

Transcriptomic Analysis in the Hippocampus and Retina of Tg2576 AD Mice Reveals Defective Mitochondrial Oxidative Phosphorylation and Recovery by Tau 12A12mAb Treatment
 , and
, and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals, Ethical Approval and Tissue Collection
2.2. RNA Extraction and Sequencing
2.3. Transcriptomic Data Analysis
2.4. Validation by Reverse Transcription-Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR)
3. Results
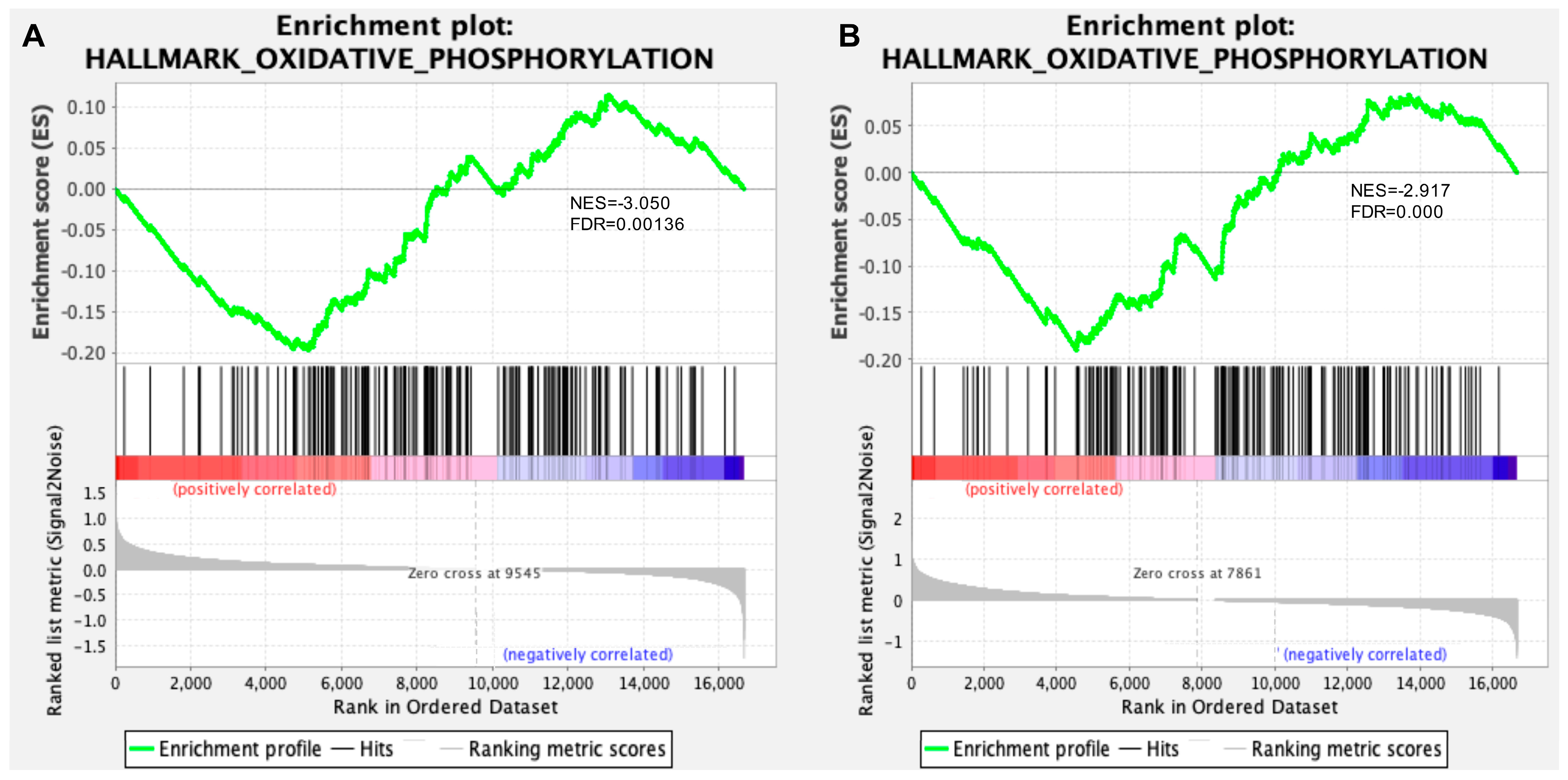
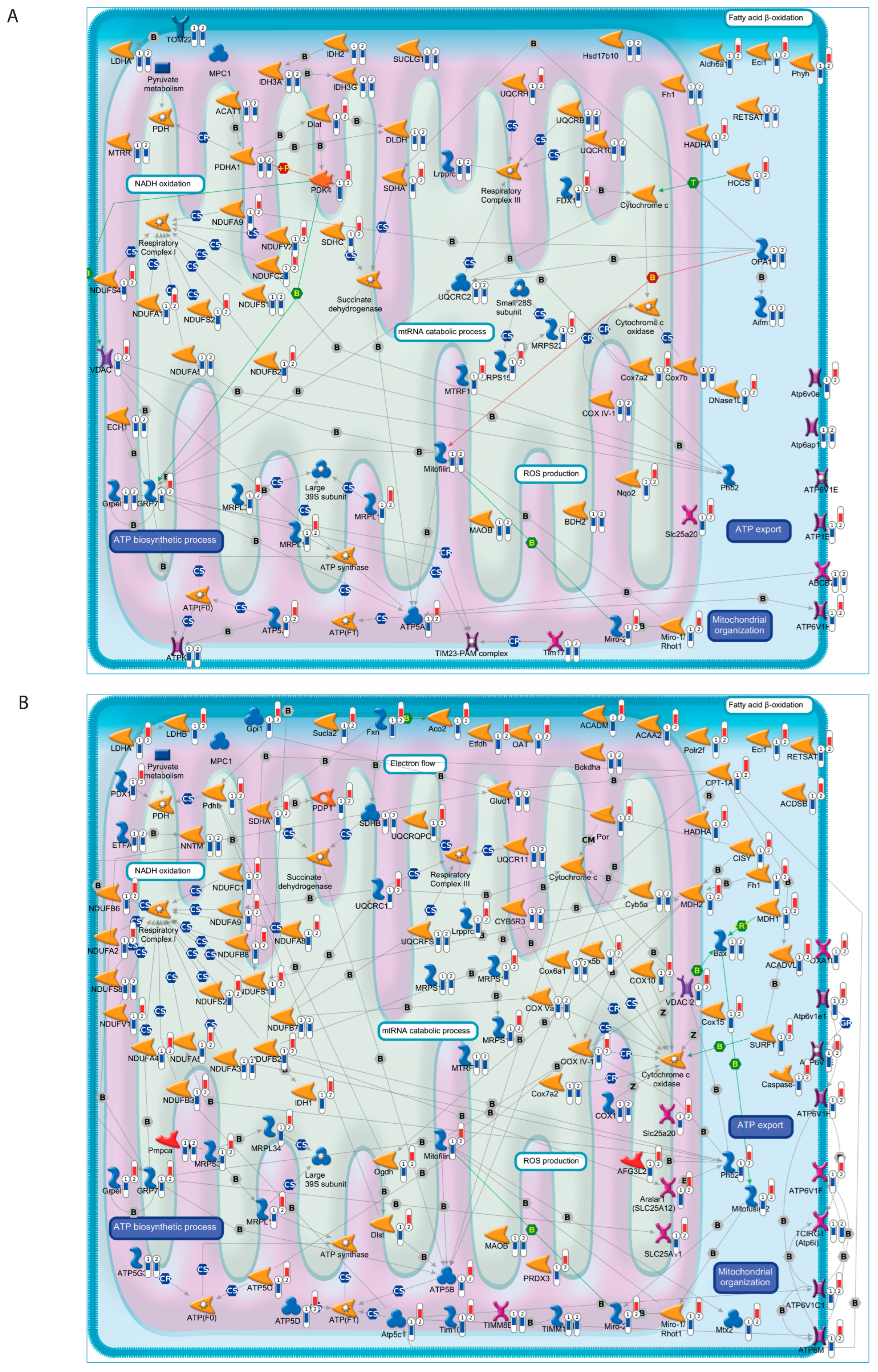
3.1. Transcriptomic Analysis of Hippocampus and Retina from Symptomatic Tg2576 AD Mice Revealed Defective Mitochondrial Bioenergetics
3.2. Defective Mitochondrial Bioenergetics Are Recovered by 12A12mAb-Mediated Neuroprotection in Both the Hippocampus and Retina of the AD Mouse Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2015 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2015, 11, 332–384. [Google Scholar] [CrossRef]
- Murphy, M.P.; Levine, H. Alzheimer’s Disease and the Amyloid-β Peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef]
- Kocahan, S.; Doǧan, Z. Mechanisms of Alzheimer’s Disease Pathogenesis and Prevention: The Brain, Neural Pathology, N-methyl-D-aspartate Receptors, Tau Protein and Other Risk Factors. Clin. Psychopharmacol. Neurosci. 2017, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and “wingmen”. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Austad, S.N.; Ballinger, S.; Buford, T.W.; Carter, C.S.; Smith, D.L.; Darley-Usmar, V.; Zhang, J. Targeting whole body metabolism and mitochondrial bioenergetics in the drug development for Alzheimer’s disease. Acta Pharm. Sin. B 2022, 12, 511–531. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef]
- Swerdlow, R.H. The Mitochondrial Hypothesis: Dysfunction, Bioenergetic Defects, and the Metabolic Link to Alzheimer’s Disease. Int. Rev. Neurobiol. 2020, 154, 207–233. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Voos, W. Mitochondria as potential targets in Alzheimer disease therapy: An update. Front. Pharmacol. 2019, 10, 902. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Kovács, R. Mitochondria and neuronal activity. Am. J. Physiol.-Cell Physiol. 2007, 292, 641–657. [Google Scholar] [CrossRef] [PubMed]
- Trigo, D.; Avelar, C.; Fernandes, M.; Sá, J.; da Cruz e Silva, O. Mitochondria, energy, and metabolism in neuronal health and disease. FEBS Lett. 2022, 596, 1095–1110. [Google Scholar] [CrossRef] [PubMed]
- Lezi, E.; Swerdlow, R.H. Mitochondria in Neurodegeneration. Adv. Exp. Med. Biol. 2012, 942, 269–286. [Google Scholar] [CrossRef]
- Jadiya, P.; Garbincius, J.F.; Elrod, J.W. Reappraisal of metabolic dysfunction in neurodegeneration: Focus on mitochondrial function and calcium signaling. Acta Neuropathol. Commun. 2021, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Marmolejo-Garza, A.; Medeiros-Furquim, T.; Rao, R.; Eggen, B.J.; Boddeke, E.; Dolga, A.M. Transcriptomic and epigenomic landscapes of Alzheimer’s disease evidence mitochondrial-related pathways. Biochim. Biophys. Acta-Mol. Cell Res. 2022, 1869, 119326. [Google Scholar] [CrossRef]
- Harerimana, N.V.; Paliwali, D.; Romero-Molina, C.; Bennett, D.A.; Pa, J.; Goate, A.; Swerdlow, R.H.; Andrews, S.J. The role of mitochondrial genome abundance in Alzheimer’s disease. Alzheimer’s Dement. 2023, 19, 2069–2083. [Google Scholar] [CrossRef]
- Mani, S.; Swargiary, G.; Singh, M.; Agarwal, S.; Dey, A.; Ojha, S.; Jha, N.K. Mitochondrial defects: An emerging theranostic avenue towards Alzheimer’s associated dysregulations. Life Sci. 2021, 285, 119985. [Google Scholar] [CrossRef]
- Piras, I.S.; Krate, J.; Delvaux, E.; Nolz, J.; Mastroeni, D.F.; Persico, A.M.; Jepsen, W.M.; Beach, T.G.; Huentelman, M.J.; Coleman, P.D.; et al. Transcriptome Changes in the Alzheimer’s Disease Middle Temporal Gyrus: Importance of RNA Metabolism and Mitochondria-Associated Membrane Genes. J. Alzheimer’s Dis. 2019, 70, 691–713. [Google Scholar] [CrossRef]
- Mastroeni, D.; Khdour, O.M.; Delvaux, E.; Nolz, J.; Olsen, G.; Berchtold, N.; Cotman, C.; Hecht, S.M.; Coleman, P.D. Nuclear but not mitochondrial-encoded oxidative phosphorylation genes are altered in aging, mild cognitive impairment, and Alzheimer’s disease. Alzheimer’s Dement. 2017, 13, 510–519. [Google Scholar] [CrossRef]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Reddy, P.H.; Tripathi, R.; Troung, Q.; Tirumala, K.; Reddy, T.P.; Anekonda, V.; Shirendeb, U.P.; Calkins, M.J.; Reddy, A.P.; Mao, P.; et al. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim. Biophys. Acta-Mol. Basis Dis. 2012, 1822, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Lunnon, K.; Keohane, A.; Pidsley, R.; Newhouse, S.; Riddoch-Contreras, J.; Thubron, E.B.; Devall, M.; Soininen, H.; Kłoszewska, I.; Mecocci, P.; et al. Mitochondrial genes are altered in blood early in Alzheimer’s disease. Neurobiol. Aging 2017, 53, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Gillardon, F.; Rist, W.; Kussmaul, L.; Vogel, J.; Berg, M.; Danzer, K.; Kraut, N.; Hengerer, B. Proteomic and functional alterations in brain mitochondria from Tg2576 mice occur before amyloid plaque deposition. Proteomics 2007, 7, 605–616. [Google Scholar] [CrossRef]
- Varghese, M.; Zhao, W.; Wang, J.; Cheng, A.; Qian, X.; Chaudhry, A.; Ho, L.; Pasinetti, G.M. Mitochondrial bioenergetics is defective in presymptomatic Tg2576 AD mice. Transl. Neurosci. 2011, 2, 1–5. [Google Scholar] [CrossRef]
- Reddy, P.H.; McWeeney, S.; Park, B.S.; Manczak, M.; Gutala, R.V.; Partovi, D.; Jung, Y.; Yau, V.; Searles, R.; Mori, M.; et al. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: Up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum. Mol. Genet. 2004, 13, 1225–1240. [Google Scholar] [CrossRef]
- Amadoro, G.; Latina, V.; Corsetti, V.; Calissano, P. N-terminal tau truncation in the pathogenesis of Alzheimer’s disease (AD): Developing a novel diagnostic and therapeutic approach. Biochim. Biophys. Acta-Mol. Basis Dis. 2020, 1866, 165584. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH(2)-tau fragment and Aβ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging 2012, 33, 833.e1–833.e25. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Stringaro, A.; Colone, M.; D’aguanno, S.; Meli, G.; Ciotti, M.; Sancesario, G.; Cattaneo, A.; Bussani, R.; et al. A NH2 Tau Fragment Targets Neuronal Mitochondria at AD Synapses: Possible Implications for Neurodegeneration. J. Alzheimer’s Dis. 2010, 21, 445–470. [Google Scholar] [CrossRef]
- Latina, V.; Giacovazzo, G.; Cordella, F.; Balzamino, B.O.; Micera, A.; Varano, M.; Marchetti, C.; Malerba, F.; Florio, R.; Ercole, B.B.; et al. Systemic delivery of a specific antibody targeting the pathological N-terminal truncated tau peptide reduces retinal degeneration in a mouse model of Alzheimer’s Disease. Acta Neuropathol. Commun. 2021, 9, 38. [Google Scholar] [CrossRef]
- Atlante, A.; Amadoro, G.; Bobba, A.; de Bari, L.; Corsetti, V.; Pappalardo, G.; Marra, E.; Calissano, P.; Passarella, S. A peptide containing residues 26–44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim. Biophys. Acta-Bioenerg. 2008, 1777, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, V.; Florenzano, F.; Atlante, A.; Bobba, A.; Ciotti, M.T.; Natale, F.; Della Valle, F.; Borreca, A.; Manca, A.; Meli, G.; et al. NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: Implications in Alzheimer’s disease. Hum. Mol. Genet. 2015, 24, 3058–3081. [Google Scholar] [CrossRef]
- Szabo, L.; Eckert, A.; Grimm, A. Insights into Disease-Associated Tau Impact on Mitochondria. Int. J. Mol. Sci. 2020, 21, 6344. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.J.; Jara, C.; Quintanilla, R.A. Contribution of Tau Pathology to Mitochondrial Impairment in Neurodegeneration. Front. Neurosci. 2018, 12, 441. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Bai, F. The association of tau with mitochondrial dysfunction in Alzheimer’s disease. Front. Neurosci. 2018, 12, 163. [Google Scholar] [CrossRef]
- Torres, A.; Rivera, B.; Polanco, C.; Jara, C.; Tapia-Rojas, C. Phosphorylated tau as a toxic agent in synaptic mitochondria: Implications in aging and Alzheimer’s disease. Neural Regen. Res. 2022, 17, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Latina, V.; Calissano, P. A long story for a short peptide: Therapeutic efficacy of a cleavage-specific tau antibody. Neural Regen. Res. 2021, 16, 2417–2419. [Google Scholar] [CrossRef]
- Latina, V.; Atlante, A.; Malerba, F.; La Regina, F.; Balzamino, B.O.; Micera, A.; Pignataro, A.; Stigliano, E.; Cavallaro, S.; Calissano, P.; et al. The Cleavage-Specific Tau 12A12mAb Exerts an Anti-Amyloidogenic Action by Modulating the Endocytic and Bioenergetic Pathways in Alzheimer’s Disease Mouse Model. Int. J. Mol. Sci. 2023, 24, 9683. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative Memory Deficits, Aβ Elevation, and Amyloid Plaques in Transgenic Mice. Science 1996, 274, 99–103. [Google Scholar] [CrossRef]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Corsetti, V.; Borreca, A.; Latina, V.; Giacovazzo, G.; Pignataro, A.; Krashia, P.; Natale, F.; Cocco, S.; Rinaudo, M.; Malerba, F.; et al. Passive immunotherapy for N-truncated tau ameliorates the cognitive deficits in two mouse Alzheimer’s disease models. Brain Commun. 2020, 2, fcaa039. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef]
- Dong, Y.; Brewer, G.J. Global Metabolic Shifts in Age and Alzheimer’s Disease Mouse Brains Pivot at NAD+/NADH Redox Sites. J. Alzheimer’s Dis. 2019, 71, 119–140. [Google Scholar] [CrossRef]
- Newington, J.T.; Rappon, T.; Albers, S.; Wong, D.Y.; Rylett, R.J.; Cumming, R.C. Overexpression of Pyruvate Dehydrogenase Kinase 1 and Lactate Dehydrogenase A in Nerve Cells Confers Resistance to Amyloid and Other Toxins by Decreasing Mitochondrial Respiration and Reactive Oxygen Species Production. J. Biol. Chem. 2012, 287, 37245–37258. [Google Scholar] [CrossRef] [PubMed]
- Perry, E.K.; Perry, R.H.; Tomlinson, B.E.; Blessed, G.; Gibson, P.H. Coenzyme a-acetylating enzymes in Alzheimer’s disease: Possible cholinergic ‘compartment’ of pyruvate dehydrogenase. Neurosci. Lett. 1980, 18, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Rigotto, G.; Valente, G.; Giorgio, V.; Basso, E.; Filadi, R.; Pizzo, P. Defective Mitochondrial Pyruvate Flux Affects Cell Bioenergetics in Alzheimer’s Disease-Related Models. Cell Rep. 2020, 30, 2332–2348.e10. [Google Scholar] [CrossRef]
- Harris, R.A.; Tindale, L.; Lone, A.; Singh, O.; Macauley, S.L.; Stanley, M.; Holtzman, D.M.; Bartha, R.; Cumming, R.C. Aerobic Glycolysis in the Frontal Cortex Correlates with Memory Performance in Wild-Type Mice But Not the APP/PS1 Mouse Model of Cerebral Amyloidosis. J. Neurosci. 2016, 36, 1871–1878. [Google Scholar] [CrossRef] [PubMed]
- Ryu, W.I.; Bormann, M.K.; Shen, M.; Kim, D.; Forester, B.; Park, Y.; So, J.; Seo, H.; Sonntag, K.C.; Cohen, B.M. Brain cells derived from Alzheimer’s disease patients have multiple specific innate abnormalities in energy metabolism. Mol. Psychiatry 2021, 26, 5702–5714. [Google Scholar] [CrossRef]
- Park, J.S.; Saeed, K.; Jo, M.H.; Kim, M.W.; Lee, H.J.; Park, C.B.; Lee, G.; Kim, M.O. LDHB Deficiency Promotes Mitochondrial Dysfunction Mediated Oxidative Stress and Neurodegeneration in Adult Mouse Brain. Antioxidants 2022, 11, 261. [Google Scholar] [CrossRef]
- Chen, W.; Sun, X.; Zhan, L.; Zhou, W.; Bi, T. Conditional Knockout of Pdha1 in Mouse Hippocampus Impairs Cognitive Function: The Possible Involvement of Lactate. Front. Neurosci. 2021, 15, 767560. [Google Scholar] [CrossRef]
- Stacpoole, P.W. The pyruvate dehydrogenase complex as a therapeutic target for age-related diseases. Aging Cell 2012, 11, 371–377. [Google Scholar] [CrossRef]
- Rex Sheu, K.-F.; Kim, Y.-T.; Blass, J.P.; Weksler, M.E. An immunochemical study of the pyruvate dehydrogenase deficit in Alzheimer’s disease brain. Ann. Neurol. 1985, 17, 444–449. [Google Scholar] [CrossRef]
- Adav, S.S.; Park, J.E.; Sze, S.K. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol. Brain 2019, 12, 8. [Google Scholar] [CrossRef]
- Patro, S.; Ratna, S.; Yamamoto, H.A.; Ebenezer, A.T.; Ferguson, D.S.; Kaur, A.; McIntyre, B.C.; Snow, R.; Solesio, M.E. ATP Synthase and Mitochondrial Bioenergetics Dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 11185. [Google Scholar] [CrossRef]
- Trumpff, C.; Owusu-Ansah, E.; Klein, H.-U.; Lee, A.; Petyuk, V.; Wingo, T.S.; Wingo, A.P.; Thambisetty, M.; Ferrucci, L.; Seyfried, N.T.; et al. Mitochondrial Respiratory Chain Protein Co-Regulation in the Human Brain. Heliyon 2022, 8, e09353. [Google Scholar] [CrossRef]
- Galindo, M.F.; Ikuta, I.; Zhu, X.; Casadesus, G.; Jordán, J. Mitochondrial biology in Alzheimer’s disease pathogenesis. J. Neurochem. 2010, 114, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Fišar, Z.; Hroudová, J.; Zvěřová, M.; Jirák, R.; Vaněčková, M. Disruption of mitochondrial respiration and the monoamine neurotransmitter system in Alzheimer’s disease. Res. Sq. 2022; preprint. [Google Scholar] [CrossRef]
- Parihar, M.S.; Brewer, G.J. Mitoenergetic failure in Alzheimer disease. Am. J. Physiol.-Cell Physiol. 2007, 292, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D.; Parks, J.; Filley, C.M.; Kleinschmidt-Demasters, B.K. Electron transport chain defects in Alzheimer’s disease brain. Neurology 1994, 44, 1090. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Levault, K.R.; Brewer, G.J. Relative importance of redox buffers GSH and NAD(P)H in age-related neurodegeneration and Alzheimer disease-like mouse neurons. Aging Cell 2014, 13, 631–640. [Google Scholar] [CrossRef]
- Yin, F.; Sancheti, H.; Cadenas, E. Silencing of nicotinamide nucleotide transhydrogenase impairs cellular redox homeostasis and energy metabolism in PC12 cells. Biochim. Biophys. Acta-Bioenerg. 2012, 1817, 401–409. [Google Scholar] [CrossRef]
- Eckert, A.; Schmitt, K.; Götz, J. Mitochondrial dysfunction—The beginning of the end in Alzheimer’s disease? Separate and synergistic modes of tau and amyloid-toxicity. Alzheimer’s Res. Ther. 2011, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Onyango, I.G.; Dennis, J.; Khan, S.M. Mitochondrial Dysfunction in Alzheimer’s Disease and the Rationale for Bioenergetics Based Therapies. Aging Dis. 2016, 7, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.W.; Yuan, Z.; Zhao, D.Q.; Wang, Z.W.; Zhu, F.Q.; Luan, P. The effect of V-ATPase function defects in pathogenesis of Alzheimer’s disease. CNS Neurosci. Ther. 2018, 24, 837–840. [Google Scholar] [CrossRef]
- Reiss, A.B.; Ahmed, S.; Dayaramani, C.; Glass, A.D.; Gomolin, I.H.; Pinkhasov, A.; Stecker, M.M.; Wisniewski, T.; De Leon, J. The role of mitochondrial dysfunction in Alzheimer’s disease: A potential pathway to treatment. Exp. Gerontol. 2022, 164, 111828. [Google Scholar] [CrossRef]
- Collins, M.P.; Forgac, M. Regulation and function of V-ATPases in physiology and disease. Biochim. Biophys. Acta-Biomembr. 2020, 1862, 183341. [Google Scholar] [CrossRef]
- Santos-Pereira, C.; Rodrigues, L.R.; Côrte-Real, M. Emerging insights on the role of V-ATPase in human diseases: Therapeutic challenges and opportunities. Med. Res. Rev. 2021, 41, 1927–1964. [Google Scholar] [CrossRef]
- Eaton, A.F.; Merkulova, M.; Brown, D. The H+-ATPase (V-ATPase): From proton pump to signaling complex in health and disease. Am. J. Physiol.-Cell Physiol. 2021, 320, C392–C414. [Google Scholar] [CrossRef]
- Williamson, W.R.; Hiesinger, P.R. On the role of v-ATPase V0a1–dependent degradation in Alzheimer Disease. Commun. Integr. Biol. 2010, 3, 604–607. [Google Scholar] [CrossRef]
- Zhou, Z.; Bai, J.; Zhong, S.; Zhang, R.; Kang, K.; Zhang, X.; Xu, Y.; Zhao, C.; Zhao, M. Downregulation of ATP6V1A Involved in Alzheimer’s Disease via Synaptic Vesicle Cycle, Phagosome, and Oxidative Phosphorylation. Oxid. Med. Cell. Longev. 2021, 2021, 5555634. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Cho, Y.-S.; Kim, Y.; Park, J.; Yoo, S.-M.; Gwak, J.; Kim, Y.; Gwon, Y.; Kam, T.-i.; Jung, Y.-K. Endolysosomal impairment by binding of amyloid beta or MAPT/Tau to V-ATPase and rescue via the HYAL-CD44 axis in Alzheimer disease. Autophagy 2023, 19, 2318–2337. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Meng, B.; Xu, H.; Mao, Z. The emerging roles of vacuolar-type ATPase-dependent Lysosomal acidification in neurodegenerative diseases. Transl. Neurodegener. 2020, 9, 17. [Google Scholar] [CrossRef]
- Haitina, T.; Lindblom, J.; Renström, T.; Fredriksson, R. Fourteen novel human members of mitochondrial solute carrier family 25 (SLC25) widely expressed in the central nervous system. Genomics 2006, 88, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Hawking, Z.L. Alzheimer’s disease: The role of mitochondrial dysfunction and potential new therapies. Biosci. Horizons Int. J. Student Res. 2016, 9, hzw014. [Google Scholar] [CrossRef]
- Hu, C.; Tao, L.; Cao, X.; Chen, L. The solute carrier transporters and the brain: Physiological and pharmacological implications. Asian J. Pharm. Sci. 2020, 15, 131–144. [Google Scholar] [CrossRef]
- Sasarman, F.; Brunel-Guitton, C.; Antonicka, H.; Wai, T.; Shoubridge, E.A.; Allen, B.; Burelle, Y.; Charron, G.; Coderre, L.; DesRosiers, C.; et al. A Highlights from MBoC Selection: LRPPRC and SLIRP Interact in a Ribonucleoprotein Complex That Regulates Posttranscriptional Gene Expression in Mitochondria. Mol. Biol. Cell 2010, 21, 1315–1434. [Google Scholar] [CrossRef]
- Bennett, J.P.; Keeney, P.M. Alzheimer’s and Parkinson’s brain tissues have reduced expression of genes for mtDNA OXPHOS Proteins, mitobiogenesis regulator PGC-1α protein and mtRNA stabilizing protein LRPPRC (LRP130). Mitochondrion 2020, 53, 154–157. [Google Scholar] [CrossRef]
- Panchal, K.; Tiwari, A.K. Miro, a Rho GTPase genetically interacts with Alzheimer’s disease-associated genes (Tau, Aβ42 and Appl) in Drosophila melanogaster. Biol. Open 2020, 9, bio049569. [Google Scholar] [CrossRef]
- Panchal, K.; Tiwari, A.K. Miro (Mitochondrial Rho GTPase), a key player of mitochondrial axonal transport and mitochondrial dynamics in neurodegenerative diseases. Mitochondrion 2021, 56, 118–135. [Google Scholar] [CrossRef]
- Iijima-Ando, K.; Sekiya, M.; Maruko-Otake, A.; Ohtake, Y.; Suzuki, E.; Lu, B.; Iijima, K.M. Loss of Axonal Mitochondria Promotes Tau-Mediated Neurodegeneration and Alzheimer’s Disease–Related Tau Phosphorylation Via PAR-1. PLoS Genet. 2012, 8, e1002918. [Google Scholar] [CrossRef]
- Kay, L.; Pienaar, I.S.; Cooray, R.; Black, G.; Soundararajan, M. Understanding Miro GTPases: Implications in the Treatment of Neurodegenerative Disorders. Mol. Neurobiol. 2018, 55, 7352–7365. [Google Scholar] [CrossRef] [PubMed]
- Maina, M.B.; Al-Hilaly, Y.K.; Serpell, L.C. Nuclear Tau and Its Potential Role in Alzheimer’s Disease. Biomolecules 2016, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Antón-Fernández, A.; Vallés-Saiz, L.; Avila, J.; Hernández, F. Neuronal nuclear tau and neurodegeneration. Neuroscience 2023, 518, 178–184. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morello, G.; Guarnaccia, M.; La Cognata, V.; Latina, V.; Calissano, P.; Amadoro, G.; Cavallaro, S. Transcriptomic Analysis in the Hippocampus and Retina of Tg2576 AD Mice Reveals Defective Mitochondrial Oxidative Phosphorylation and Recovery by Tau 12A12mAb Treatment. Cells 2023, 12, 2254. https://doi.org/10.3390/cells12182254
Morello G, Guarnaccia M, La Cognata V, Latina V, Calissano P, Amadoro G, Cavallaro S. Transcriptomic Analysis in the Hippocampus and Retina of Tg2576 AD Mice Reveals Defective Mitochondrial Oxidative Phosphorylation and Recovery by Tau 12A12mAb Treatment. Cells. 2023; 12(18):2254. https://doi.org/10.3390/cells12182254
Chicago/Turabian StyleMorello, Giovanna, Maria Guarnaccia, Valentina La Cognata, Valentina Latina, Pietro Calissano, Giuseppina Amadoro, and Sebastiano Cavallaro. 2023. "Transcriptomic Analysis in the Hippocampus and Retina of Tg2576 AD Mice Reveals Defective Mitochondrial Oxidative Phosphorylation and Recovery by Tau 12A12mAb Treatment" Cells 12, no. 18: 2254. https://doi.org/10.3390/cells12182254