
Role of the YAP/TAZ-TEAD Transcriptional Complex in the Metabolic Control of TRAIL Sensitivity by the Mevalonate Pathway in Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents
2.3. Antibodies
2.4. Determination of Hypodiploid Apoptotic Cells
2.5. Analysis of Cell Viability
2.6. Western Blotting of Proteins
2.7. Real-Time qPCR
| GAPDH | Forward: 5′-ATGGGGAAGGTGAAGGTCG-3′ Reverse: 5′-GGGTCATTGATGGCAACAATATC-3′ |
| FLIPL | Forward:5′-CCTAGGAATCTGCGTGATAATCGA-3′ Reverse: 5′-TGGGATATACCATGCATACTGAGATG-3′ |
| FLIPS | Forward: 5′-GGGCCGAGGCAAGATAAGCAAGG-3′ Reverse: 5′-TCAGGACAATGGGCATAGGGTGT-3′ |
| CTGF | Forward: 5′-AGCTGACCTGGAAGAGAACA-3′ Reverse: 5′-CAGGCACAGGTCTTGATGAA-3′ |
| CYR61 | Hs00155479_m1 |
| HPRT1 | Hs01003267_m1 |
2.8. RNA Interference
| YAP#1: | 5′-GACAUCUUCUGGUCAGAGAdTdT-3′ |
| YAP#2: | 5′-CUGGUCAGAGAUACUUCUUdTdT-3′ |
| YAP#3: | 5′-GGUGAUACUAUCAACCAAAdTdT-3′ |
| TAZ#1: | 5′-ACGUUGACUUAGGAACUUUdTdT-3’ |
| TAZ#2: | 5´-AGGUACUUCCUCAAUCACAdTdT-3´ |
| cFLIP: | 5′-GGGACCUUCUGGAUAUUUUdTdT-3′ |
| Non-targeting control siRNA (Scr) | 5′-CUUUGGGUGAUCUACGUUAdTdT-3′ |
| siY/T#1: siYAP#1 + siTAZ#1 siY/T#2: siYAP#2 + siTAZ#2 siY/T#3: siYAP#3 + siTAZ#2 |
2.9. Retroviral and Lentiviral Vectors
| TRAIL-R2: | 5′-GATCCCCGACCCTTGTGCTCGTTGTCTTCAAGAGA GACAACGAGCACAAGGGTCTTTTTTA-3′ |
| Caspase-8: | 5′-GATCCCCGGAGCTGCTCTTCCGAATTTTCAAGAG AAATTCGGAAGAGCAGCTCCTTTTTA-3′ |
| Scrambled (Scr): | 5′-GATCCCCCTTTGGGTGATCTACGTTATTCAAGAGA TAACGTAGATCACCCAAAGTTTTTA-3′ |
2.10. Generation of A549 Cell Lines
2.11. Immunofluorescence
2.12. Assessment of Luciferase Activity
2.13. Quantitative Analysis of Caspase-8 Activity
2.14. Global Protein Synthesis Determination by the SUnSET Method
2.15. Statistics
3. Results
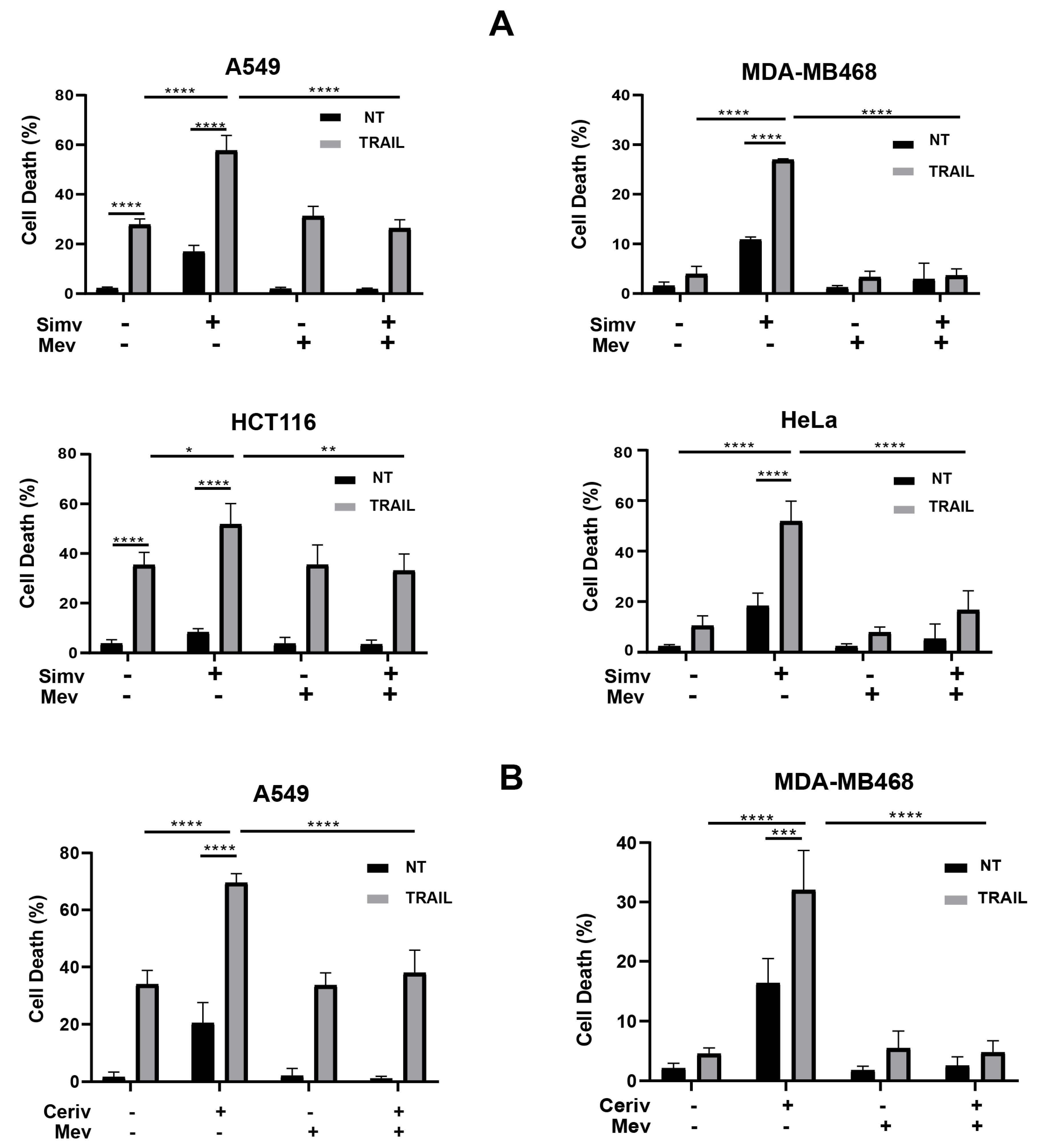
3.1. Control of TRAIL Sensitivity by the Mevalonate Pathway in Cancer Cells
3.2. Metabolic Control of YAP/TAZ Subcellular Localization and Activity by the Mevalonate Pathway in Cancer Cells
3.3. Inhibition of YAP/TAZ Signaling Mediates Statin-Induced Sensitization to TRAIL
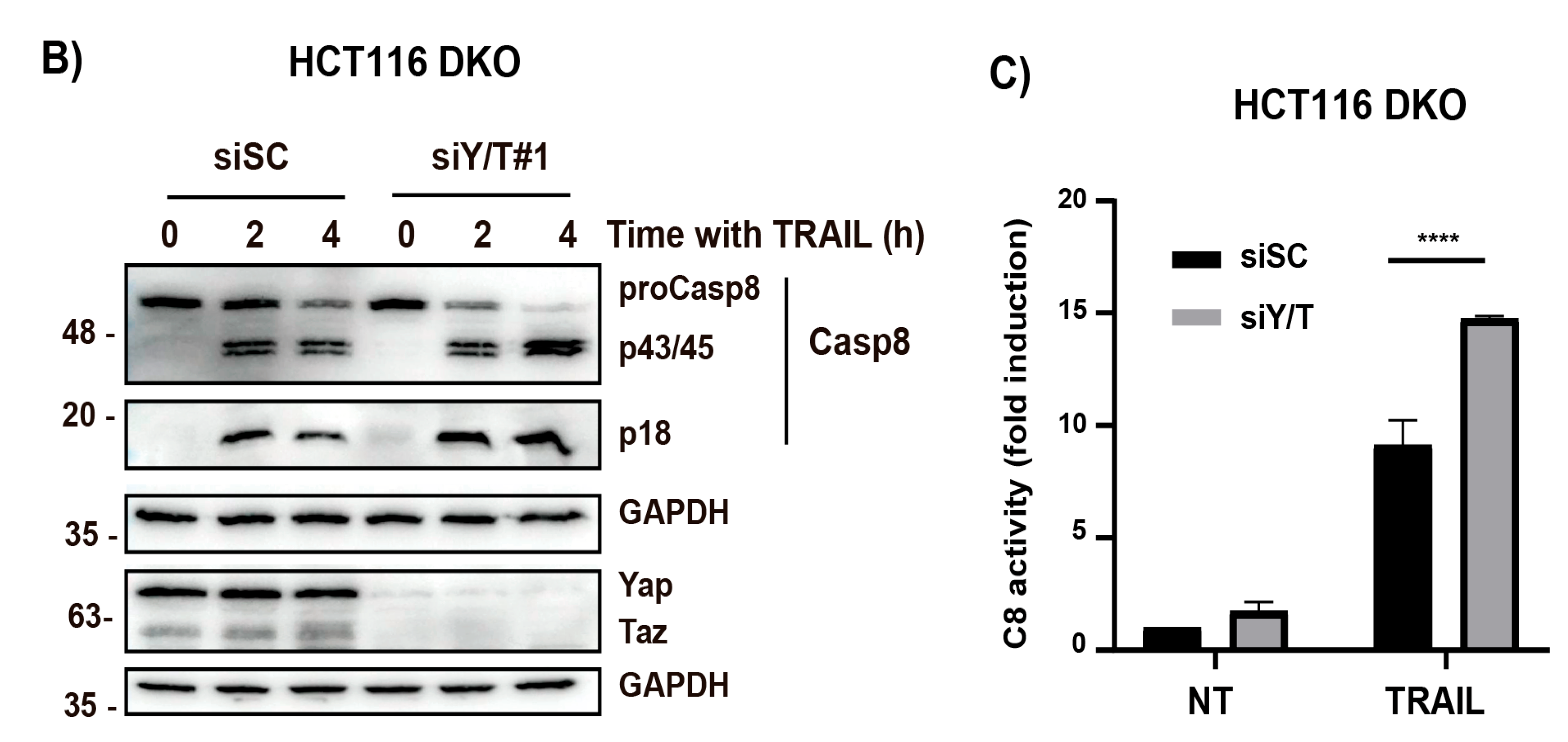
3.4. Control of TRAIL Sensitivity by the YAP/TAZ-TEAD Signaling Module
3.5. YAP/TAZ-TEAD Signaling Confers Resistance to TRAIL-Induced Apoptosis by Controlling cFLIP Levels in Cancer Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A.; et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Investig. 1999, 104, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Sprick, M.R.; Weigand, M.A.; Rieser, E.; Rauch, C.T.; Juo, P.; Blenis, J.; Krammer, P.H.; Walczak, H. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity 2000, 12, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, J.L.; Holler, N.; Reynard, S.; Vinciguerra, P.; Schneider, P.; Juo, P.; Blenis, J.; Tschopp, J. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat. Cell Biol. 2000, 2, 241–243. [Google Scholar] [CrossRef] [PubMed]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.L.; Schroter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Hietakangas, V.; Poukkula, M.; Heiskanen, K.M.; Karvinen, J.T.; Sistonen, L.; Eriksson, J.E. Erythroid differentiation sensitizes K562 leukemia cells to TRAIL-induced apoptosis by downregulation of c-FLIP. Mol. Cell Biol. 2003, 23, 1278–1291. [Google Scholar] [CrossRef]
- Kim, Y.; Suh, N.; Sporn, M.; Reed, J.C. An inducible pathway for degradation of FLIP protein sensitizes tumor cells to TRAIL-induced apoptosis. J. Biol. Chem. 2002, 277, 22320–22329. [Google Scholar] [CrossRef]
- Mauro-Lizcano, M.; Lopez-Rivas, A. Glutamine metabolism regulates FLIP expression and sensitivity to TRAIL in triple-negative breast cancer cells. Cell Death Dis. 2018, 9, 205. [Google Scholar] [CrossRef]
- Ortiz-Ferron, G.; Yerbes, R.; Eramo, A.; Lopez-Perez, A.I.; De Maria, R.; Lopez-Rivas, A. Roscovitine sensitizes breast cancer cells to TRAIL-induced apoptosis through a pleiotropic mechanism. Cell Res. 2008, 18, 664–676. [Google Scholar] [CrossRef]
- Palacios, C.; Yerbes, R.; Lopez-Rivas, A. Flavopiridol induces cellular FLICE-inhibitory protein degradation by the proteasome and promotes TRAIL-induced early signaling and apoptosis in breast tumor cells. Cancer Res. 2006, 66, 8858–8869. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef]
- Feussner, G. HMG CoA reductase inhibitors. Curr. Opin. Lipidol. 1994, 5, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. HMG-CoA reductase inhibitors for treatment of hypercholesterolemia. N. Engl. J. Med. 1988, 319, 24–33. [Google Scholar] [PubMed]
- Swanson, K.M.; Hohl, R.J. Anti-cancer therapy: Targeting the mevalonate pathway. Curr. Cancer Drug Targets 2006, 6, 15–37. [Google Scholar] [CrossRef] [PubMed]
- Edwards, P.A.; Ericsson, J. Sterols and isoprenoids: Signaling molecules derived from the cholesterol biosynthetic pathway. Annu. Rev. Biochem. 1999, 68, 157–185. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Hu, J.W.; He, X.R.; Jin, W.L.; He, X.Y. Statins: A repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef]
- Ivanov, V.N.; Hei, T.K. Regulation of apoptosis in human melanoma and neuroblastoma cells by statins, sodium arsenite and TRAIL: A role of combined treatment versus monotherapy. Apoptosis 2011, 16, 1268–1284. [Google Scholar] [CrossRef]
- Jin, Z.; Dicker, D.T.; El-Deiry, W.S. Enhanced sensitivity of G1 arrested human cancer cells suggests a novel therapeutic strategy using a combination of simvastatin and TRAIL. Cell Cycle 2002, 1, 82–89. [Google Scholar] [CrossRef]
- Liu, P.C.; Lu, G.; Deng, Y.; Wang, C.D.; Su, X.W.; Zhou, J.Y.; Chan, T.M.; Hu, X.; Poon, W.S. Inhibition of NF-kappaB Pathway and Modulation of MAPK Signaling Pathways in Glioblastoma and Implications for Lovastatin and Tumor Necrosis Factor-Related Apoptosis Inducing Ligand (TRAIL) Combination Therapy. PLoS ONE 2017, 12, e0171157. [Google Scholar]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.S.; Park, H.W.; Guan, K.L. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014, 15, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Urtasun, R.; Latasa, M.U.; Demartis, M.I.; Balzani, S.; Goni, S.; Garcia-Irigoyen, O.; Elizalde, M.; Azcona, M.; Pascale, R.M.; Feo, F.; et al. Connective tissue growth factor autocriny in human hepatocellular carcinoma: Oncogenic role and regulation by epidermal growth factor receptor/yes-associated protein-mediated activation. Hepatology 2011, 54, 2149–2158. [Google Scholar] [CrossRef]
- Diep, C.H.; Zucker, K.M.; Hostetter, G.; Watanabe, A.; Hu, C.; Munoz, R.M.; Von Hoff, D.D.; Han, H. Down-regulation of Yes Associated Protein 1 expression reduces cell proliferation and clonogenicity of pancreatic cancer cells. PLoS ONE 2012, 7, e32783. [Google Scholar] [CrossRef]
- Kim, M.; Kim, T.; Johnson, R.L.; Lim, D.S. Transcriptional co-repressor function of the hippo pathway transducers YAP and TAZ. Cell Rep. 2015, 11, 270–282. [Google Scholar] [CrossRef]
- Ma, K.; Xu, Q.; Wang, S.; Zhang, W.; Liu, M.; Liang, S.; Zhu, H.; Xu, N. Nuclear accumulation of Yes-Associated Protein (YAP) maintains the survival of doxorubicin-induced senescent cells by promoting survivin expression. Cancer Lett. 2016, 375, 84–91. [Google Scholar] [CrossRef]
- Strano, S.; Monti, O.; Pediconi, N.; Baccarini, A.; Fontemaggi, G.; Lapi, E.; Mantovani, F.; Damalas, A.; Citro, G.; Sacchi, A.; et al. The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA Damage. Mol. Cell 2005, 18, 447–459. [Google Scholar] [CrossRef]
- Lapi, E.; Di Agostino, S.; Donzelli, S.; Gal, H.; Domany, E.; Rechavi, G.; Pandolfi, P.P.; Givol, D.; Strano, S.; Lu, X.; et al. PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Mol. Cell 2008, 32, 803–814. [Google Scholar] [CrossRef]
- Xiao, Q.; Qian, Z.; Zhang, W.; Liu, J.; Hu, E.; Zhang, J.; Li, M.; Wang, J.; Kong, F.; Li, Y.; et al. Depletion of CABYR-a/b sensitizes lung cancer cells to TRAIL-induced apoptosis through YAP/p73-mediated DR5 upregulation. Oncotarget 2016, 7, 9513–9524. [Google Scholar] [CrossRef]
- Santinon, G.; Pocaterra, A.; Dupont, S. Control of YAP/TAZ Activity by Metabolic and Nutrient-Sensing Pathways. Trends Cell Biol. 2016, 26, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, Y.; Wang, H.; Zhang, Y.; Mei, L.; Fang, X.; Zhang, X.; Zhang, F.; Chen, H.; Liu, Y.; et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc. Natl. Acad. Sci. USA 2014, 111, E89–E98. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Harper, N.; Farrow, S.N.; Kaptein, A.; Cohen, G.M.; MacFarlane, M. Modulation of tumor necrosis factor apoptosis-inducing ligand- induced NF-kappa B activation by inhibition of apical caspases. J. Biol. Chem. 2001, 276, 34743–34752. [Google Scholar] [CrossRef] [PubMed]
- Yerbes, R.; Palacios, C.; Reginato, M.J.; Lopez-Rivas, A. Cellular FLIP(L) plays a survival role and regulates morphogenesis in breast epithelial cells. Biochim. Biophys. Acta 2011, 1813, 168–178. [Google Scholar] [CrossRef]
- Wiznerowicz, M.; Trono, D. Conditional suppression of cellular genes: Lentivirus vector-mediated drug-inducible RNA interference. J. Virol. 2003, 77, 8957–8961. [Google Scholar] [CrossRef]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef]
- Duarte, J.A.; de Barros, A.L.B.; Leite, E.A. The potential use of simvastatin for cancer treatment: A review. Biomed. Pharmacother. 2021, 141, 111858. [Google Scholar] [CrossRef]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.; Bardet, A.F.; Roma, G.; Bergling, S.; Clay, I.; Ruchti, A.; Agarinis, C.; Schmelzle, T.; Bouwmeester, T.; Schubeler, D.; et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet. 2015, 11, e1005465. [Google Scholar] [CrossRef] [PubMed]
- Troilo, A.; Benson, E.K.; Esposito, D.; Garibsingh, R.A.; Reddy, E.P.; Mungamuri, S.K.; Aaronson, S.A. Angiomotin stabilization by tankyrase inhibitors antagonizes constitutive TEAD-dependent transcription and proliferation of human tumor cells with Hippo pathway core component mutations. Oncotarget 2016, 7, 28765–28782. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Park, J.; Feng, A.; Awasthi, P.; Wang, Z.; Chen, Q.; Iglesias-Bartolome, R. YAP1/TAZ-TEAD transcriptional networks maintain skin homeostasis by regulating cell proliferation and limiting KLF4 activity. Nat. Commun. 2020, 11, 1472. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, J.P.; Marsters, S.A.; Pitti, R.M.; Gurney, A.; Skubatch, M.; Baldwin, D.; Ramakrishnan, L.; Gray, C.L.; Baker, K.; Wood, W.I.; et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 1997, 277, 818–821. [Google Scholar] [CrossRef]
- Martin-Perez, R.; Palacios, C.; Yerbes, R.; Cano-Gonzalez, A.; Iglesias-Serret, D.; Gil, J.; Reginato, M.J.; Lopez-Rivas, A. Activated ERBB2/HER2 licenses sensitivity to apoptosis upon endoplasmic reticulum stress through a PERK-dependent pathway. Cancer Res. 2014, 74, 1766–1777. [Google Scholar] [CrossRef]
- Caro-Maldonado, A.; Tait, S.W.; Ramirez-Peinado, S.; Ricci, J.E.; Fabregat, I.; Green, D.R.; Munoz-Pinedo, C. Glucose deprivation induces an atypical form of apoptosis mediated by caspase-8 in Bax-, Bak-deficient cells. Cell Death Differ. 2010, 17, 1335–1344. [Google Scholar] [CrossRef]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef]
- Martin-Perez, R.; Yerbes, R.; Mora-Molina, R.; Cano-Gonzalez, A.; Arribas, J.; Mazzone, M.; Lopez-Rivas, A.; Palacios, C. Oncogenic p95HER2/611CTF primes human breast epithelial cells for metabolic stress-induced down-regulation of FLIP and activation of TRAIL-R/Caspase-8-dependent apoptosis. Oncotarget 2017, 8, 93688–93703. [Google Scholar] [CrossRef]
- McComb, S.; Chan, P.K.; Guinot, A.; Hartmannsdottir, H.; Jenni, S.; Dobay, M.P.; Bourquin, J.P.; Bornhauser, B.C. Efficient apoptosis requires feedback amplification of upstream apoptotic signals by effector caspase-3 or -7. Sci. Adv. 2019, 5, eaau9433. [Google Scholar] [CrossRef]
- Wilson, T.R.; McLaughlin, K.M.; McEwan, M.; Sakai, H.; Rogers, K.M.; Redmond, K.M.; Johnston, P.G.; Longley, D.B. c-FLIP: A key regulator of colorectal cancer cell death. Cancer Res. 2007, 67, 5754–5762. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.A.; Powley, I.R.; Jukes-Jones, R.; Horn, S.; Feoktistova, M.; Fairall, L.; Schwabe, J.W.; Leverkus, M.; Cain, K.; MacFarlane, M. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol. Cell 2016, 61, 834–849. [Google Scholar] [CrossRef] [PubMed]
- Mora-Molina, R.; Stohr, D.; Rehm, M.; Lopez-Rivas, A. cFLIP downregulation is an early event required for endoplasmic reticulum stress-induced apoptosis in tumor cells. Cell Death Dis. 2022, 13, 111. [Google Scholar] [CrossRef] [PubMed]
- Yerbes, R.; Mora-Molina, R.; Fernandez-Farran, F.J.; Hiraldo, L.; Lopez-Rivas, A.; Palacios, C. Limiting glutamine utilization activates a GCN2/TRAIL-R2/Caspase-8 apoptotic pathway in glutamine-addicted tumor cells. Cell Death Dis. 2022, 13, 906. [Google Scholar] [CrossRef]
- Tuckow, A.P.; Jefferson, S.J.; Kimball, S.R.; Jefferson, L.S. Simvastatin represses protein synthesis in the muscle-derived C(2)C(1)(2) cell line with a concomitant reduction in eukaryotic initiation factor 2B expression. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E564–E570. [Google Scholar] [CrossRef]
- Fukazawa, T.; Fujiwara, T.; Uno, F.; Teraishi, F.; Kadowaki, Y.; Itoshima, T.; Takata, Y.; Kagawa, S.; Roth, J.A.; Tschopp, J.; et al. Accelerated degradation of cellular FLIP protein through the ubiquitin-proteasome pathway in p53-mediated apoptosis of human cancer cells. Oncogene 2001, 20, 5225–5231. [Google Scholar] [CrossRef]
- von Karstedt, S.; Montinaro, A.; Walczak, H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 2017, 17, 352–366. [Google Scholar] [CrossRef]
- Lemke, J.; von Karstedt, S.; Zinngrebe, J.; Walczak, H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014, 21, 1350–1364. [Google Scholar] [CrossRef]
- Montinaro, A.; Walczak, H. Harnessing TRAIL-induced cell death for cancer therapy: A long walk with thrilling discoveries. Cell Death Differ. 2023, 30, 237–249. [Google Scholar] [CrossRef]
- Carr, R.M.; Qiao, G.; Qin, J.; Jayaraman, S.; Prabhakar, B.S.; Maker, A.V. Targeting the metabolic pathway of human colon cancer overcomes resistance to TRAIL-induced apoptosis. Cell Death Discov. 2016, 2, 16067. [Google Scholar] [CrossRef]
- Munoz-Pinedo, C.; Ruiz-Ruiz, C.; Ruiz de Almodovar, C.; Palacios, C.; Lopez-Rivas, A. Inhibition of glucose metabolism sensitizes tumor cells to death receptor-triggered apoptosis through enhancement of death-inducing signaling complex formation and apical procaspase-8 processing. J. Biol. Chem. 2003, 278, 12759–12768. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, L.M.; Fox, J.P.; Higgins, C.A.; Majkut, J.; Sessler, T.; McLaughlin, K.; McCann, C.; Roberts, J.Z.; Crawford, N.T.; McDade, S.S.; et al. A revised model of TRAIL-R2 DISC assembly explains how FLIP(L) can inhibit or promote apoptosis. EMBO Rep. 2020, 21, e49254. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Perez, T.; Ortiz-Ferron, G.; Lopez-Rivas, A. Mitotic arrest and JNK-induced proteasomal degradation of FLIP and Mcl-1 are key events in the sensitization of breast tumor cells to TRAIL by antimicrotubule agents. Cell Death Differ. 2010, 17, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Shirley, S.; Micheau, O. Targeting c-FLIP in cancer. Cancer Lett. 2013, 332, 141–150. [Google Scholar] [CrossRef]
- Hassanzadeh, A.; Farshdousti Hagh, M.; Alivand, M.R.; Akbari, A.A.M.; Shams Asenjan, K.; Saraei, R.; Solali, S. Down-regulation of intracellular anti-apoptotic proteins, particularly c-FLIP by therapeutic agents; the novel view to overcome resistance to TRAIL. J. Cell. Physiol. 2018, 233, 6470–6485. [Google Scholar] [CrossRef]
- Humphreys, L.; Espona-Fiedler, M.; Longley, D.B. FLIP as a therapeutic target in cancer. FEBS J. 2018, 285, 4104–4123. [Google Scholar] [CrossRef]
- Dey, A.; Varelas, X.; Guan, K.L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, L.; Gong, Z.; Shen, L.; Kao, C.; Hock, J.M.; Sun, L.; Li, X. Lovastatin enhances adenovirus-mediated TRAIL induced apoptosis by depleting cholesterol of lipid rafts and affecting CAR and death receptor expression of prostate cancer cells. Oncotarget 2015, 6, 3055–3070. [Google Scholar] [CrossRef]
- Guruswamy, S.; Rao, C.V. Synergistic effects of lovastatin and celecoxib on caveolin-1 and its down-stream signaling molecules: Implications for colon cancer prevention. Int. J. Oncol. 2009, 35, 1037–1043. [Google Scholar]
- De Miguel, D.; Gallego-Lleyda, A.; Martinez-Ara, M.; Plou, J.; Anel, A.; Martinez-Lostao, L. Double-Edged Lipid Nanoparticles Combining Liposome-Bound TRAIL and Encapsulated Doxorubicin Showing an Extraordinary Synergistic Pro-Apoptotic Potential. Cancers 2019, 11, 1948. [Google Scholar] [CrossRef]
- Pobbati, A.V.; Kumar, R.; Rubin, B.P.; Hong, W. Therapeutic targeting of TEAD transcription factors in cancer. Trends Biochem. Sci. 2023, 48, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, A.; Kaneko, K.J.; Shu, H.; Zhao, Y.; DePamphilis, M.L. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 2001, 15, 1229–1241. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Yousfi, Y.; Mora-Molina, R.; López-Rivas, A.; Yerbes, R. Role of the YAP/TAZ-TEAD Transcriptional Complex in the Metabolic Control of TRAIL Sensitivity by the Mevalonate Pathway in Cancer Cells. Cells 2023, 12, 2370. https://doi.org/10.3390/cells12192370
El Yousfi Y, Mora-Molina R, López-Rivas A, Yerbes R. Role of the YAP/TAZ-TEAD Transcriptional Complex in the Metabolic Control of TRAIL Sensitivity by the Mevalonate Pathway in Cancer Cells. Cells. 2023; 12(19):2370. https://doi.org/10.3390/cells12192370
Chicago/Turabian StyleEl Yousfi, Younes, Rocío Mora-Molina, Abelardo López-Rivas, and Rosario Yerbes. 2023. "Role of the YAP/TAZ-TEAD Transcriptional Complex in the Metabolic Control of TRAIL Sensitivity by the Mevalonate Pathway in Cancer Cells" Cells 12, no. 19: 2370. https://doi.org/10.3390/cells12192370
APA StyleEl Yousfi, Y., Mora-Molina, R., López-Rivas, A., & Yerbes, R. (2023). Role of the YAP/TAZ-TEAD Transcriptional Complex in the Metabolic Control of TRAIL Sensitivity by the Mevalonate Pathway in Cancer Cells. Cells, 12(19), 2370. https://doi.org/10.3390/cells12192370