1. Introduction
Cardiovascular disease (CVD) is higher by up to tenfold in obese, diabetic adults than in the age-matched population [
1]. Unlocking the intricate relationship between metabolic diseases and vascular dysfunction through the study of intermediate molecular events is therefore of great importance. It is widely acknowledged that fat depots are important secretory organs that release numerous bioactive molecules and that a significant portion of this secretome is found in adipocyte-derived extracellular vesicles (adiposomes) [
2,
3,
4]. In the current study, we propose a novel role for adiposomes as a conveyor of the adipocyte-dysregulated metabolites to endothelial cells, causing endothelial dysfunction. Adiposomes are lipid envelopes that carry genetic materials, proteins, lipids, and other metabolites [
5], and their production increases in metabolic diseases [
6,
7], contributing to inflammation and insulin resistance [
8,
9]. Yet, mechanisms of adiposome interaction with endothelial cells have remained to be explored. Therefore, this study will pave the way to examine biological messages that can be transferred by adiposomes to endothelial cells. In addition, it will reveal some of the effects of these adiposomes on endothelial cell structure and function.
Obesity and insulin resistance are characterized by changes in sphingolipid metabolism and an induced synthesis of glycosphingolipids (GSLs), which may provide a common pathway linking excess nutrients and inflammation to increased cardiovascular risk [
10]. GSLs are a family of active lipids generated via the glycosylation of ceramide [
11]. The generation of GSLs is augmented in inflamed tissues and is driven by the local milieu of high glucose and inflammation-mediated induction of the activity of GSL synthesis enzymes [
12,
13]. High levels of GSLs were shown to be associated with obesity, diabetes, and cardiovascular diseases [
14,
15]. GSLs were reported to promote atherosclerosis, retinopathy, and neuropathy in diabetic models [
16,
17,
18]. Furthermore, the content of lactosyl ceramides in atherosclerotic lesions was found to be 10- to 50-fold higher than the low-density lipoproteins [
19]. However, the critical barrier to progress in this area is that the method of delivery of excess GSLs to endothelial cells and the precise mechanism that mediates their impact on vascular function are poorly understood. GSLs are among the most deleterious lipid classes since they induce aberrant signaling when deposited in cells incompatible with lipid storage, such as endothelial cells.
Previous studies reported the tendency of GSLs to accumulate in surface caveolae [
20]. The latter are 50 to 100 nm plasma membrane invaginations abundant in endothelial cells and critical for lipid homeostasis, vesicular trafficking, and signal transduction [
21]. In endothelial cells, caveolae cover more than 50% of the cell surface [
21]. The major protein component in the caveolar bulb is caveolin-1 (cav-1). Cav-1 and other caveolar proteins represent a docking site for endothelial nitric oxide synthase (eNOS) and other proteins involved in angiogenesis, atherogenesis, adhesion, junctional permeability, and signal transduction, such as Src kinase [
21]. Cav-1 has been identified as a Src kinase substrate [
22]. Tyrosine 14 (Y14) in cav-1 is phosphorylated by Src kinase and has been linked to surface caveolae fission and internalization [
23,
24]. Accordingly, interfering with Src kinase activation inhibited cav-1 phosphorylation and caveolar internalization in endothelial cells [
25]. Interestingly, prolonged cav-1 phosphorylation leads to polyubiquitination and degradation, which is thought to contribute to caveolar reduction on the endothelial cell surface [
26].
Previous research by our team reported a caveolar loss from the endothelial cell surface in response to Src activation and reciprocal regulation of cav-1 and eNOS in endothelial cells. These studies showed induced phosphorylation of eNOS in response to caveolar loss and subsequently detrimental effects on endothelial cell function, such as oxidative stress and nitrosylation of critical endothelial proteins [
27]. In the current study, we sought to examine the lipid content of adiposomes produced by obese diabetic individuals versus lean, healthy controls. Also, we sought to investigate the effect of obese diabetic adiposomes on (1) endothelial caveolae and the associated signaling and (2) on vasoreactivity of adipose tissue arterioles isolated from lean, healthy individuals.
Adiposome production by metabolically challenged adipocytes is a quality control mechanism for maintaining cellular homeostasis and eliminating unrequired metabolites such as GSLs [
28]. This spillover of lipid metabolites into the circulation is followed by ectopic deposition in cells incompatible with lipid storage, such as endothelial cells [
29]. Unlike the protein and nucleic acid cargo, the lipid content in extracellular vesicles and its implication in vascular function has been far less explored. Accordingly, the current research may contribute to a better knowledge of new risk factors for the development of CVD in obese diabetic patients, as well as new therapeutic targets.
2. Materials and Methods
2.1. Study Subjects
Adipose tissue (AT) biopsies were obtained at the University of Illinois Hospital from 15 obese-diabetic subjects (body mass index (BMI) higher than 30 kg/m2) and 15 non-obese controls (BMI less than 25 kg/m2) during bariatric and elective surgeries. Adults below 50 years of age were recruited to eliminate potential detrimental effects of aging on arterial function, accelerated after 50 years of age. The criteria for T2D were confirmed use of diabetes medication and a fasting glucose level of ≥126 mg/dL. Exclusion criteria included current smoking, pregnancy, prior metabolic surgery, current liver, kidney, or heart disease, malignancy, and acute or chronic inflammatory diseases. The methodologies and procedures employed in this study were approved by the Institutional Review Board of the University of Illinois (Protocol # 2017-1125) and were found to be in compliance with the latest version of the Declaration of Helsinki.
2.2. Adipose Tissue Processing and Endothelial Cell and Adiposome Isolation
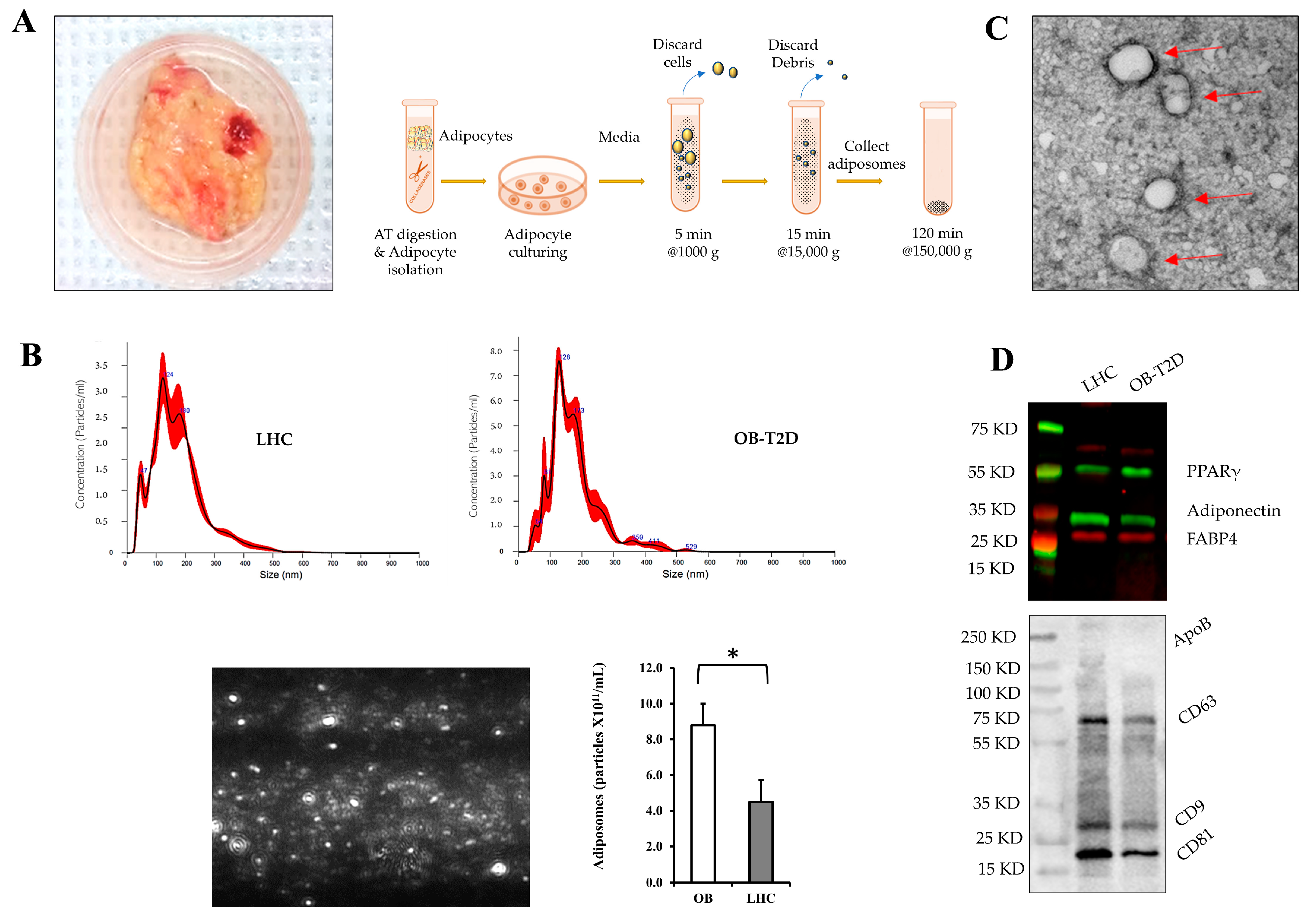
Surgically obtained fresh AT biopsies were washed in sterile Medium 199 (M199) (Gibco, Waltham, MA, USA) and minced into small pieces around 5–10 mg each (1–2 mm
3), using a sterile scalpel and sharp scissors. Collagenase solution (2–3 mL per 1 g of AT) that consists of 1 mg/mL Type 1 collagenase (Worthington) and 4% BSA in M199 medium was added to the minced AT followed by incubation for 15 min in a cell culture incubator. Tissues were then incubated in a 37 °C water bath for 60 min with frequent shaking. The digested AT were filtered through a nylon mesh filter and centrifuged at a low speed (500×
g for 1 min) [
30]. Floating adipocytes were collected, washed three times, and cultured in M199 medium with 5% exosome-free fetal bovine serum (FBS) and 1% penicillin/streptomycin for 48–72 h. Cells were cultured with and without the glucosylceramide synthase inhibitor (1R,2R)-nonanoic acid [2-(2′,3′-dihydro-benzo[1,4]dioxin-6′-yl)-2-hydroxy-1-pyrrolidin-1-ylmethyl-ethyl]-amide-l-tartaric acid salt (Genz-123346; 1 μmol/L) purchased from Sigma-Aldrich (St. Louis, MO, USA). Culture media were centrifuged at 4 °C at 1000×
g for 5 min, followed by centrifugation at 15,000×
g for 15 min. The resulting supernatant was then filtered through a 0.45 μm syringe-driven filter to remove cellular debris, apoptotic bodies, and microvesicles. Filtered samples were then ultracentrifuged at 150,000×
g for 2 h to pellet adiposomes. Adiposomes were then resuspended in PBS and measured using nanoparticle tracking analysis (NTA) to evaluate particle size and concentration. NTA was carried out with a NanoSight NS300 Analyzer (Malvern Instruments Ltd., Malvern, UK), software version 2.3, screen gain 4, and camera level 10. Five 1 min movies were produced for each sample, with the analysis screen gain set to 10 and the detection threshold set to 4 [
31].
Following AT digestion, the stromal vascular fraction (SVF) was used to isolate endothelial cells. Red blood cell lysis buffer was used to purify the SVF for better binding to magnetic beads. Each sample was resuspended in an appropriate amount of CD31 human MicroBeads (human, Miltenyi Biotec, Cat#130-091-935) and FcR Blocking Reagent based on the provided protocol by the manufacturer. Samples were then incubated for 30 min at a cold temperature (4 °C) followed by centrifugation at 300× g for 5 min, repeated washing steps, and finally, cells were resuspended in the provided washing buffer. Samples were loaded in the LS columns (Miltenyi Biotec, Gaithersburg, MD, USA) that were placed in the magnetic field of MACS MultiStand (Miltenyi Biotec) equipped with QuadroMACS™ Separator (Miltenyi Biotec). Columns were then washed 3 to 4 times, removed from the magnetic separator, and placed in a 15 mL falcon tube. CD31-positive endothelial cells were then flushed out of the column using an appropriate volume of washing buffer.
2.3. Endothelial Cell Culture and Treatment
Human adipose microvascular endothelial cells (HAMECs) were obtained from Lonza Walkersville Inc. (Walkersville, MD, USA), and passages 3 through 6 were used in the following experiments. Cells were maintained in ECM media with L-glutamine supplemented with 5% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Experiments were performed using passages from 2 to 6. All cell culture media were purchased from ScienCell Research Laboratories (Carlsbad, CA, USA). Cells were cultured in a CO2 incubator at 37 °C. Cells cultured in 35 mm dishes were treated with adiposome particles (5 × 1011) or GM3 (500 nmol/L) for periods that ranged from 30 min to 24 h with and without the Src kinase inhibitor, PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine; 1 µmol/L) purchased from Sigma-Aldrich (St. Louis, MO, USA). All experiments were conducted in triplicates, and the findings were expressed as the mean ± standard error (SE).
2.4. Isolation of Endothelial Cell Total Cell Lysate and Cytoplasmic/Membranous Protein Fractions
The extraction of total cell lysate was performed using RIPA lysis buffer, which is composed of various components including 150 mM NaCl, 20 mM Tris-HCl (pH 7.5), 1 mM EGTA, 1 mM Na2 EDTA, 1% NP-40, 2.5 mM sodium pyrophosphate, 1% sodium deoxycholate, 1 mM Na3VO4, 1 mM b-glycerophosphate, and 1 µg/mL leupeptin (Cell Signaling, Danvers, MA, USA). Additionally, the lysis buffer was supplemented with a protease and phosphatase inhibitor mixture (MS-SAFE) obtained from Sigma-Aldrich (St. Louis, MO, USA). Membranous and cytoplasmic protein fractions were isolated via a Subcellular Protein Fractionation Kit (ThermoFisher Scientific, Waltham, MA, USA). Briefly, the cell pellet was washed and left to dry, followed by adding the membranous and cytoplasmic extraction buffers following the recommended volume and centrifugation steps in the kit’s manual. First, the cytoplasmic extraction buffer was added and incubated at 4 °C for 10 min, followed by centrifugation at 500× g for 5 min and transfer of the cytoplasmic supernatant to a clean tube. During the second phase, the membranous extraction buffer was introduced and allowed to incubate at a temperature of 4 °C for a duration of 10 min. Subsequently, centrifugation was performed at a force of 3000× g for a period of 5 min. The membrane extract was then carefully transferred to a clean tube.
2.5. Lipid Extraction and Mass Spectrometry Analysis
Lipid extraction from adiposomes was performed using the Folch method. Briefly, ice-cold methanol and chloroform (1:2, v/v) were added directly to the samples (adiposome pellet). For non-targeted lipid analysis in adiposomes, the lipid internal standards, EquiSPLASH LIPIDOMIX mixture (Avanti Polar Lipids, Birmingham, AL, USA) was used. Samples were sonicated, vortexed, and kept on ice for 30 min, followed by the addition of ice-cold chloroform/methanol/water (8:4:3, v/v/v) and centrifugation at 10,000 rpm for 5 min at 4 °C to induce phase separation. The lower phase (organic layer) was collected and evaporated under a nitrogen stream at 30 °C. The extracted lipids were resuspended in methanol and chloroform (1:1, v/v) and transferred to a liquid chromatography vial with a 200 μL glass insert. A quality control sample was generated by combining a portion of each individual sample. The lipid extracts were subjected to analysis using an Agilent 6545 Q-TOF LC-MS system, which was operated with the Agilent Mass Hunter acquisition software version B.08.00. The mass spectrometer was utilized in a 2 GHz extended dynamic range mode, utilizing precursor ion analysis for conducting relative quantification tests in positive ion mode. Calibration was conducted using internal references. The composition of mobile phase A consisted of a mixture of 90% water and 10% methanol (v/v), supplemented with 10 mM ammonium acetate and 0.5 mM ammonium fluoride. On the other hand, mobile phase B was composed of a mixture in a ratio of 5:3:2. The mixture consists of isopropanol, methanol, and acetonitrile in a volumetric ratio, supplemented with 10 mM ammonium acetate and 0.5 mM ammonium fluoride. The lipid extracts that had been resuspended were introduced onto a 2.1 × 100 mm Agilent Poroshell C18, 2.7 μm column (Agilent Technologies Inc., Santa Clara, CA, USA) for separation. This was achieved using an Agilent 1290 UPLC system, employing a gradient consisting of 70% B from 0 to 1 min, 86% B from 3.5 to 10 min, and 100% B from 11 to 17 min. The flow rate used was 300 μL/min. A uniform post-column equilibration duration of 5 min was employed for all experimental runs. The experimental conditions were defined by the following parameters: the temperature of the gas was set at 200 °C, the flow rate of the drying gas was maintained at 11 L/min, the nebulizer pressure was set to 35 psi, the temperature of the sheath gas was maintained at 350 °C, the flow rate of the sheath gas was set at 12 L/min, the voltage applied to the VCap was 3000 V, and the fragmentor voltage was set to 145 V. Data were gathered for the purpose of relative quantification, with a scan speed of 4 MS spectra per second. The construction of the lipid database involved the utilization of pooled samples. A repetitive MS/MS workflow was executed using the Mass Hunter acquisition software version B.08.00. This workflow was applied to numerous injections of the pooled samples, with a scan speed of 10 MS and 3 MS/MS spectra per second.
Lipid raft isolation from endothelial cells was performed using Minute™ Plasma Membrane-Derived Lipid Raft Isolation Kit (Invent Biotechnologies Inc., Plymouth, MN, USA). Lipid extraction from isolated lipid rafts was performed as described above. For targeted lipidomics in endothelial cell membranes, a mixture of C16 Lactosyl (β) Ceramide (d18:1/16:0) and C16 Ganglioside GM3-d9 (d18:1/16:0-d9) standards for LacCer and GM3 targeted analyses (Avanti Polar Lipids, Birmingham, AL, USA) was used. Five microliters of the calibrator/sample were injected into an AB SCIEX 5500 QTRAP system coupled to an Agilent 1290 UPLC system. At a flow rate of 300 L/min, all samples were eluted from an Agilent Poroshell 120 SB-AQ 2.7 m, 2.1 × 100 mm column (P/N 685775-914). The column compartment was maintained at 40 °C. LC elution began with 50% mobile phase A (0.1% FA in H2O) for 1 min, followed by a linear gradient increase in mobile phase B (0.1% FA in ACN) from 50% to 100% over the same period. The column was maintained at 100% B for 7 min before being re-equilibrated to its initial state of 50% A for 3 min. The autosampler temperature was kept at 4 °C. MS data were acquired by MRM scan at positive mode. ESI spray voltage and source temperature were maintained at 4.5 kV and 500 °C, respectively. Different chain length combinations of positively charged LacCer and GM3 were detected by monitoring their transition to signature product ions.
The raw LC-MS/MS data were utilized to generate a fragmentation-based (MS/MS) library. This library included m/z precursors and retention times for all lipids that were identified through the use of the Lipid Annotator software version B.07.01 (Agilent Technologies Inc., Santa Clara, CA, USA). The software was configured with specific settings, namely the inclusion of lipid species for positive ionization ([M + H]+, [M + Na]+, [M + NH4]+, [M + H − H2O]+, [M + Na − H2O]+, [M + NH4 − H2O]+), a Q-Score threshold of 60 or higher, and a mass deviation limit of 10 ppm or less. The LC-MS data files were subjected to processing using the Profinder program (version B.10.00, Agilent Technologies Inc., Santa Clara, CA, USA). In this study, we extracted molecular characteristics for peaks with counts ≥ 5000 in the positive mode. Specifically, we focused on ions such as [M + H]+, [M + Na]+, [M + NH4]+, [M + H − H2O]+, [M + Na − H2O]+, and [M + NH4 − H2O]+. To perform this extraction, we employed an isotopic model that accounted for common organic compounds without halogens. The compound list obtained was subsequently refined by applying additional filters, including a minimum absolute height of 10,000 counts, a minimum quality score of 60, and the presence of two or more isotopes in the compounds. Furthermore, the retention period for each compound was adjusted to within a range of ±0.1 min, employing a mass accuracy window of ≤5.0 parts per million (ppm). The peaks were then quantified using the Agile integrator within the Profinder program. The retention time and fragmentation matching of each integrated peak were individually evaluated. Subsequently, the processed data file was exported and subsequently imported into the Mass Profiler Professional program (version 15.1, Agilent Technologies Inc., Santa Clara, CA, USA), wherein each data set was subjected to separate analysis. The compound abundance results were subjected to baseline correction using the median abundance and afterward adjusted based on the lipid class in the internal standard (IS).
2.6. Biotin Switch Assay
Biotin Switch Assay was used to detect cav-1 S-nitrosylation following previously published methods [
26,
32]. Briefly, endothelial cells were cultured in a serum-free medium for 3 to 5 h and then treated with adiposome particles (5 × 10
11) with and without L-NAME or PP2 for 15 min. Endothelial cells were then harvested in lysis buffer, and 25% SDS along with 10% S-Methyl methanethiosulfonate (Sigma Aldrich; St. Louis, MO, USA) were added to the cell lysates to block thiol groups. Subsequently, ascorbate was utilized to facilitate the conversion of S-nitroso (SNO) groups to free thiol groups. This was followed by protein biotinylation via biotin-HPDP (ThermoFisher Scientific, Waltham, MA, USA). The biotinylated protein was pulled down via streptavidin beads and resolved by SDS–PAGE. As loading controls, we used the same samples prior to immunoprecipitation. A similar protocol was used to test cav-1 S-nitrosylation in AT-isolated endothelial cells.
2.7. Western Blotting and Cav-1 Oligomerization
Thermo Fisher Scientific’s Pierce BCA Protein Assays were used to measure the total protein concentration, which was then transferred to polyvinylidene fluoride (PVDF) after electrophoresis on 4–12% Bis-Tris gradient gels. Membranes were subsequently blocked using Intercept (TBS) Blocking Buffer (LI-COR Biosciences; Lincoln, NE, USA) and incubated in primary monoclonal antibodies (total cav-1, phospho-cav-1 Tyr14, total eNOS, phospho-eNOS Ser1177, and NADPH oxidase (Nox2) (Cell Signaling, Danvers, MA, USA), total Src, phospho-Src Tyr416 (Thermo Fisher Scientific, Waltham, MA, USA) at 4 °C overnight. Following this, infrared IRDye-labeled secondary antibodies (LI-COR Biosciences) were applied for 1 h at room temperature and protected from light. The membranes were washed with TBS + 0.1% Tween-20, dried at room temperature, and imaged in the appropriate channel (700 nm for IRDye680TM antibodies, 800 nm for IRDye800TM antibodies) using an Odyssey Clx infrared imaging system [
33]. The quantification of images was performed using Image Studio ver. 4.0 (LI-COR Biosciences) to determine the intensity of phosphorylated protein bands relative to the expression of their corresponding total proteins.
Low-temperature SDS-PAGE was used to detect eNOS dimers utilizing the previously reported methods [
34]. Briefly, total proteins were incubated at 37 °C for 5 min in 1X Laemmli buffer without 2-mercaptoethanol. The samples were then separated on Bio-Rad 4–12% Bis-Tris gradient electrophoresis. Before electrophoresis, gels, and buffers were equilibrated at 4 °C, and during electrophoresis, the gel temperature was maintained at <15 °C by placing the buffer vessel in an ice bath. The samples were then transferred to PVDF membranes for Western blotting. For cav-1 oligomerization, endothelial cells were cultured in a serum-free medium for 3 to 4 h and treated with adiposome with and without L-NAME or PP2 for indicated times. Samples were heated at 60 °C in a loading buffer plus DTT. Finally, samples were gel electrophoresed for Western blot analysis.
2.8. Confocal Microscopy
For adiposome uptake assay, adiposomes were labeled with BODIPY™ 493/503 (4,4-Difluoro-1,3,5,7,8-Pentamethyl-4-Bora-3a,4a-Diaza-s-Indacene) (ThermoFisher Scientific, Waltham, MA, USA) using the manufacturer protocol. Endothelial cells were cultured in glass-bottom 35 mm dishes and treated with adiposomes for 30 to 60 min. Treated cells were washed and fixed with 4% paraformaldehyde for 15 min at room temperature, followed by washing, permeabilization with 0.01% tween 20, and blocking non-specific binding with 1% bovine serum albumin (BSA) for 1 h. Cells were then incubated with the primary monoclonal antibodies, VE-cadherin or total cav-1 (Cell Signaling, Danvers, MA, USA) diluted in 1% BSA blocking buffer for 1 h at room temperature. After washing, cells were incubated with fluorophore-conjugated secondary antibodies for 1 h at room temperature. Finally, 30 μL of VECTASHIELD® Antifade Mounting Medium (Vector Laboratories, Burlingame, CA, USA) was added. The cells were enclosed by a coverslip and captured using a 63 × 1.2 N.A. objective on a Zeiss LSM 510 META microscope (Carl Zeiss MicroImaging, Oberkochen, Germany). The imaging process involved utilizing a 488-excitation laser line and adjusting the pinhole to reach 1 Airy unit.
2.9. Electron Microscopy
For adiposome visualization, adiposomes were fixed with 2% Paraformaldehyde for 5 min and loaded onto thin formvar/carbon film coated 200 mesh copper EM grids and immediately stained with filtered 1% uranyl acetate. Excess stain was removed, and the grid was rinsed with water, dried for 10 min at room temperature, and examined by JEM-3010 transmission electron microscope. For caveolae visualization, treated endothelial cells were detached, washed, pelleted, and then fixed in 2.5% glutaraldehyde (Sigma Aldrich, St. Louis, MO, USA) for 1 h at room temperature. Cell pellets were washed with 1X phosphate buffer saline (PBS) for 5 min, then fixed with 1% citric acid for 1 h. Following fixation, cell pellets were dehydrated in gradient ethanol (50%, 70%, 90%, and 100%) and then stained with OsO4. For penetration and polymerization, cell pellets were embedded in Epon® Resin 828 (Sigma Aldrich). Ultrathin sections of 70–80 nm in thickness were obtained from the cell pellets using the Leica EM UC7 ultra-microtome, manufactured by Leica Microsystems Inc. in Buffalo Grove, IL, USA. These sections were then placed on a 100 mesh grid and subjected to a double staining procedure involving immersion in a 4% uranyl acetate solution for 3 min, followed by immersion in a 2% lead citrate solution for 20 min. The slices were examined using a JEM-3010 transmission electron microscope (JEOL USA, Inc., Peabody, MA, USA).
2.10. Adiposome Uptake and Lipid Fusion Assays
Adiposomes were fluorescently labeled using BODIPY TR ceramide (Life Technologies, Carlsbad, CA, USA), a red fluorescent stain with absorption/emission wavelengths of 589 and 617 nm, respectively. Briefly, 1 mmol/L stock solution of the BODIPY stain was prepared, and one μL was added to every 100 μL of adiposome samples to reach a final concentration of 10 μmol/L. Adiposomes were incubated with the BODIPY stain at 37 °C for 20 min, followed by filtration through Exosome Spin Columns (MW 3000, Life Technologies) to remove excess unbound stain. For adiposome uptake assays, HAMECs were incubated for 30 min with fluorescently labeled adiposome particles (8 × 10
9). Cells were washed, fixed, labeled with VE-cadherin to define cell boundaries, and imaged using a Zeiss LSM 710 confocal microscope. For lipid fusion assays, adiposomes were labeled with octadecyl rhodamine B chloride (R18, ThermoFisher Scientific, Waltham, MA, USA) (20 μM, 1 h, room temperature) following previously published protocols for labeling extracellular vesicles [
35]. To remove the unbound stain, labeled adiposomes were purified by gel filtration (Sephadex G-75). Ten micrograms of labeled adiposomes were then incubated with unlabeled cultured endothelial cells that were serum-starved for 2–3 h. The fusion of labeled adiposomes and unlabeled membranes is expected to dilute the probe and subsequently increase fluorescence. Therefore, 10–15 min after adiposome incubation with endothelial cells, cells were imaged using a fluorescence microscope (Carl Zeiss MicroImaging, Oberkochen, Germany) with an excitation/emission wavelength of 560/590 nm. Images were analyzed using the ImageJ Particle Analysis module.
2.11. Nitric Oxide and Peroxynitrite Measurements
Nitric oxide (NO) generation was assessed by measuring the levels of nitrates and nitrites (stable metabolites of NO) in the cell culture media. Cell culture media from treated cells were collected and centrifuged to remove cell debris and detached cells. Nitrates and nitrites in the cell culture media were measured using the Griess assay kit (Cayman Chemicals, Ann Arbor, MI, USA) [
36]. Briefly, the assay begins by converting all nitrates into nitrites using the supplied nitrate reductase enzyme, followed by the sequential addition of Griess reagents I and II, which convert nitrites into a dark purple azo compound. The developed color intensity was then measured at 540 nm, as recommended by the manufacturer, using a microplate reader. Nitrite concentration calculations were performed using the generated nitrate standard curve. For peroxynitrite measurements, Peroxynitrite Assay (Abcam) was used. Cultured cells were pre-stained with a peroxynitrite sensor green (10×) at 37 °C for 60 min protected from light, followed by incubation with adiposomes with and without PP2 or Tetrahydrobiopterin (BH4) for the recommended periods. The fluorescence intensity was quantified using a microplate reader, with excitation and emission wavelengths set at 490 nm and 530 nm, respectively.
2.12. Shear Stress Experiments
Endothelial cells were cultured in fibronectin-coated ibidi 4 chamber μ-slide (ibidi USA, Inc., Fitchburg, WI, USA) at a density of 10
5 cells/well. Cells were incubated for 8 to 10 h to allow them to adhere to the bottom of the slide. The chamber slides were subsequently interconnected sequentially within a flow configuration consisting of a peristaltic pump, pulse dampener, and requisite silicone tubing and fittings, adhering to the comprehensive procedure outlined by Lane et al. [
37] in their published protocol. The cells were initially exposed to the parallel flow condition by passing media through them at a flow rate determined to provide an average shear stress of 5 dynes/cm
2, as calculated using the following formula:
where Q represents the desired flow rate, τ represents the target shear stress acting tangentially on the cells, w represents the width of the flow chamber, h represents the height of the flow chamber, and μ represents the viscosity of the perfusate (cell culture medium). The viscosity (μ) of the medium typically employed is 0.9 centipoise (cP), which is equivalent to 0.009 g per cubic centimeter per second (g cm
−1 s
−1). A commonly employed range for the goal shear stress (τ) is 15–20 dynes/cm
2, which is representative of the average arterial shear stress. The dimensions of our chamber are conventionally characterized by a width (w) of 46 mm and a height (h) ranging from 2 to 2.5 mm. The aforementioned procedure was carried out until cellular alignment was observed in the direction of fluid flow, which typically occurred within a timeframe of 4 to 6 h. After the conditioning process, the endothelial cells were exposed to shear stress at an average magnitude of 15 to 20 dynes/cm
2 for a duration of 2 to 4 h. The flowing media contains the designated treatment (adiposomes or GM3 with and without PP2). Cells were imaged via Bright-field microscopy then they were collected for RNA isolation and analysis of gene expression.
2.13. Fluorescently Labeled Albumin Uptake in Endothelial Cells
For this experiment, endothelial cells were cultured on fibronectin-coated glass-bottom 35 mm dishes. Cells that were preconditioned with the designated treatments were incubated with 10 μg/mL Texas Red conjugated bovine serum albumin (BSA) in basal media containing 0.1 mg/mL unlabeled BSA. The cells underwent a process of acid washing using a buffer with a pH of 2.5, followed by a wash with Hank’s balanced salt solution (HBSS) to eliminate any surface-bound BSA. Subsequently, the cells were fixed using 4% paraformaldehyde and stained with cav-1 and DAPI. Finally, the cells were photographed using the procedures outlined in
Section 2.8.
2.14. Measurements of Microvascular Flow-Induced Dilation and NO and ROS Production
Samples of AT were dissected in order to isolate tiny arterioles. These arterioles were subsequently cleared of any excess connective tissue and fat. As we previously reported, the internal diameter of dissected vessels was assessed in relation to a progressively escalating pressure gradient. The vessels were cannulated using glass microcapillaries that were placed into an organ perfusion chamber. The microcapillaries were secured by tying the vascular ends using nylon Ethilon monofilament. Subsequently, the organ chamber was relocated to an inverted microscope that was linked to video microscopy to observe and assess the vasoreactivity of arterioles. The terminals of the cannulated arterioles were linked to two reservoirs that contained Krebs buffer. The manipulation of the reservoirs in a symmetrical manner, involving upward and downward movements, resulted in the generation of an intraluminal pressure gradient ranging from 10 to 100 cm of water (cm H
2O). The Krebs buffer solution was supplemented with a combination of oxygen (21%), carbon dioxide (5%), and nitrogen (74%), while its pH and temperature were upheld at 7.4 and 37 °C, respectively. After conducting baseline measurements, the cannulated vessels were subjected to constriction using endothelin (ET-1) at a concentration of 10
−6 mol/L. Only vessels that exhibited a constriction of more than 30% were included in further measurements. The measurement of arteriolar vasoreactivity was conducted under two conditions: with the presence and absence of the endothelial nitric oxide synthase inhibitor, L-NG-Nitro arginine methyl ester (L-NAME, 10
−4 mol/L). The calculation of vasodilation % involved the normalization of arteriolar diameter at each pressure gradient to the diameter observed after the administration of ET-1. For the Ad-cav-1 transfection experiments, a custom-designed adenoviral vector expressing cav-1 with VE-Cadherin endothelial cell-specific promoter was constructed in Vector BioLabs (Malvern, PA, USA). Isolated arterioles were intramurally transfected with the cav-1 vector (100 MOI) or an empty vector for 8 h at 37 °C following previously published protocols [
38,
39]. cav-1 overexpression in arterioles transfected with Ad-cav-1 versus an empty vector was verified via Western blotting techniques. The generated NO and ROS by isolated arterioles were measured as we previously described [
36]. using the NO Detection Kit (Enzo Life Sciences, Farmingdale, NY, USA) and the ROS indicator (2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA). In cannulated arterioles, measurements were made in response to a pressure gradient of about 60 cm H
2O. Arterioles underflow were excised, cleaned, and mounted on microscope slides after being treated with the NO and ROS detection reagents. At 650 and 495 nm, respectively, images were captured using a fluorescent microscope (Eclipse TE 2000, Nikon, Japan). Every experiment followed the same incubation, staining, and imaging procedures. Using Image J software version 1.8.0 (NIH, Bethesda, MD, USA), images were then examined for fluorescence intensity. The fluorescent signal was then represented in arbitrary units.
2.15. Statistical Analysis
The obtained data were representative of triplicates from three different experiments and were expressed as the average value ± the standard error. The data were subjected to statistical analysis using the Student’s t-test or one-way ANOVA, followed by a post-hoc analysis using Bonferroni’s method where deemed suitable. Statistical significance was determined by considering results with a p-value less than 0.05.
4. Discussion
The primary findings of the present study indicate that adiposomes derived from individuals with obesity and type 2 diabetes (OB-T2D) contain higher levels of glycosphingolipids (GSLs) compared with those from lean, healthy controls. These GSLs are believed to contribute, at least in part, to the detrimental effects of OB-T2D adiposomes on endothelial cells. Furthermore, when OB-T2D adiposomes were introduced to endothelial cells, they induced detachment of cell surface caveolae through the activation of Src kinase and phosphorylation of cav-1. This effect had a negative impact on endothelial nitric oxide synthase (eNOS) function, resulting in eNOS uncoupling, reduced nitric oxide (NO) production, and increased generation of peroxynitrites. These molecular changes were also reflected in the impaired functionality of endothelial cells, characterized by a loss of response to shear stress and reduced intake of albumin. Finally, the adiposomes of OB-T2D were found to have a detrimental effect on flow-induced dilation (FID) and nitric oxide (NO) production in healthy arterioles.
The endocrine roles of AT have primarily been ascribed to adipokines, a diverse group of soluble bioactive molecules that are released from adipocytes, including but not limited to leptin and adiponectin [
47]. Relatively recently, adipocyte-derived extracellular vesicles (adiposomes) have been discovered [
48]. These vesicles include a wide range of lipids, proteins, and genetic material, suggesting an additional mechanism for endocrine signaling originating from AT. Dysfunctional adipocytes with dysregulated metabolism in obese and diabetic AT may release adiposomes with an altered cargo of lipids and other biological content. This content may promote vascular dysfunction upon interaction with endothelial cells. Therefore, comprehensive studies that explore the potential role of adiposomes in predisposing obese, diabetic individuals to cardiovascular diseases (CVD) are required.
The association between dysregulated lipid metabolism in AT and cardiovascular risk has been established [
49]. However, the mechanism by which the dysregulated lipid milieu is transported to vascular tissues has primarily been attributed to soluble factors, neglecting the potential role of adiposomes. A significant portion of secretions originating from AT are encapsulated within adiposomes, and prior research has indicated that this method of packaging helps protect the cargo and improves its chance of reaching distant cells and tissues [
50]. However, due to technical difficulties in isolating and characterizing human adiposomes, our understanding of their mechanistic contribution to CVD remains limited to findings from few in vitro studies [
51].
It is worth mentioning that one-third of the extracellular vesicle (EV) volume is lipids [
52]; however, unlike the protein and nucleic acid cargo, the lipid content in EVs has been far less explored, and its implication in vascular function has not been investigated. In the current study, we analyzed the differences in the lipid content of adiposomes between OB-T2D patients and lean, healthy controls (LHCs). Our findings demonstrated significantly higher levels of GSLs in the OB-T2D adiposomes, on top of which were lactosylceramide and GM3. GSLs are generated via the glycosylation of ceramide. The latter is a bioactive sphingolipid, and its generation in inflamed tissues is mainly mediated by sphingomyelinase (SMase) [
53], which increases in chronic inflammatory illnesses such as obesity, diabetes, and CVD [
14,
15]. Glycosylation of ceramides is upregulated in diabetic patients and is driven by the local milieu of high glucose and inflammation-mediated induction of enzyme activity [
12,
13]. The attachment of glucose to ceramide via glucosylceramide synthase (GCS) yields glucosylceramide (GluCer), which can be converted to lactosylceramide (LacCer) by the enzyme glucosylceramide β4-galactosyltransferase (B4GalT). From LacCer, more complex GSLs, such as globosides and gangliosides, can be synthesized via different sialyltransferases and galactosyltransferases [
40].
GSLs serve as secondary messengers in various signaling pathways and are implicated in the pathological consequences of T2D, such as retinopathy, neuropathy, atherosclerosis, and coronary artery diseases [
16,
17,
18]. However, the critical barrier to progress in this area is that the precise mechanism that mediates GSL impact on vascular function is poorly understood. These GSLs are glycosylated lipids that were shown to be induced under conditions of hyperglycemia and inflammation. So, indeed, adiposomes reflect the internal environment and metabolic status of dysfunctional AT, which lends significance to the investigation of the biological messages that are carried and transferred by adiposomes to other cells, such as endothelial cells.
In order to gain insight into the impact of adiposomes on endothelial cells, we conducted the current investigation to assess the structural and functional consequences observed in endothelial cells following treatment with adiposomes. Compared with healthy adiposomes, GSL-rich adiposomes from OB-T2D patients were able to more efficiently incorporate GSLs such as lacCer and GM3 into the endothelial cell membrane. In addition, these GSL-rich adiposomes tended to accumulate in caveolae-rich regions, which led to their detachment from the cell surface or diminution in size. Previous studies have shown that GSLs such as LacCer are internalized by a caveolae-associated mechanism [
54]. This finding may explain the observed propensity of GSLs to accumulate in locations abundant in caveolae.
Even though caveolae are typically associated with lipids on the cell surface, they undergo short-range cycles of fission and fusion, and their biogenesis and stability on the cell surface are dependent on the tightly regulated balance of their lipid composition. Hubert et al. [
20] demonstrated that synthetic GSL-rich fusogenic liposomes reduced the diameter of caveolar necks in HeLa cells, leading to caveolar fission and confirming the fact that caveolar stability is highly sensitive to changes in the plasma membrane lipid composition. However, no comparable research on endothelial cells has been conducted. Consequently, we believe that our study is one of the first to examine the effect of GSL-rich adiposomes isolated from OB-T2D patients on the lipid content of endothelial membranes and caveolar fission. The lack of effect of GSL-depleted adiposomes further supported the role of GSLs in inducing these effects.
Inhibition of glucosylceramide synthase (GCS) in LHC adiposomes did not have the same significant impact on GSL levels as in OB-T2D adiposomes. We believe this is due to the low baseline levels of GSLs in LHCs. Previous studies have documented the increased activity of the GCS enzyme under conditions of inflammation and hyperglycemia observed in obese diabetic patients, lending support to this assumption [
12,
55]. It can be deduced that the outcome of GCS inhibition is dependent on baseline levels of GSLs and GCS activity, a trait also observed in lipid-lowering drugs that do not substantially lower lipid levels in individuals with normal lipid profiles.
Cav-1 is an essential structural element of caveolae, and its phosphorylation by Src kinase is correlated with an increase in caveolae-mediated fission from the cell surface. We previously showed that cav-1 phosphorylation at tyr-14 induces the separation of adjacent negatively charged N-terminal phosphotyrosine residues. This event facilitated the swelling of caveolae structures, ultimately resulting in their detachment from the plasma membrane [
56]. The present investigation revealed that adiposomes derived from OB-T2D individuals elicited a more pronounced activation of Src kinase and cav-1 phosphorylation compared with adiposomes obtained from healthy control subjects. A significant reduction in cav-1 phosphorylation and caveolar detachment was observed when Src kinase activity was suppressed in endothelial cells. These findings imply that the influence of adiposomes on cav-1 and caveolae is facilitated by the activation of Src kinase.
Caveolae are essential structures in endothelial cells, as they serve to concentrate a diverse array of transmembrane signaling molecules (signalosomes) that play a regulatory role in endothelial cell functions [
21]. Caveolae serve as a scaffold for the inactive form of eNOS, ensuring the maintenance of well-regulated cycles of eNOS activation and inhibition and subsequently, proper vasoreactivity. Caveola-eNOS interactions are governed by stimuli such as shear stress, which results in eNOS dissociation from caveolae and subsequent phosphorylation and activation [
57]. The loss or reduction in surface caveolae is anticipated to disrupt these tightly regulated cycles. In the present study, the adiposome-induced fission of caveolae from the endothelial cell surface was associated with an increase in eNOS phosphorylation. However, the induction of eNOS phosphorylation was not accompanied by an increase in nitric oxide production. Instead, peroxynitrate production was stimulated, indicating an increase in superoxides and other reactive oxygen species. Indeed, the data presented in this study clearly indicate notable elevations in NADPH oxidase (Nox2) activity and peroxynitrite generation, which were observed concurrently with the stimulation of eNOS phosphorylation. Moreover, eNOS monomerization increased in response to GSL-rich adiposomes from OB-T2D patients.
These observations collectively refer to a unique phenomenon known as eNOS uncoupling, in which eNOS produces highly reactive superoxide, which, in the presence of little NO, induces the production of the more reactive molecule, peroxynitrite [
58]. However, it is unclear whether eNOS uncoupling is a consequence or cause of induced oxidative stress and where the process begins. Previous research uncovered discrepancies between eNOS phosphorylation and uncoupling, leading researchers to conclude that eNOS phosphorylation at ser1177 is not always accompanied by NO production [
59]. According to our research, the induction of eNOS uncoupling by OB-T2D adiposomes was found to be disrupted by inhibitors of Src kinase, as well as by external replenishment with BH
4. The latter is a necessary co-factor that facilitates eNOS activity and NO production from L-arginine. Oxidation of BH
4 results in transmutation to BH
2, which disturbs the normal electron flow and promotes superoxide formation, resulting in a vicious circle of induced oxidative stress [
60].
The overproduction of reactive oxygen species (ROS), particularly peroxynitrites, plays a significant role in the promotion of S-nitrosylation of cellular proteins [
61]. In the present investigation, we detected a notable augmentation in the S-nitrosylation levels of cav-1. Given that the assembly of cav-1 protein into caveolae necessitates oligomerization, our objective was to examine the oligomer-to-monomer ratio of this protein [
62]. Our data demonstrated a decrease in this ratio, which is anticipated to be a consequential result of cav-1 S-nitrosylation. This notion is confirmed by prior research from our group and others indicating impairments in the capacity of cav-1 to oligomerize when it undergoes nitrosylation [
63,
64]. It is anticipated that these effects will impede the process of caveolar construction and exacerbate the deficiency of caveolae on the cell surface.
In addition to eNOS signaling, caveolae contain a variety of mechanosensitive molecules, including mechanosensitive ion channels, G-protein coupled receptors (GPCRS), extracellular signal-regulated kinases (ERK), receptor tyrosine kinases (RTK), and others [
65,
66,
67]. These proteins convert shear stress into cell signaling through multiple mechanisms, including the induced expression of the transcription factors KLF2 and KLF4. These factors transcribe genes that modify endothelial cell morphology and physiology in response to shear stress [
68]. In this investigation, the shear response of endothelial cells treated with OB-T2D adiposomes was found to be impaired. These impairments were observed at the morphological and molecular levels, as indicated by the lack of cell streaming and the absence of induced mRNA expression of the KLF2 and KLF4 transcription factors, respectively. These effects were not observed in response to GSL-depleted OB-T2D adiposomes and were eliminated by the Src kinase inhibitor, PP2. However, more in-depth research on the effect of adiposomes on shear stress-related signaling pathways is required.
In order to investigate additional functional implications of caveolar loss generated by obesity-related type 2 diabetes (OB-T2D), we analyzed albumin uptake. A substantial body of research supports the crucial role of endothelial cell caveolae in the effective uptake of albumin [
69]. In fact, the current study demonstrated that GSL-rich adiposomes from OB-T2D patients decreased endothelial albumin uptake, corroborating the impairment of caveola-mediated endocytosis. The latter refers to a mechanism by which endothelial cells facilitate the absorption of several molecules, including insulin. Consequently, the disruption of this process is anticipated to result in many adverse outcomes. In addition, several drugs, including the chemotherapeutic agent paclitaxel, are administered in albumin-bound form [
70]. Consequently, the inability of endothelial cells to absorb albumin will result in decreased bioavailability and bioefficacy of these medications. These findings have clinical relevance for the current investigation and may help explain the treatment resistance observed in obese diabetic patients in response to certain medications. Moreover, these findings pave the way for a new field of research in which adiposomes and their cargo could be examined as a potential cause of therapeutic resistance in certain diseases such as obesity and diabetes.
The observed alterations in the Src/cav-1/eNOS axis in cultured endothelial cells were confirmed in AT-isolated human endothelial cells. Compared with healthy controls, endothelial cells from OB-T2D patients demonstrated elevated levels of phosphorylated Src kinase, cav-1, and eNOS. In addition, the cav-1 protein in the endothelial cells of OB-T2D patients was significantly more S-nitrosylated and had a reduced oligomer-to-monomer ratio. Furthermore, targeted lipidomics analysis revealed increased levels of LacCer and GM3 species in OB-T2D patients’ endothelial cells compared with healthy controls. Notably, GM3 and LacCer exhibit distinct subspecies that differ with respect to their sphingosine base structure, fatty acid chain length, double bonds, and other related characteristics. Consequently, there is typically substantial variation among individuals, which explains the lack of an exact match between the LacCer and GM3 subspecies differentially detected in human endothelial cells and those observed in in vitro-cultured human endothelial cells. In conclusion, these findings provide support for the credibility of our in vitro experiments and suggest that the observed ex vivo impact of adiposomes on cultured endothelial cells closely mirrors their actions within the human body.
Arteriolar flow-induced dilation (FID) is a technique utilized by our group and others to test microvascular vasoreactivity in human arterioles promptly after their removal from the body. These arterioles are expected to be directly affected by the molecular changes in the AT. Also, these small arterioles are the primary site of peripheral resistance and blood pressure regulation, making these studies clinically relevant and highly impactful. Moreover, our team and others have refined the methodologies for manipulating gene expression in these arterioles, making them well-suited for conducting mechanistic studies [
38]. In our previous studies, we demonstrated an impaired FID of AT arterioles isolated from obese or OB-T2D individuals compared with healthy counterparts [
36]. In the current investigation, we observed FID impairments in healthy arterioles incubated with OB-T2D adiposomes, an effect not observed in response to GSL-depleted adiposomes and partially suppressed by cav-1 transfection (overexpression). To the best of our knowledge, this is the first study to test the effect of human-isolated adiposomes on arteriolar FID. These findings are novel, and we believe they pave the way for further mechanistic studies of the effect of adiposomes on arteriolar reactivity. Furthermore, these findings can point to adiposomal cargo as a potential mechanistic link between metabolic disorders and vascular dysfunction.

 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}