Sci. 2022, 23, 14159. [Google Scholar] [CrossRef]
- Di Sante, G.; Page, J.; Jiao, X.; Nawab, O.; Cristofanilli, M.; Skordalakes, E.; Pestell, R.G. Recent advances with cyclin-dependent kinase inhibitors: Therapeutic agents for breast cancer and their role in immuno-oncology. Expert Rev. Anticancer Ther. 2019, 19, 569–587. [Google Scholar] [CrossRef]
- Zazo, S.; Gonzalez-Alonso, P.; Martin-Aparicio, E.; Chamizo, C.; Luque, M.; Sanz-Alvarez, M.; Minguez, P.; Gomez-Lopez, G.; Cristobal, I.; Carames, C.; et al. Autocrine CCL5 Effect Mediates Trastuzumab Resistance by ERK Pathway Activation in HER2-Positive Breast Cancer. Mol. Cancer Ther. 2020, 19, 1696–1707. [Google Scholar] [CrossRef]
- Uzhachenko, R.V.; Bharti, V.; Ouyang, Z.; Blevins, A.; Mont, S.; Saleh, N.; Lawrence, H.A.; Shen, C.; Chen, S.C.; Ayers, G.D.; et al. Metabolic modulation by CDK4/6 inhibitor promotes chemokine-mediated recruitment of T cells into mammary tumors. Cell Rep. 2021, 35, 108944. [Google Scholar] [CrossRef]
- Pestell, T.G.; Jiao, X.; Kumar, M.; Peck, A.R.; Prisco, M.; Deng, S.; Li, Z.; Ertel, A.; Casimiro, M.C.; Ju, X.; et al. Stromal cyclin D1 promotes heterotypic immune signaling and breast cancer growth. Oncotarget 2017, 8, 81754–81775. [Google Scholar] [CrossRef]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin. Cancer Res. 2016, 22, 1499–1509. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.B.; Hernandez, R.; Carlson, P.; Grudzinski, J.; Bates, A.M.; Jagodinsky, J.C.; Erbe, A.; Marsh, I.R.; Arthur, I.; Aluicio-Sarduy, E.; et al. Low-dose targeted radionuclide therapy renders immunologically cold tumors responsive to immune checkpoint blockade. Sci. Transl. Med. 2021, 13, eabb3631. [Google Scholar] [CrossRef] [PubMed]
- Herrera, F.G.; Ronet, C.; Ochoa de Olza, M.; Barras, D.; Crespo, I.; Andreatta, M.; Corria-Osorio, J.; Spill, A.; Benedetti, F.; Genolet, R.; et al. Low-Dose Radiotherapy Reverses Tumor Immune Desertification and Resistance to Immunotherapy. Cancer Discov. 2022, 12, 108–133. [Google Scholar] [CrossRef] [PubMed]
- Barsoumian, H.B.; Ramapriyan, R.; Younes, A.I.; Caetano, M.S.; Menon, H.; Comeaux, N.I.; Cushman, T.R.; Schoenhals, J.E.; Cadena, A.P.; Reilly, T.P.; et al. Low-dose radiation treatment enhances systemic antitumor immune responses by overcoming the inhibitory stroma. J. Immunother. Cancer 2020, 8, e000537. [Google Scholar] [CrossRef]
- Samanta, D.; Park, Y.; Ni, X.; Li, H.; Zahnow, C.A.; Gabrielson, E.; Pan, F.; Semenza, G.L. Chemotherapy induces enrichment of CD47+/CD73+/PDL1+ immune evasive triple-negative breast cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1239–E1248. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Opzoomer, J.W.; Sosnowska, D.; Anstee, J.E.; Spicer, J.F.; Arnold, J.N. Cytotoxic Chemotherapy as an Immune Stimulus: A Molecular Perspective on Turning Up the Immunological Heat on Cancer. Front. Immunol. 2019, 10, 1654. [Google Scholar] [CrossRef]
- Gilmore, E.; McCabe, N.; Kennedy, R.D.; Parkes, E.E. DNA Repair Deficiency in Breast Cancer: Opportunities for Immunotherapy. J. Oncol. 2019, 2019, 4325105. [Google Scholar] [CrossRef]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djw199. [Google Scholar] [CrossRef] [PubMed]
- Blattner, C.; Fleming, V.; Weber, R.; Himmelhan, B.; Altevogt, P.; Gebhardt, C.; Schulze, T.J.; Razon, H.; Hawila, E.; Wildbaum, G.; et al. CCR5+ Myeloid-Derived Suppressor Cells Are Enriched and Activated in Melanoma Lesions. Cancer Res. 2018, 78, 157–167. [Google Scholar] [CrossRef]
- Yang, L.; Wang, B.; Qin, J.; Zhou, H.; Majumdar, A.P.N.; Peng, F. Blockade of CCR5-mediated myeloid derived suppressor cell accumulation enhances anti-PD1 efficacy in gastric cancer. Immunopharmacol. Immunotoxicol. 2018, 40, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Williford, J.M.; Ishihara, J.; Ishihara, A.; Mansurov, A.; Hosseinchi, P.; Marchell, T.M.; Potin, L.; Swartz, M.A.; Hubbell, J.A. Recruitment of CD103+ dendritic cells via tumor-targeted chemokine delivery enhances efficacy of checkpoint inhibitor immunotherapy. Sci. Adv. 2019, 5, eaay1357. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Dent, R.; Pusztai, L.; McArthur, H.; Kummel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; et al. Event-free Survival with Pembrolizumab in Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2022, 386, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Zhang, H.; Barrios, C.H.; Saji, S.; Jung, K.H.; Hegg, R.; Koehler, A.; Sohn, J.; Iwata, H.; Telli, M.L.; et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): A randomised, double-blind, phase 3 trial. Lancet 2020, 396, 1090–1100. [Google Scholar] [CrossRef]
- Sinn, B.V.; Loibl, S.; Hanusch, C.A.; Zahm, D.M.; Sinn, H.P.; Untch, M.; Weber, K.; Karn, T.; Becker, C.; Marme, F.; et al. Immune-related Gene Expression Predicts Response to Neoadjuvant Chemotherapy but not Additional Benefit from PD-L1 Inhibition in Women with Early Triple-negative Breast Cancer. Clin. Cancer Res. 2021, 27, 2584–2591. [Google Scholar] [CrossRef]
- Hemmatazad, H.; Berger, M.D. CCR5 is a potential therapeutic target for cancer. Expert Opin. Ther. Targets 2021, 25, 311–327. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCT | Trial Title | Drug(s) | n | Phase | Status | Results |
|---|---|---|---|---|---|---|
| NCT03274804 | Combined PD-1 and CCR5 Inhibition for the Treatment of Refractory Microsatellite Stable mCRC (PICCASSO) | Maraviroc (CCR5 antagonist) + Pembrolizumab (PD-1 inhibitor) | 20 | I | Completed | Median survival from ~6 mo to >9 mo Median PFS: 2.1 mo (95% CI 1.68–2.30) Median OS: 9.83 mo (95% CI 5.59–20.02) |
| NCT03838367 | Leronlimab (PRO 140) Combined With Carboplatin in Patients With CCR5+ mTNBC | Leronlimab (CCR5 mAb) + Carboplatin | 48 | Ib/II | Active, not recruiting | Phase 1b: 8/10 patients—stable or regressed 72% decrease in CAML 30 days post tx linked to 300% increase in mean PFS + 450% increase in OS (12 mo) |
| NCT04504942 | Basket Study of Leronlimab (PRO 140) in Patients With CCR5+ Locally Advanced or Metastatic Solid Tumors | Leronlimab (CCR5 mAb) | 30 | II | Active, not recruiting | Pooled data (n = 19) “>75% improved mPFS 6.1 mo (95%CI 2.3–7.5) & mOS 12+ mo (95%CI 5.5–12+)” Reduced circulating TACs in 75% (n = 21/28) pts (strong predictor of improved survival) |
| NCT03631407 | Safety and Efficacy of Vicriviroc (MK-7690) in Combination With Pembrolizumab (MK-3475) in Participants With Advanced/Metastatic Microsatellite Stable (MSS) Colorectal Cancer (CRC) (MK-7690-046) | Vicriviroc (CCR5 antagonist) + Pembrolizumab (PD-1 inhibitor) | 41 | II | Completed | Vicriviroc Dose Level DL1: 150 mg (n = 20) DL2: 250 mg (n = 20) mORR DL1: 5% (95% CI 0.1–24.9) DL2: 5% (95% CI 0.1–24.9) mPFS DL1: 4.0 mo (95% CI 2.7–5.6) DL2: 4.9 mo (95% CI 3.1–8.0) OS DL1: 4.6 mo (95% CI 2.7–12.6) DL2: 5.3 mo (95% CI 3.2–8.0) Abort Tx due to AE DL1: 4/20 DL2: 7/20 |
| NCT01736813 | CCR5-blockade in Metastatic Colorectal Cancer | Maraviroc (CCR5 antagonist) | 12 | I | Completed | 5/11 pts re-exposed to chemotherapy 3 of those 5: ORR favorable to response rates in pts with mCRC, on or after the third line of chemotherapy, 5–10%. PET-MRI image from 1 pt with advanced-stage mCRC refractory to standard chemotherapy showed clear tumor shrinkage after maraviroc treatment |
| NCT03767582 | Trial of Neoadjuvant and Adjuvant Nivolumab and BMS-813160 With or Without GVAX for Locally Advanced Pancreatic Ductal Adenocarcinomas | BMS-813160 (CCR2/5 dual antagonist) + Nivolumab (PD-1 mAb) +/− GVAX | I: 30 | I/II | I: Completed II: Recruiting | Phase I: 9/13 pts proceeded to immunotherapy after neoadjuvant chemotherapy + rad 3 pts received treatment at DL1 6 pts at DL2. No DLTs observed grade 3+ AE: 1 pt |
| NCT04721301 | Ipilimumab, Maraviroc and Nivolumab in Advanced Metastatic Colorectal and Pancreatic Cancer the LUMINESCENCE Trial | Maraviroc (CCR5 antagonist) + Ipilimumab (CTLA-4 mAb) + Nivolumab (PD-1 mAb) | I | Active, not recruiting | ||
| NCT04123379 | Neoadjuvant Nivolumab With CCR2/5-inhibitor or Anti-IL-8) for Non-small Cell Lung Cancer (NSCLC) or Hepatocellular Carcinoma (HCC) | BMS-813160 (CCR2/5 dual antagonist) + Nivolumab (PD-1 mAb) + BMS-986253 (IL-8 mAb) | II | Recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamid, R.; Alaziz, M.; Mahal, A.S.; Ashton, A.W.; Halama, N.; Jaeger, D.; Jiao, X.; Pestell, R.G. The Role and Therapeutic Targeting of CCR5 in Breast Cancer. Cells 2023, 12, 2237. https://doi.org/10.3390/cells12182237
Hamid R, Alaziz M, Mahal AS, Ashton AW, Halama N, Jaeger D, Jiao X, Pestell RG. The Role and Therapeutic Targeting of CCR5 in Breast Cancer. Cells. 2023; 12(18):2237. https://doi.org/10.3390/cells12182237
Chicago/Turabian StyleHamid, Rasha, Mustafa Alaziz, Amanpreet S. Mahal, Anthony W. Ashton, Niels Halama, Dirk Jaeger, Xuanmao Jiao, and Richard G. Pestell. 2023. "The Role and Therapeutic Targeting of CCR5 in Breast Cancer" Cells 12, no. 18: 2237. https://doi.org/10.3390/cells12182237
APA StyleHamid, R., Alaziz, M., Mahal, A. S., Ashton, A. W., Halama, N., Jaeger, D., Jiao, X., & Pestell, R. G. (2023). The Role and Therapeutic Targeting of CCR5 in Breast Cancer. Cells, 12(18), 2237. https://doi.org/10.3390/cells12182237