
Development of New Genome Editing Tools for the Treatment of Hyperlipidemia
1
VU LSC-EMBL Partnership Institute for Genome Editing Technologies, Life Sciences Center, Vilnius University, LT-10257 Vilnius, Lithuania
2
Institute of Biochemistry, Life Science Center, Vilnius University, LT-10257 Vilnius, Lithuania
Cells 2023, 12(20), 2466; https://doi.org/10.3390/cells12202466
Submission received: 12 September 2023
/
Revised: 10 October 2023
/
Accepted: 12 October 2023
/
Published: 16 October 2023
(This article belongs to the Section Cell and Gene Therapy)
Abstract
:Hyperlipidemia is a medical condition characterized by high levels of lipids in the blood. It is often associated with an increased risk of cardiovascular diseases such as heart attacks and strokes. Traditional treatment approaches for hyperlipidemia involve lifestyle modifications, dietary changes, and the use of medications like statins. Recent advancements in genome editing technologies, including CRISPR-Cas9, have opened up new possibilities for the treatment of this condition. This review provides a general overview of the main target genes involved in lipid metabolism and highlights the progress made during recent years towards the development of new treatments for dyslipidemia.
1. Introduction
Genome editing refers to the modification of an organism’s DNA to alter its genetic information. One of the most promising genome editing tools is CRISPR-Cas9, which stands for clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated Protein 9 (Cas-9). CRISPR-Cas9 allows scientists to make precise changes in the DNA sequence by targeting specific genes and introducing modifications [1,2]. In the context of hypercholesterolemia, the main goal is to target genes involved in cholesterol metabolism to reduce the levels of low-density lipoprotein (LDL) cholesterol in the blood. Since the liver plays a critical role in the production and clearance of lipoprotein particles, genome editing strategies are optimized to target genes within hepatocytes. For instance, adeno-associated virus (AAV) vectors based on serotype 8 have a specific tropism for the liver and have been used in several studies for somatic genome editing in mice [3,4]. Lipid nanoparticles (LNP) are also delivery vehicles for CRISPR-Cas9 editing and are efficiently taken up by hepatocytes due to their ability to interact with serum proteins [5].
Researchers have been exploring the use of genome editing to develop novel therapies as an alternative to the existing ones including statins, ezetimibe, PCSK9 (proprotein convertase subtilisin/kexin type 9) inhibitors, niacin, bile acid sequestrants, fibrates and bempedoic acid [6,7,8,9]. Statins were first introduced for the treatment of high cholesterol levels in the late 1980s. The first statin to be approved for clinical use was lovastatin (Mevacor) in 1987. Following the approval of lovastatin, other statins were subsequently developed and introduced for the treatment of hypercholesterolemia. Some of the commonly prescribed statins include simvastatin (Zocor), atorvastatin (Lipitor), pravastatin (Pravachol), and rosuvastatin (Crestor). While statins are considered safe and effective, there are different observed side effects associated with their use, including muscle pain and weakness, gastrointestinal symptoms, and liver enzyme abnormalities, which led to the development of alternative or complementary therapies [10,11]. Ezetimibe is often considered a valid option for individuals who cannot tolerate statins or require additional LDL reduction [12]. This medication acts by reducing the absorption of cholesterol from the intestine, selectively inhibiting the protein Niemann-Pick C1-Like 1 (NPC1L1), which is responsible for transporting cholesterol from the intestine into the bloodstream [13]. In clinical studies, when used as a monotherapy, ezetimibe was able to reduce LDL cholesterol by 18% [14], while when used in combination with statins, it provides a variable reduction according to the statin used, sample size, and dosage [15,16].
PCSK9 inhibitors (evolocumab and alirocumab) are monoclonal antibodies that specifically target PCSK9 [17,18]. When injected, these antibodies bind to circulating PCSK9 molecules, preventing them from interacting with LDL receptors (LDLRs). By blocking the interaction between PCSK9 and LDLRs, PCSK9 inhibitors hamper the internalization of LDLRs, allowing them to remain on the surface of cells and increasing the liver’s capacity to capture LDL particles from the bloodstream [19,20]. However, there are still several obstacles to the clinical use of PCSK9 inhibitors including the appearance of several side effects and the cost of its clinical use in relation to its effectiveness [21]. The use of small interfering RNA (siRNA) represents another strategy to inhibit the internalization of LDLRs. Inclisiran specifically targets the 3` UTR of PCSK9 mRNA, entering hepatocytes through asialoglycoprotein receptors and leading to an increased expression of LDLR receptors in the membranes [22]. The results on its efficacy are based mainly on three clinical trials named ORION-9, ORION-10, and ORION-11 showing a decrease in LDL-C, lipoprotein(a) [Lp(a)], and triglycerides (TG). All ORION clinical trials are reviewed by Katsiki and colleagues [23]. The long-term safety and efficacy of inclisiran will be evaluated in the ongoing trial ORION-4, which will determine the clinical relevance of this promising new treatment [8]. Evinacumab is another monoclonal antibody that pharmacologically inhibits angiopoietin-like 3 (ANGPTL3) and was recently approved by the FDA as a complementary agent to other LDL-C-lowering therapies for patients with homozygous familial hypercholesterolemia (HoFH). The binding of evinacumab to ANGPTL3 preserves the function of lipoproteins and endothelial lipase, leading to a decrease in total cholesterol (TC), TG, and LDL-C [24]. Similar results were observed with the apolipoprotein C3 (ApoC3) inhibitor olezarsen, a hepatocyte-targeted, GalNAc-modified antisense oligonucleotide that decreases the plasma levels of ApoC3 and consequently reduces triglycerides levels in subjects with high cardiovascular risk [25,26]. Pelacarsen is an antisense oligonucleotide covalently bonded to GalNAc, which prevents the production of apoliporotein(a) [Apo(a)]. Apo(a) is encoded by the LPA gene and should not be confused with members of the apolipoprotein A family, encoded by different genes (i.e., APOA1, APOA2). The binding of Apo(a) to ApoB100 on LDL leads to the formation of Lp(a). Phase 1 and phase 2 pelacarsen clinical trials showed a considerable decrease in the serum level of Lp(a) [27,28]. Olpasiran is a siRNA that blocks the assembly of Lp(a) by inhibiting the translation of Apo(a) in the hepatocytes. Several clinical trials proved its efficiency and safety, promoting the development of additional siRNA in order to reduce Lp(a) blood levels [29,30,31].
Niacin was considered a powerful drug for the treatment of lipid abnormalities, acting by decreasing fatty acid mobilization from adipose tissue and by inhibiting triglyceride synthesis [32]. However, two large randomized clinical studies have recently shown disappointing results, leading to the conclusion that there are no effective benefits to adding niacin to existing statins therapy for patients with high cardiovascular risk [33,34]. Limitations in the design of these two clinical trials as well as the possibilities for usage of niacin for specific types of dyslipidemias are described in a study by Zeman and colleagues [35]. Bile acid sequestrants such as cholestyramine, cholestipol, or colesevelam, due to their high level of charged molecules, bind to negatively charged bile acids in the intestine, inhibiting cholesterol absorption. A growing amount of evidence suggests that they play a role not only in lipid but also in glucose metabolism [36,37]. Bile acid sequestrants can be used as monotherapy or in combination with statins or ezetimibe. Moreover, since they are not absorbed in the gastrointestinal tract, they have limited toxicity [38].
Treatment with fibrates results in a substantial decrease in plasma triglycerides and is also associated with a slight reduction in LDL cholesterol [39,40]. The effects of fibrates are related to alterations in the transcription of genes encoding for proteins that control lipoprotein metabolism. More specifically, the primary target of fibrates is the peroxisome-proliferator-activated receptor alpha (PPAR-alpha). The binding of fibrates to PPAR-alpha receptors induces their activation and the formation of a complex between PPAR-alpha and the retinoid X receptor (RXR). This PPAR-alpha/RXR complex binds to specific DNA sequences known as peroxisome proliferator response elements (PPREs) in the promoter regions of target genes, leading to the upregulation of genes involved in lipid metabolism, especially those responsible for fatty acid oxidation in the liver and muscles [41,42]. Bempedoic acid is a novel LDL-cholesterol-lowering agent, inhibiting adenosine triphosphate citrate lyase, an enzyme involved in the cholesterol synthesis pathway, upstream from the HMG-CoA reductase [12]. Several clinical trials have demonstrated the efficiency of this drug when used as a monotherapy or in combination with other lipid-lowering therapies [43,44,45]. In Figure 1, a schematic outline of the above-described drugs’ mechanisms for lowering serum lipids is presented.
2. PCSK9 Gene Editing Strategies in Mice
The PCSK9 gene plays a role in regulating cholesterol levels by controlling the number of LDL receptors on the surface of liver cells. To become fully functional, PCSK9 protein undergoes a maturation process involving several steps: PCSK9 is synthesized in the endoplasmic reticulum (ER) in the form of inactive zymogen, called PreProPCSK9. PreProPCSK9 is composed of five parts: a signal peptide, the pro-domain, the catalytic domain, the hinge region, and the C-terminal domain. The protein is then subjected to autocatalytic cleavage in the ER in order to lose its signal peptide, and it becomes ProPCSK9. ProPCSK9 is then transported to the trans-Golgi network (TGN), where it undergoes proteolysis to form mature PCSK9 [46,47]. Only the mature form is then transported in endosomes and secreted into the circulation, where it binds to the LDL receptor on the surface of hepatocytes. The PCSK9-LDLR complex is internalized into the cell via endocytosis. Inside the cell, the LDLR is targeted for lysosomal degradation instead of being recycled to the cell surface for further use. In this way, with fewer functional LDL receptors available on the cell surface, the liver becomes less efficient at clearing LDL cholesterol from the bloodstream [48]. This leads to higher levels of LDL cholesterol in the blood and to the potential risk of cardiovascular diseases. Autosomal dominant variants of the PCSK9 gene can lead to a condition called familial hypercholesterolemia (FH), which is characterized by high levels of LDL cholesterol and an increased risk of cardiovascular disease [49,50]. PCSK9 genome editing strategies aim at loss-of-functions mutations that are always associated with reduced plasma levels of LDL-C in nature [51,52,53,54]. Researchers have used CRISPR-Cas9 to disrupt or modify the PCSK9 gene, effectively reducing the production of PCSK9 protein and the levels of LDL cholesterol [55,56]. In the first in vivo study in mice, conducted by Ding and colleagues, the authors selected a gRNA-targeting exon 1 of mouse PCSK9 and generated an adenovirus expressing this gRNA and Cas9. As soon as 3 to 4 days after injection of the adenovirus, P mutagenesis had occurred in more than half the mice, resulting in decreased PCSK9 levels and reductions in TC of up to 40%. No off-target effects were observed [57]. A few years later, in a mouse model with humanized hepatocytes, adenovirus was used again as a vector to deliver gRNA-targeting exon 1 of PCSK9. After a few days, almost 50% mutagenesis was observed, with a reduction in human levels of PCSK9 of 52%. Interestingly, mouse PCSK9 protein levels increased, probably as a compensatory mechanism, and no effect on TC was observed [58]. The limitation of these two studies was related to the use of adenovirus as a vector, since their long persistence and immunogenicity in the host prevent the potential therapeutic applications in humans [59,60]. AAV-mediated delivery of the CRISPR-Cas9 system has shown high gene targeting efficacy in vivo and a lower immunogenicity emerging as an alternative delivery method for the CRISPR-Cas9 system to various cell types, tissues, and organs [61,62]. Ran and colleagues used an adeno-associated virus and a Cas9 orthologue from Staphylococcus aureus (for its smaller size) instead of the Streptococcus pyogenes Cas9. The mutagenesis observed in PCSK9 gene was greater than 40%, the reduction in PCSK9 levels reached 95%, and the reduction in cholesterol levels was 40% [63]. Since long-term expression of Cas9 in target cells creates concerns related to toxicity and appearance of off-target effects, a self-cleaving AAV-CRISPR-Cas9 system was developed. This system can effectively eliminate Cas9 protein expression without compromising the editing efficacy of the PCSK9 gene and reducing the off-target effects [64]. A completely different approach was used in a 2017 study, where newly developed lipid-like nanoparticles (LLNs) were used successfully to deliver Cas9 RNA- and gRNA-targeting PCSK9 in the livers of mice [65,66]. The targeting was effective for both episomal and chromosomal DNA, and since Cas9 mRna/protein and gRNA were degraded within one day in mice, this methodology provides a temporarily controllable way to achieve in vivo genome editing. A strategy termed selective organ targeting (SORT) was developed to allow lipid nanoparticles to be engineered for precise delivery of different cargoes including mRNA, Cas9 mRNA/single-guide RNA (sgRNA), and Cas9 ribonucleoprotein (RNP). When this methodology was used for PCSK9 targeting, a significant indel induction at the PCSK9 locus (~60%), corresponding to a ~100% reduction in PCSK9 levels in the liver and blood, was observed [67]. Lipid fats were also used as a delivery method in a study where chemically modified gRNA was developed. These modifications did not inhibit the interaction between gRNA and Cas9, while maintaining or even enhancing the genome editing activity, leading to a >80% editing of in the liver with a single injection [68].
To address the lack of effective models to test the efficacy of techniques to target human PCSK9, Carreras and colleagues developed a liver-specific human PCSK9 knockin mouse model (hPCSK9-KI) [69]. Human PCSK9 was expressed in the liver from hPCSK9-KI but not from their wild-type littermates, whereas the expression of endogenous mouse PCSK9 mRNA was comparable in the liver between the two types of mice. In this model, CRISPR-Cas9-mediated genome editing of human PCSK9 decreased plasma levels of human but not mouse PCSK9, and in parallel, it reduced the plasma concentrations of cholesterol, while genome editing of mouse PCSK9 did not affect cholesterol levels. Base editing using a guide RNA that targeted both human and mouse PCSK9 reduced the plasma levels of human and mouse PCSK9 and cholesterol levels. Therefore, this model can be used for the evaluation of genome-/base-editing therapies to regulate the expression of PCSK9 and consequently the blood cholesterol levels.
3. PCSK9 Gene Editing Strategies in Non-Human Primates
The first in vivo gene editing study in non-human primates (NHPs) for PCSK9 was carried out using an AAV vector expressing an engineered meganuclease targeting PCSK9 [70]. Meganucleases have been more challenging to engineer for new target sequences, making them less versatile than CRISPR-Cas9; however, advances in protein engineering have improved their flexibility [71,72]. The meganucleases were used in Rhesus macaques, and a reduction in PCSK9 levels of up to 84% was observed, while LDL-C reduction was up to 60%. Several off-target effects were registered during the study, as well as the induction of an immune response [70]. To overcome these unwanted effects, related to the use of an AAV vector, lipid nanoparticles were used as a delivery system [73]. CRISPR base editors, delivered in vivo using lipid nanoparticles, can efficiently target the PCSK9 gene in Macaca fascicularis. A near-complete knockdown of PCSK9 in the liver after a single infusion of lipid nanoparticles was achieved. This led to a reduction in blood levels of PCSK9 and low-density lipoprotein cholesterol of approximately 90% and 60%, respectively. Compared with Wang and colleagues’ study, no significant off-target activity was observed. Lipid nanoparticles were also used for the delivery of mRNA encoding an adenine base editor (ABE) and a single-guide RNA targeting PCSK9 [74]. ABEs consist of a catalytically impaired Cas9 protein fused with an adenine deaminase and a modified gRNA. The modified gRNA guides the ABE to the target DNA sequence. The adenine deaminase enzyme then chemically modifies the adenine base in the DNA to become inosine, which is recognized as guanine by cellular machinery. During DNA replication, the complementary cytosine is added to the modified adenine, leading to a G•C base pair conversion [75]. PCSK9 base-editing in Cynomolgus monkeys occurred in a mean of 26%, while the reduction in PCSK9 protein was 32% and 14% for LDL-C. In this study as well, no off-target editing was observed. VERVE-101, an investigational CRISPR base editing therapy, consists of an mRNA for an adenine base editor and a gRNA targeting the PCSK9 gene assembled in a lipid nanoparticle delivery system [76]. Liver biopsies 14 days after Cynomolgus monkeys were given a single intravenous infusion of a vehicle control or VERVE-101 at a dose of 0.75 mg/kg or 1.5 mg/kg showed PCSK9 editing of 46% (0.75 mg/kg) and 70% (1.5 mg/kg). The related reduction in low-density lipoprotein cholesterol was 49% (0.75 mg/kg) and 69% (1.5 mg/kg). These promising results led Verve Therapeutics to start human clinical trials using VERVE-101 in patients with FH [77].
4. ANGPTL3 Gene Editing Strategies in Mice
ANGPTL3 is a gene that produces a protein involved in lipid metabolism. ANGPTL3 plays an important role in regulating triglycerides and cholesterol blood levels via the inhibition of lipoprotein lipase and endothelial lipase enzymes activity [78,79]. Loss-of-function mutations in this gene have been associated with lower LDL cholesterol levels and a reduced risk of cardiovascular diseases [80,81]. The potential benefits of targeting ANGPTL3 were confirmed in a study, using antisense oligonucleotide. An effective reduction in ANGLPT3 protein levels was achieved, and the same was achieved for TG and LDL cholesterol [82]. Intravenous injection of a specific monoclonal antibody in dyslipidemic C57BL/6 mice also reduced TG, LDL-C, and HDL-C levels in the blood [83]. Scientists have recently explored the use of CRISPR-Cas9 to disrupt or modify the ANGPTL3 gene to mimic the effects of these beneficial mutations and reduce LDL cholesterol levels. The base-editing approach was tested, which allows us to alter specific nucleotides in the DNA sequence without generating double-strand breaks (Figure 2) [84]. The authors produced an adenoviral vector expressing base editor 3 targeting ANGPTL3 and injected this vector into C57BL/6J mice. This resulted in reduced plasma ANGPTL3, triglyceride, and TC levels (49%, 31%, and 19%, respectively) [85]. The effect was even bigger when hyperlipidemic LDLR knockout mice were injected (triglycerides reduction by 56% and cholesterol reduction by 51%). Interestingly, this study also compared the effects of targeting ANGPLT3 versus PCSK9. ANGPTL3-targeted therapy is a more potent triglycerides-lowering therapy, whereas PCSK9-targeted therapy is a more potent LDL-lowering therapy. Moreover, inhibiting both ANGPTL3 and PCSK9 did not result in any synergistic or additive effects. Lipid nanoparticles were also used for the delivery of Cas9 mRNA and gRNA for CRISPR-Cas9-based genome editing of ANGPLT3 in mice. This delivery system has reduced delivery efficiency compared to viral vectors, but possesses less undesired insertional mutagenesis and potential biosafety issues [86,87]. Liver-specific knockdown of ANGPTL3 resulted in a profound lowering of LDL-C and triglycerides levels. No evidence of off-target mutagenesis was detected, nor of any liver toxicity, and the genome editing retains a therapeutically relevant level for at least 100 days after the injection of a single dose [88]. In another study, where base editing of ANGPTL3 via AAV delivery was used in C57BL/6J mice, the authors managed to achieve a near-complete knockout of the ANGPTL3 protein in the circulation and a reduction in serum levels of triglyceride and TC by 58% and 61%, respectively [89]. Evaluation of liver toxicity was also conducted, showing no significant changes in the levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) and no T cells infiltration or general sign of inflammation. While the above-mentioned study uses a dual-AAV base editor, Davis and colleagues developed a single-AAV adenine base editor system that supports robust editing in vivo and has a broad targeting capability [90]. The use of a single AAV vector for delivery guarantees a maximum editing efficiency, making it the best option when targeting non-liver tissues, or when toxicity limits AAV dosage. In mice, single-AAV-encoded ABE led to a knockdown of both PCSK9 and ANGPTL3 > 90% and to a reduction in circulating cholesterol.
5. ANGPTL3 Gene Editing Strategies in Non-Human Primates
An alternative approach to the use of lipid nanoparticles as a delivery system is a multi-valent N-acetylgalactosamine (GalNAc)-targeting ligand, which allows for uptake via the asialoglycoprotein receptor (ASGPR) pathway [91]. Delivery via ASGPR has several positive aspects: the receptor is highly expressed in the liver but not in other tissues of the body, induces an immediate endocytosis of the drug candidate when bound by GalNAc, and is rapidly recycled to the hepatocyte surface [92]. This methodology was selected for CRISPR base editing therapy targeting the ANGPTL3 gene in NHPs. A mean liver ANGPTL3 editing of 61% was observed in the six LDLR-deficient NHPs treated with the GalNAc-LNPs, corresponding to a reduction in blood ANGPTL3 protein of 89%. Circulating LDL-C also fell by 35%, a stable reduction for three months after treatment [93]. Liver toxicity tests registered only a transient increase in ALT and AST. In WT NHPs, a mean reduction in blood ANGPTL3 protein of 90% was noted for animals treated with the GalNAc-LNP versus 75% in those treated with a standard LNP. However, WT NHPs showed minimal changes in LDL-C despite a significant ANGPTL3 reduction. This is in line with prior preclinical data on NHPs of a monoclonal antibody targeting ANGPTL3 [83]. Based on this study, currently, VERVE Therapeutics is conducting a trial with its candidate VERVE-201 in a larger 34 NHPs sample.
6. LDLR Gene Editing Strategies in Mice
The LDLR gene encodes a receptor that plays a crucial role in regulating cholesterol levels in the body by allowing cells to take up cholesterol-rich LDL particles from the blood. Several cases of FH are related to mutations of the LDLR gene [94,95]. Mutations in LDLR can impair LDLR activity at different levels and are classified according to their phenotypic behavior as class 1 (no protein synthesis), class 2 (partial or complete retention of LDLR in the endoplasmic reticulum), class 3 (defective binding to apolipoprotein B), class 4 (defective endocytosis), and class 5 (decreased LDLR turnover ability) [96]. The role of the LDLR gene in lipid metabolism was also investigated using CRISPR-Cas9. AAV-CRISPR-Cas9 was used to disrupt the hepatic LDLR gene in adult mice, leading to severe hypercholesterolemia and atherosclerotic lesion in the aortas of C57BL/6J mice [97]. Similar observations were made in another study, where an LDLRE208X mutant knockin mouse model was generated. This model is based on an E207X nonsense point mutation in LDLR, observed in individuals with FH, and led to severe atherosclerosis as a consequence of the total depletion of LDLR expression [98]. However, when the mutant LDLRE208X strain was treated with AAV-CRISPR-Cas9, LDLR expression was partially restored, and the signs of atherosclerosis were mitigated, highlighting the potential use of CRISPR-Cas9 in the treatment of the HoFH [99]. Greig and colleagues used LDLR−/− mouse to test their AAV8 vectors expressing both murine and human versions of LDLR [100]. These vectors were previously used in double knockout mouse models, resulting in a complete correction of hypercholesterolemia [101,102]. Minimal levels of toxicity and inflammation response (cytokines production) were observed in the study, while a stable reduction in cholesterol was achieved with the lowest doses of LDLR vectors.
7. LDLR Gene Editing Strategies in Other Animal Models
Rabbits and hamsters have been widely used as animal models in the study of atherosclerosis because they have similar lipoprotein metabolism to humans and are more susceptible to atherosclerosis [103,104]. LDLR-KO rabbits with biallelic mutations were created to induce spontaneous hypercholesterolemia and atherosclerosis on a normal chow diet. Analysis of their plasma lipids showed an increase in triglycerides and a parallel decrease in HDL-C [105]. LDLR-KO hamsters can be induced by microinjecting CRISPR-Cas9 components into fertilized eggs for the development of hypercholesterolemia and hyperlipidemia models [106]. In line with the FH patients with LDLR gene mutations who have severe hypercholesterolemia in their homozygous form and a moderate hypercholesterolemia in the heterozygous form, LDLR −/− hamsters exhibit a severe form of hypercholesterolemia, while LDLR +/− hamsters exhibit a moderate form. This behavior differs compared with other species, including mice, where in the heterozygous form, there is never a significant increase in cholesterol levels, making hamsters an optimal tool for research on human atherosclerosis [107,108].
8. Apolipoproteins Gene Editing Strategies in Mice
Apolipoproteins are protein components associated with lipoproteins that have several functions, including stabilizing the structure of lipoproteins, serving as ligands for cellular receptors, and participating in enzymatic reactions [109]. Apolipoproteins can be classified into two subgroups: the soluble apolipoproteins including ApoA1, A2, A4, C1, C2, C3, and E, and the insoluble forms like ApoB100 and ApoB48. Alterations in their expression levels or spatial structure are closely related to a variety of diseases [110]. ApoA1 is the primary structural component of HDL particles and is a key mediator of plasma cholesterol transport and cholesterol homeostasis, interacting with several transporters and receptors [111,112]. De Giorgi and colleagues targeted the APOA1 locus with AAV delivery of CRISPR-Cas9 in mice and achieved rates between 6% and 16% of targeted hepatocytes, with no evidence of toxicity. In this study, improved expression of transgenic proteins from the APOA1 locus enhanced the expression of ApoE, reducing plasma lipids in a model of hypercholesterolemia [113]. The APOB gene provides instructions for the production of apolipoprotein B, a protein that is essential for the assembly and transport of LDL cholesterol in the bloodstream. Mutations in the APOB gene can cause familial hypercholesterolemia, a genetic disorder characterized by extremely high LDL cholesterol levels [114]. Mice treated with AAV-CRISPR vectors to disrupt the APOB gene showed a significant decrease in plasma cholesterol and were protected from atherosclerosis [3]. However, the treatment exacerbated hepatic fat accumulation, resulting in a microvesicular steatosis, as observed in humans with naturally occurring loss-of-function mutations in APOB [115]. Fat accumulation was also observed in another study where the APOB gene was targeted, using hepatocyte-tropic AAV-8 serotype [63].
9. Apolipoproteins Gene Editing Strategies in Other Animal Models
ApoC3 is another key regulator of plasma triglycerides and is found on chylomicrons, VLDL, LDL, and HDL particles. Recent studies have shown that ApoC3 levels are an independent risk factor for cardiovascular diseases (CVD) [116,117]. Despite the overexpression of human ApoC3 significantly accelerated atherosclerotic development in mice, the protective effect of ApoC3 deficiency on atherogenesis was not observed in KO mice [118]. However, inactivation of the APOC3 gene by CRISPR-Cas9 in hamsters, led to a decrease in plasma tryglicerides and to an enhanced conversion of VLDL into LDL. When the hamsters were fed with a high-cholesterol diet, a clear reduction in atherosclerotic lesions was observed [119]. APOC3 KO rabbits were also generated and displayed triglyceride levels 50% lower than those of the age-matched control group when given a normal chow diet. When fed with a high-fat diet, the APOC3 KO rabbits showed limited atherosclerotic lesions, while the WT rabbits had obvious atherosclerotic lesions, as well as increased intima thickening, collagen content, and levels of inflammatory cytokines (IL-1β and TNF-α) [120]. The limitations of this study were the usage of only three APOC3 KO rabbits without a consistent genotype, suggesting that further in vivo studies are required to elucidate the impact of APOC3 knockout on hyperlipidemia. A summary of animal model studies targeting genes associated with dyslipidemia is presented in Table 1.
10. Off-Target Effects in CRISPR-Cas9 Gene Editing
A major concern in the application of CRISPR-Cas9 gene editing technologies is the occurrence of off-target effects. Off-target effects are defined as unintended cleavage and mutations at untargeted genomic sites showing similarity with the target sequence [121]. Several studies have shown that Cas9 binds to unintended genomic sites and creates double-strand breaks, leading to undesired outcomes [122,123]. These off-target sites are often gRNA-dependent, since Cas9 is known to tolerate up to three mismatches between the gRNA and the genomic DNA [124]. However, recent findings suggest that gRNA-independent off-target effects could also occur with base editors as a consequence of random deamination [125,126]. Off-targeting can cause severe problems for the host organism, since it could lead to chromosomal rearrangements, loss of functional gene activity, or activation of oncogenes [127,128]. It is therefore crucial for the future therapeutic use of CRISPR-Cas9 gene editing to limit the occurrence of off-target effects by designing a well-engineered CRISPR system with high on-targeting efficiency. There are several strategies that are currently used to achieve this goal including the increase in nucleases cleavage specificity. In recent years, several new Cas9 proteins have been developed, such as Sniper-Cas9 and HypaCas9 [129,130]. Alternatively, a mutated form of Cas9 acts as a nickase (nCas9), where one of the endonuclease domains is catalytically inactivated. This leads to a cut in just one of the two DNA strands, creating a single-strand break. A second nCas9 targeting the opposite strand completes the double-strand break. This variant of Cas9 is able to reduce off-target effects by up to 1500 times compared with its wild-type form, but it is limited by the need for two appropriately spaced gRNAs acting on opposite strands [124,131]. Another strategy is to modify the gRNA: several studies show that the specificity of Cas9 activity can be increased by extending or truncating gRNA [132,133]. Additional decreases in unwanted mutagenesis are observed when the modified gRNA is associated with Cas9 nickase [134]. The selection of an appropriate delivery method for Cas9/gRNA is crucial not only for inducing low immunogenicity in the host, but also because it can profoundly affect the occurrence of off-target effects [135]. AAV-based gene delivery is known to last for years in terminally differentiated cells or exhibits a higher tendency to induce unwanted off-target effects over time [136,137] to trigger an immune response [138]. In contrast, LNP-delivered Cas9 are rapidly degraded in vivo, decreasing the opportunity for off-target activity during in vivo genome editing [88,139]. Choosing an appropriate off-target detection method such as biased and unbiased methods with predictive on-target and off-target sites is a necessary step in preventing the occurrence of undesired mutagenesis. Differences among the several off-target detection methods are outside the scope of this manuscript, but several reviews extensively describe the available tools [140,141].
11. Conclusions
Hyperlipidemia is definitely a suitable condition to address with genome editing, since preclinical studies have demonstrated that the targeting of genes associated with cholesterol levels is feasible. Currently, there are already several attractive targets for liver-directed genome editing that could lower lipid blood levels and prevent/reduce CVD. Mutations in different genes have been shown to cause monogenic dyslipidemia, including LDLRAP1 mutations that are involved in autosomal recessive hypercholesterolemia, ABCG5/ABCG8, involved in sitosterolemia, or LMF1, which is associated with familial chylomicronemia syndrome [56]. These genes are new potential candidates for CRISPR-Cas9 genome editing. The limitation of an appropriate delivery system is nowadays averted by the use of effective specific viral vectors and nanoparticles. These newly developed vectors aim at reducing the innate and adaptive cellular responses observed in the past, including those towards particular Cas9 nucleases [142,143]. However, the permanent nature of DNA changes caused by gene editing requires a careful evaluation of this technology before it can be broadly used to treat CVD diseases. Clinical trials of novel therapeutics in humans will require constant surveillance to enable the identification of any undesirable effects and to evaluate the long-term efficacy of the treatment in terms of plasma lipid levels and markers of atherosclerosis lesions. Indeed, the choice of some therapeutic targets such as PCSK9 or ANGPLT3 seems justified by the good overall health condition of individuals with spontaneous deficiency in these genes [81,144,145,146]. The assessment of off-target effects remains the biggest challenge, since the human genome is different to tested animal models, and since inter-individual differences are also possible. A combination of in silico prediction and testing of induced pluripotent stem-cell-derived hepatocytes are among the used approaches for detection of these off-target effects.
Funding
G.P. receives funding from the European Regional Development Fund under grant agreement number 01.2.2-CPVA-V-716-01-0001 with the Central Project Management Agency (CPVA), Lithuania.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
G.P. thanks Stephen Knox Jones Jr. for feedback on the manuscript.
Conflicts of Interest
The author declares no conflict of interests.
Abbreviations
| AAV: | Adeno-associated virus |
| ABE: | Adenine base editor |
| ALT: | Alanine aminotransferase |
| ANGPTL3: | Angiopoietin like 3 |
| Apo(a): | Apoliporotein (a) |
| ApoA1: | Apolipoprotein A1 |
| ApoB: | Apolipoprotein B |
| ApoC3: | Apolipoprotein C3 |
| ASGPR: | Asialoglycoprotein receptor |
| AST: | Aspartate aminotransferase |
| Cas9: | CRISPR-associated protein 9 |
| CRISPR: | Clustered regularly interspaced short palindromic repeats |
| CVD: | Cardiovascular diseases |
| ER: | Endoplasmic reticulum |
| FDA: | Food and Drug Administration |
| FH: | Familial hypercholesterolemia |
| GalNac: | Multi-valent N-AcetylGalactosamine |
| gRNA: | guide RNA |
| HDL: | High-density lipoprotein |
| HMG-CoA: | β-Hydroxy β-methylglutaryl-CoA |
| HoFH: | Homozygous familial hypercholesterolemia |
| KO: | Knockout |
| LDL: | Low-density lipoprotein |
| LDLR: | Low-density lipoprotein receptor |
| LLN: | Lipid-like nanoparticle |
| LNP: | Lipid nanoparticle |
| Lp(a): | Lipoprotein A |
| NHP: | Non-human primates |
| PCSK9: | Proprotein convertase subtilisin/kexin type 9 |
| PPAR-alpha: | Peroxisome proliferator-activated receptor alpha |
| RNP: | Ribonucleoprotein |
| RXR: | Retinoid X receptor |
| siRNA: | Small interfering RNA |
| TC: | Total cholesterol |
| TG: | Triglycerides |
| VLDL: | Very low density lipoprotein |
| WT: | Wild-type |
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Karimian, A.; Azizian, K.; Parsian, H.; Rafieian, S.; Shafiei-Irannejad, V.; Kheyrollah, M.; Yousefi, M.; Majidinia, M.; Yousefi, B. CRISPR/Cas9 technology as a potent molecular tool for gene therapy. J. Cell. Physiol. 2019, 234, 12267–12277. [Google Scholar] [CrossRef]
- Jarrett, K.E.; Lee, C.M.; Yeh, Y.-H.; Hsu, R.H.; Gupta, R.; Zhang, M.; Rodriguez, P.J.; Lee, C.S.; Gillard, B.K.; Bissig, K.-D.; et al. Somatic genome editing with CRISPR/Cas9 generates and corrects a metabolic disease. Sci. Rep. 2017, 7, 44624. [Google Scholar] [CrossRef]
- Yin, H.; Song, C.-Q.; Dorkin, J.R.; Zhu, L.J.; Li, Y.; Wu, Q.; Park, A.; Yang, J.; Suresh, S.; Bizhanova, A.; et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol. 2016, 34, 328–333. [Google Scholar] [CrossRef]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef]
- Ahn, C.H.; Choi, S.H. New drugs for treating dyslipidemia: Beyond statins. Diabetes Metab. J. 2015, 39, 87–94. [Google Scholar] [CrossRef]
- Brautbar, A.; Ballantyne, C.M. Pharmacological strategies for lowering LDL cholesterol: Statins and beyond. Nat. Rev. Cardiol. 2011, 8, 253–265. [Google Scholar] [CrossRef]
- Muscoli, S.; Ifrim, M.; Russo, M.; Candido, F.; Sanseviero, A.; Milite, M.; Di Luozzo, M.; Marchei, M.; Sangiorgi, G.M. Current Options and Future Perspectives in the Treatment of Dyslipidemia. J. Clin. Med. 2022, 11, 4716. [Google Scholar] [CrossRef]
- Nurmohamed, N.S.; Stroes, E.S.G. Working towards full eradication of lipid-driven cardiovascular risk? Neth. Heart J. 2022, 30, 15–24. [Google Scholar] [CrossRef]
- Schirris, T.J.J.; Renkema, G.H.; Ritschel, T.; Voermans, N.C.; Bilos, A.; van Engelen, B.G.M.; Brandt, U.; Koopman, W.J.H.; Beyrath, J.D.; Rodenburg, R.J.; et al. Statin-Induced Myopathy Is Associated with Mitochondrial Complex III Inhibition. Cell Metab. 2015, 22, 399–407. [Google Scholar] [CrossRef]
- Lim, G.B. Dyslipidaemia: Balancing the benefits and risks of statin therapy. Nat. Rev. Cardiol. 2016, 13, 632–633. [Google Scholar] [CrossRef]
- Bardolia, C.; Amin, N.S.; Turgeon, J. Emerging Non-statin Treatment Options for Lowering Low-Density Lipoprotein Cholesterol. Front. Cardiovasc. Med. 2021, 8, 789931. [Google Scholar] [CrossRef]
- Garcia-Calvo, M.; Lisnock, J.; Bull, H.G.; Hawes, B.E.; Burnett, D.A.; Braun, M.P.; Crona, J.H.; Davis, H.R.J.; Dean, D.C.; Detmers, P.A.; et al. The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1). Proc. Natl. Acad. Sci. USA 2005, 102, 8132–8137. [Google Scholar] [CrossRef]
- Pandor, A.; Ara, R.M.; Tumur, I.; Wilkinson, A.J.; Paisley, S.; Duenas, A.; Durrington, P.N.; Chilcott, J. Ezetimibe monotherapy for cholesterol lowering in 2,722 people: Systematic review and meta-analysis of randomized controlled trials. J. Intern. Med. 2009, 265, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Nodari, S.; Rocca, P.; Saporetti, A.; Bettari, L.; Foresti, A.L.; Tanghetti, E.; Metra, M.; Dei Cas, L. The combination of Ezetimibe and Statin: A new treatment for hypercholesterolemia. Heart Int. 2007, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Descamps, O.; Tomassini, J.E.; Lin, J.; Polis, A.B.; Shah, A.; Brudi, P.; Hanson, M.E.; Tershakovec, A.M. Variability of the LDL-C lowering response to ezetimibe and ezetimibe + statin therapy in hypercholesterolemic patients. Atherosclerosis 2015, 240, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Ghasempour, G.; Zamani-Garmsiri, F.; Shaikhnia, F.; Soleimani, A.A.; Fard, S.R.H.; Leila, J.; Teimuri, S.; Parvaz, N.; Mohammadi, P.; Najafi, M. Efficacy and Safety of Alirocumab and Evolocumab as Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) Inhibitors in Familial Hypercholesterolemia: A Systematic Review and Meta-Analysis. Curr. Med. Chem. 2023, 31, 223–241. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, M.; Peterson, K.; Holzhammer, B.; Fazio, S. A Systematic Review of PCSK9 Inhibitors Alirocumab and Evolocumab. J. Manag. Care Spec. Pharm. 2016, 22, 641q–653q. [Google Scholar] [CrossRef]
- Kuzmich, N.; Andresyuk, E.; Porozov, Y.; Tarasov, V.; Samsonov, M.; Preferanskaya, N.; Veselov, V.; Alyautdin, R. PCSK9 as a Target for Development of a New Generation of Hypolipidemic Drugs. Molecules 2022, 27, 434. [Google Scholar] [CrossRef]
- Lagace, T.A. PCSK9 and LDLR degradation: Regulatory mechanisms in circulation and in cells. Curr. Opin. Lipidol. 2014, 25, 387–393. [Google Scholar] [CrossRef]
- Choi, J.Y.; Na, J.O. Pharmacological Strategies beyond Statins: Ezetimibe and PCSK9 Inhibitors. J. Lipid. Atheroscler. 2019, 8, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Katsiki, N.; Vrablik, M.; Banach, M.; Gouni-Berthold, I. Inclisiran, Low-Density Lipoprotein Cholesterol and Lipoprotein (a). Pharmaceuticals 2023, 16, 577. [Google Scholar] [CrossRef]
- Kosmas, C.E.; Bousvarou, M.D.; Sourlas, A.; Papakonstantinou, E.J.; Peña Genao, E.; Echavarria Uceta, R.; Guzman, E. Angiopoietin-Like Protein 3 (ANGPTL3) Inhibitors in the Management of Refractory Hypercholesterolemia. Clin. Pharmacol. 2022, 14, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Karwatowska-Prokopczuk, E.; Tardif, J.-C.; Gaudet, D.; Ballantyne, C.M.; Shapiro, M.D.; Moriarty, P.M.; Baum, S.J.; Amour, E.S.; Alexander, V.J.; Xia, S.; et al. Effect of olezarsen targeting APOC-III on lipoprotein size and particle number measured by NMR in patients with hypertriglyceridemia. J. Clin. Lipidol. 2022, 16, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; Karwatowska-Prokopczuk, E.; Amour, E.S.; Ballantyne, C.M.; Shapiro, M.D.; Moriarty, P.M.; Baum, S.J.; Hurh, E.; Bartlett, V.J.; Kingsbury, J.; et al. Apolipoprotein C-III reduction in subjects with moderate hypertriglyceridaemia and at high cardiovascular risk. Eur. Heart J. 2022, 43, 1401–1412. [Google Scholar] [CrossRef]
- Tsimikas, S.; Viney, N.J.; Hughes, S.G.; Singleton, W.; Graham, M.J.; Baker, B.F.; Burkey, J.L.; Yang, Q.; Marcovina, S.M.; Geary, R.S.; et al. Antisense therapy targeting apolipoprotein(a): A randomised, double-blind, placebo-controlled phase 1 study. Lancet 2015, 386, 1472–1483. [Google Scholar] [CrossRef]
- Viney, N.J.; van Capelleveen, J.C.; Geary, R.S.; Xia, S.; Tami, J.A.; Yu, R.Z.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M.; et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): Two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef]
- Koren, M.J.; Moriarty, P.M.; Baum, S.J.; Neutel, J.; Hernandez-Illas, M.; Weintraub, H.S.; Florio, M.; Kassahun, H.; Melquist, S.; Varrieur, T.; et al. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein(a). Nat. Med. 2022, 28, 96–103. [Google Scholar] [CrossRef]
- Sohn, W.; Winkle, P.; Neutel, J.; Wu, Y.; Jabari, F.; Terrio, C.; Varrieur, T.; Wang, J.; Hellawell, J. Pharmacokinetics, Pharmacodynamics, and Tolerability of Olpasiran in Healthy Japanese and Non-Japanese Participants: Results from a Phase I, Single-dose, Open-label Study. Clin. Ther. 2022, 44, 1237–1247. [Google Scholar] [CrossRef]
- Rider, D.A.; Eisermann, M.; Löffler, K.; Aleku, M.; Swerdlow, D.I.; Dames, S.; Hauptmann, J.; Morrison, E.; Lindholm, M.W.; Schubert, S.; et al. Pre-clinical assessment of SLN360, a novel siRNA targeting LPA, developed to address elevated lipoprotein (a) in cardiovascular disease. Atherosclerosis 2022, 349, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Ganji, S.H.; Kamanna, V.S.; Kashyap, M.L. Niacin and cholesterol: Role in cardiovascular disease (review). J. Nutr. Biochem. 2003, 14, 298–305. [Google Scholar] [CrossRef] [PubMed]
- The HPS2-THRIVE Collaborative Group. Effects of extended-release niacin with laropiprant in high-risk patients. N. Engl. J. Med. 2014, 371, 203–212. [Google Scholar] [CrossRef]
- The AIM-HIGH Investigators. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.; Vecka, M.; Perlík, F.; Hromádka, R.; Staňková, B.; Tvrzická, E. Niacin in the Treatment of Hyperlipidemias in Light of New Clinical Trials: Has Niacin Lost its Place? Med. Sci. Monit. 2015, 21, 2156–2162. [Google Scholar] [PubMed]
- Hansen, M.; Sonne, D.P.; Knop, F.K. Bile acid sequestrants: Glucose-lowering mechanisms and efficacy in type 2 diabetes. Curr. Diab. Rep. 2014, 14, 482. [Google Scholar] [CrossRef]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef]
- Feng, Y.; Li, Q.; Ou, G.; Yang, M.; Du, L. Bile acid sequestrants: A review of mechanism and design. J. Pharm. Pharmacol. 2021, 73, 855–861. [Google Scholar] [CrossRef]
- Abourbih, S.; Filion, K.B.; Joseph, L.; Schiffrin, E.L.; Rinfret, S.; Poirier, P.; Pilote, L.; Genest, J.; Eisenberg, M.J. Effect of fibrates on lipid profiles and cardiovascular outcomes: A systematic review. Am. J. Med. 2009, 122, 962.E1–962.E8. [Google Scholar] [CrossRef]
- Kim, N.H.; Kim, S.G. Fibrates Revisited: Potential Role in Cardiovascular Risk Reduction. Diabetes Metab. J. 2020, 44, 213–221. [Google Scholar] [CrossRef]
- Fazio, S.; Linton, M.F. The role of fibrates in managing hyperlipidemia: Mechanisms of action and clinical efficacy. Curr. Atheroscler. Rep. 2004, 6, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Dallongeville, J.; Auwerx, J.; Schoonjans, K.; Leitersdorf, E.; Fruchart, J.-C. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998, 98, 2088–2093. [Google Scholar] [CrossRef] [PubMed]
- Bilen, O.; Ballantyne, C.M. Bempedoic Acid (ETC-1002): An Investigational Inhibitor of ATP Citrate Lyase. Curr. Atheroscler. Rep. 2016, 18, 61. [Google Scholar] [CrossRef]
- Ray, K.K.; Bays, H.E.; Catapano, A.L.; Lalwani, N.D.; Bloedon, L.T.; Sterling, L.R.; Robinson, P.L.; Ballantyne, C.M. Safety and Efficacy of Bempedoic Acid to Reduce LDL Cholesterol. N. Engl. J. Med. 2019, 380, 1022–1032. [Google Scholar] [CrossRef]
- Nissen, S.E.; Lincoff, A.M.; Brennan, D.; Ray, K.K.; Mason, D.; Kastelein, J.J.; Thompson, P.D.; Libby, P.; Cho, L.; Plutzky, J.; et al. Bempedoic Acid and Cardiovascular Outcomes in Statin-Intolerant Patients. N. Engl. J. Med. 2023, 388, 1353–1364. [Google Scholar] [CrossRef]
- Sundararaman, S.S.; Döring, Y.; van Der Vorst, E.P. PCSK9: A Multi-Faceted Protein That Is Involved in Cardiovascular Biology. Biomedicines 2021, 9, 793. [Google Scholar] [CrossRef]
- Xia, X.-D.; Peng, Z.-S.; Gu, H.-M.; Wang, M.; Wang, G.-Q.; Zhang, D.-W. Regulation of PCSK9 Expression and Function: Mechanisms and Therapeutic Implications. Front. Cardiovasc. Med. 2021, 8, 764038. [Google Scholar] [CrossRef]
- Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Molecular biology of PCSK9: Its role in LDL metabolism. Trends Biochem. Sci. 2007, 32, 71–77. [Google Scholar] [CrossRef]
- Defesche, J.C.; Gidding, S.S.; Harada-Shiba, M.; Hegele, R.A.; Santos, R.D.; Wierzbicki, A.S. Familial hypercholesterolaemia. Nat. Rev. Dis. Primers 2017, 3, 17093. [Google Scholar] [CrossRef] [PubMed]
- Di Taranto, M.D.; Benito-Vicente, A.; Giacobbe, C.; Uribe, K.B.; Rubba, P.; Etxebarria, A.; Guardamagna, O.; Gentile, M.; Martín, C.; Fortunato, G. Identification and in vitro characterization of two new PCSK9 Gain of Function variants found in patients with Familial Hypercholesterolemia. Sci. Rep. 2017, 7, 15282. [Google Scholar] [CrossRef] [PubMed]
- Leren, T.P.; Berge, K.E. Identification of mutations in the apolipoprotein B-100 gene and in the PCSK9 gene as the cause of hypocholesterolemia. Clin. Chim. Acta 2008, 397, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Berge, K.E.; Ose, L.; Leren, T.P. Missense mutations in the PCSK9 gene are associated with hypocholesterolemia and possibly increased response to statin therapy. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1094–1100. [Google Scholar] [CrossRef]
- Garvie, C.W.; Fraley, C.V.; Elowe, N.H.; Culyba, E.K.; Lemke, C.T.; Hubbard, B.K.; Kaushik, V.K.; Daniels, D.S. Point mutations at the catalytic site of PCSK9 inhibit folding, autoprocessing, and interaction with the LDL receptor. Protein Sci. 2016, 25, 2018–2027. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Sasaki, K.; Tada, H. Familial hypobetalipoproteinemia caused by homozygous loss-of-function mutations in PCSK9: A case report. J. Clin. Lipidol. 2022, 16, 596–600. [Google Scholar] [CrossRef]
- Katzmann, J.L.; Cupido, A.J.; Laufs, U. Gene Therapy Targeting PCSK9. Metabolites 2022, 12, 70. [Google Scholar] [CrossRef] [PubMed]
- Stankov, S.; Cuchel, M. Gene editing for dyslipidemias: New tools to “cut” lipids. Atherosclerosis 2023, 368, 14–24. [Google Scholar] [CrossRef]
- Ding, Q.; Strong, A.; Patel, K.M.; Ng, S.-L.; Gosis, B.S.; Regan, S.N.; Cowan, C.A.; Rader, D.J.; Musunuru, K. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ. Res. 2014, 115, 488–492. [Google Scholar] [CrossRef]
- Wang, X.; Raghavan, A.; Chen, T.; Qiao, L.; Zhang, Y.; Ding, Q.; Musunuru, K. CRISPR-Cas9 Targeting of PCSK9 in Human Hepatocytes In Vivo-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 783–786. [Google Scholar] [CrossRef]
- Zhu, J.; Huang, X.; Yang, Y. Innate immune response to adenoviral vectors is mediated by both Toll-like receptor-dependent and -independent pathways. J. Virol. 2007, 81, 3170–3180. [Google Scholar] [CrossRef]
- Mingozzi, F.; High, K.A. Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef]
- Mingozzi, F.; High, K.A. Therapeutic in vivo gene transfer for genetic disease using AAV: Progress and challenges. Nat. Rev. Genet. 2011, 12, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Kotterman, M.A.; Chalberg, T.W.; Schaffer, D.V. Viral Vectors for Gene Therapy: Translational and Clinical Outlook. Annu. Rev. Biomed. Eng. 2015, 17, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Su, J.; Liu, Y.; Jin, X.; Zhong, X.; Mo, L.; Wang, Q.; Deng, H.; Yang, Y. In vivo PCSK9 gene editing using an all-in-one self-cleavage AAV-CRISPR system. Mol. Ther. Methods Clin. Dev. 2021, 20, 652–659. [Google Scholar] [CrossRef]
- Jiang, C.; Mei, M.; Li, B.; Zhu, X.; Zu, W.; Tian, Y.; Wang, Q.; Guo, Y.; Dong, Y.; Tan, X. A non-viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo. Cell Res. 2017, 27, 440–443. [Google Scholar] [CrossRef]
- Li, B.; Luo, X.; Deng, B.; Wang, J.; McComb, D.W.; Shi, Y.; Gaensler, K.M.L.; Tan, X.; Dunn, A.L.; Kerlin, B.A.; et al. An Orthogonal Array Optimization of Lipid-like Nanoparticles for mRNA Delivery in Vivo. Nano Lett. 2015, 15, 8099–8107. [Google Scholar] [CrossRef]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef]
- Yin, H.; Song, C.-Q.; Suresh, S.; Wu, Q.; Walsh, S.; Rhym, L.H.; Mintzer, E.; Bolukbasi, M.F.; Zhu, L.J.; Kauffman, K.; et al. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat. Biotechnol. 2017, 35, 1179–1187. [Google Scholar] [CrossRef]
- Carreras, A.; Pane, L.S.; Nitsch, R.; Madeyski-Bengtson, K.; Porritt, M.; Akcakaya, P.; Taheri-Ghahfarokhi, A.; Ericson, E.; Bjursell, M.; Perez-Alcazar, M.; et al. In vivo genome and base editing of a human PCSK9 knock-in hypercholesterolemic mouse model. BMC Biol. 2019, 17, 4. [Google Scholar] [CrossRef]
- Wang, L.; Breton, C.; Warzecha, C.C.; Bell, P.; Yan, H.; He, Z.; White, J.; Zhu, Y.; Li, M.; Buza, E.L.; et al. Long-term stable reduction of low-density lipoprotein in nonhuman primates following in vivo genome editing of PCSK9. Mol. Ther. 2021, 29, 2019–2029. [Google Scholar] [CrossRef]
- Silva, G.; Poirot, L.; Galetto, R.; Smith, J.; Montoya, G.; Duchateau, P.; Paques, F. Meganucleases and other tools for targeted genome engineering: Perspectives and challenges for gene therapy. Curr. Gene Ther. 2011, 11, 11–27. [Google Scholar] [CrossRef]
- Tröder, S.E.; Zevnik, B. History of genome editing: From meganucleases to CRISPR. Lab. Anim. 2022, 56, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Rothgangl, T.; Dennis, M.K.; Lin, P.J.; Oka, R.; Witzigmann, D.; Villiger, L.; Qi, W.; Hruzova, M.; Kissling, L.; Lenggenhager, D.; et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat. Biotechnol. 2021, 39, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.G.; Mazzola, A.M.; Braun, M.C.; Platt, C.; Vafai, S.B.; Kathiresan, S.; Rohde, E.; Bellinger, A.M.; Khera, A.V. Efficacy and Safety of an Investigational Single-Course CRISPR Base-Editing Therapy Targeting PCSK9 in Nonhuman Primate and Mouse Models. Circulation 2023, 147, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Verve takes base editors into humans. Nat. Biotechnol. 2022, 40, 1159.
- Hassan, M. ANGPLT3: A novel modulator of lipid metabolism. Glob. Cardiol. Sci. Pract. 2017, 2017, e201706. [Google Scholar] [CrossRef]
- Su, X.; Peng, D. New insights into ANGPLT3 in controlling lipoprotein metabolism and risk of cardiovascular diseases. Lipids Health Dis. 2018, 17, 12. [Google Scholar] [CrossRef]
- Dewey, F.E.; Gusarova, V.; Dunbar, R.L.; O’dushlaine, C.; Schurmann, C.; Gottesman, O.; McCarthy, S.; Van Hout, C.V.; Bruse, S.; Dansky, H.M.; et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 211–221. [Google Scholar] [CrossRef]
- Stitziel, N.O.; Khera, A.V.; Wang, X.; Bierhals, A.J.; Vourakis, A.C.; Sperry, A.E.; Natarajan, P.; Klarin, D.; Emdin, C.A.; Zekavat, S.M.; et al. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. J. Am. Coll. Cardiol. 2017, 69, 2054–2063. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.J.; Lee, R.G.; Brandt, T.A.; Tai, L.-J.; Fu, W.; Peralta, R.; Yu, R.; Hurh, E.; Paz, E.; McEvoy, B.W.; et al. Cardiovascular and Metabolic Effects of ANGPTL3 Antisense Oligonucleotides. N. Engl. J. Med. 2017, 377, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Gusarova, V.; Alexa, C.A.; Wang, Y.; Rafique, A.; Kim, J.H.; Buckler, D.; Mintah, I.J.; Shihanian, L.M.; Cohen, J.C.; Hobbs, H.H.; et al. ANGPTL3 blockade with a human monoclonal antibody reduces plasma lipids in dyslipidemic mice and monkeys. J. Lipid Res. 2015, 56, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, A.C.; Evitt, N.H.; Lv, W.; Musunuru, K. Reduced Blood Lipid Levels with In Vivo CRISPR-Cas9 Base Editing of ANGPTL3. Circulation 2018, 137, 975–977. [Google Scholar] [CrossRef]
- Glass, Z.; Lee, M.; Li, Y.; Xu, Q. Engineering the Delivery System for CRISPR-Based Genome Editing. Trends Biotechnol. 2018, 36, 173–185. [Google Scholar] [CrossRef]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Qiu, M.; Glass, Z.; Chen, J.; Haas, M.; Jin, X.; Zhao, X.; Rui, X.; Ye, Z.; Li, Y.; Zhang, F.; et al. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of Angptl3. Proc. Natl. Acad. Sci. USA 2021, 118, e2020401118. [Google Scholar] [CrossRef]
- Zuo, Y.; Zhang, C.; Zhou, Y.; Li, H.; Xiao, W.; Herzog, R.W.; Xu, J.; Zhang, J.; Chen, Y.E.; Han, R. Liver-specific in vivo base editing of Angptl3 via AAV delivery efficiently lowers blood lipid levels in mice. Cell Biosci. 2023, 13, 109. [Google Scholar] [CrossRef]
- Davis, J.R.; Wang, X.; Witte, I.P.; Huang, T.P.; Levy, J.M.; Raguram, A.; Banskota, S.; Seidah, N.G.; Musunuru, K.; Liu, D.R. Efficient in vivo base editing via single adeno-associated viruses with size-optimized genomes encoding compact adenine base editors. Nat. Biomed. Eng. 2022, 6, 1272–1283. [Google Scholar] [CrossRef]
- Nair, J.K.; Willoughby, J.L.S.; Chan, A.; Charisse, K.; Alam, R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef]
- Paunovska, K.; Loughrey, D.; Dahlman, J.E. Drug delivery systems for RNA therapeutics. Nat. Rev. Genet. 2022, 23, 265–280. [Google Scholar] [CrossRef]
- Kasiewicz, L.N.; Biswas, S.; Beach, A.; Ren, H.; Dutta, C.; Mazzola, A.M.; Rohde, E.; Chadwick, A.; Cheng, C.; Garcia, S.P.; et al. GalNAc-Lipid nanoparticles enable non-LDLR dependent hepatic delivery of a CRISPR base editing therapy. Nat. Commun. 2023, 14, 2776. [Google Scholar] [CrossRef] [PubMed]
- Soutar, A.K.; Naoumova, R.P. Mechanisms of disease: Genetic causes of familial hypercholesterolemia. Nat. Clin. Pract. Cardiovasc. Med. 2007, 4, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, H.H.; Brown, M.S.; Goldstein, J.L. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum. Mutat. 1992, 1, 445–466. [Google Scholar] [CrossRef] [PubMed]
- Tolleshaug, H.; Goldstein, J.L.; Schneider, W.J.; Brown, M.S. Posttranslational processing of the LDL receptor and its genetic disruption in familial hypercholesterolemia. Cell 1982, 30, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Jarrett, K.E.; Lee, C.; De Giorgi, M.; Hurley, A.; Gillard, B.K.; Doerfler, A.M.; Li, A.; Pownall, H.J.; Bao, G.; Lagor, W.R. Somatic Editing of Ldlr with Adeno-Associated Viral-CRISPR Is an Efficient Tool for Atherosclerosis Research. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1997–2006. [Google Scholar] [CrossRef]
- Sun, X.M.; Patel, D.D.; Webb, J.C.; Knight, B.L.; Fan, L.M.; Cai, H.J.; Soutar, A.K. Familial hypercholesterolemia in China. Identification of mutations in the LDL-receptor gene that result in a receptor-negative phenotype. Arterioscler. Thromb. 1994, 14, 85–94. [Google Scholar] [CrossRef]
- Zhao, H.; Li, Y.; He, L.; Pu, W.; Yu, W.; Li, Y.; Wu, Y.-T.; Xu, C.; Wei, Y.; Ding, Q.; et al. In Vivo AAV-CRISPR/Cas9-Mediated Gene Editing Ameliorates Atherosclerosis in Familial Hypercholesterolemia. Circulation 2020, 141, 67–79. [Google Scholar] [CrossRef]
- Greig, J.A.; Limberis, M.P.; Bell, P.; Chen, S.J.; Calcedo, R.; Rader, D.J.; Wilson, J.M. Nonclinical Pharmacology/Toxicology Study of AAV8.TBG.mLDLR and AAV8.TBG.hLDLR in a Mouse Model of Homozygous Familial Hypercholesterolemia. Hum. Gene Ther. Clin. Dev. 2017, 28, 28–38. [Google Scholar] [CrossRef]
- Kassim, S.H.; Li, H.; Bell, P.; Somanathan, S.; Lagor, W.; Jacobs, F.; Billheimer, J.; Wilson, J.M.; Rader, D.J.; Greig, J.A.; et al. Adeno-associated virus serotype 8 gene therapy leads to significant lowering of plasma cholesterol levels in humanized mouse models of homozygous and heterozygous familial hypercholesterolemia. Hum. Gene Ther. 2013, 24, 19–26. [Google Scholar] [CrossRef]
- Kassim, S.H.; Li, H.; Vandenberghe, L.H.; Hinderer, C.; Bell, P.; Marchadier, D.; Wilson, A.; Cromley, D.; Redon, V.; Yu, H.; et al. Gene therapy in a humanized mouse model of familial hypercholesterolemia leads to marked regression of atherosclerosis. PLoS ONE 2010, 5, e13424. [Google Scholar] [CrossRef]
- Fan, J.; Kitajima, S.; Watanabe, T.; Xu, J.; Zhang, J.; Liu, E.; Chen, Y.E. Rabbit models for the study of human atherosclerosis: From pathophysiological mechanisms to translational medicine. Pharmacol. Ther. 2015, 146, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Gisterå, A.; Ketelhuth, D.F.; Malin, S.G.; Hansson, G.K. Animal Models of Atherosclerosis-Supportive Notes and Tricks of the Trade. Circ. Res. 2022, 130, 1869–1887. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Yuan, T.; Wang, Y.; Zhang, T.; Yuan, Y.; Wu, D.; Zhou, M.; He, Z.; Lu, Y.; Chen, Y.; et al. Spontaneous severe hypercholesterolemia and atherosclerosis lesions in rabbits with deficiency of low-density lipoprotein receptor (LDLR) on exon 7. EBioMedicine 2018, 36, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Gao, M.; Wang, Y.; Lin, X.; Yang, L.; Cong, N.; An, X.; Wang, F.; Qu, K.; Yu, L.; et al. LDL Receptor Gene-ablated Hamsters: A Rodent Model of Familial Hypercholesterolemia with Dominant Inheritance and Diet-induced Coronary Atherosclerosis. EBioMedicine 2018, 27, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, S.; Brown, M.S.; Goldstein, J.L.; Gerard, R.D.; Hammer, R.E.; Herz, J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J. Clin. Investig. 1993, 92, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Xian, X.; Wang, Y.; Liu, G. Genetically Engineered Hamster Models of Dyslipidemia and Atherosclerosis. Methods Mol. Biol. 2022, 2419, 433–459. [Google Scholar]
- Zhou, Y.; Luo, G. Apolipoproteins, as the carrier proteins for lipids, are involved in the development of breast cancer. Clin. Transl. Oncol. 2020, 22, 1952–1962. [Google Scholar] [CrossRef]
- Liu, T.; Chen, J.M.; Zhang, D.; Zhang, Q.; Peng, B.; Xu, L.; Tang, H. ApoPred: Identification of Apolipoproteins and Their Subfamilies With Multifarious Features. Front. Cell Dev. Biol. 2020, 8, 621144. [Google Scholar] [CrossRef]
- Zhao, G.J.; Yin, K.; Fu, Y.C.; Tang, C.K. The interaction of ApoA-I and ABCA1 triggers signal transduction pathways to mediate efflux of cellular lipids. Mol. Med. 2012, 18, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Mangaraj, M.; Nanda, R.; Panda, S. Apolipoprotein A-I: A Molecule of Diverse Function. Indian J. Clin. Biochem. 2016, 31, 253–259. [Google Scholar] [CrossRef] [PubMed]
- De Giorgi, M.; Li, A.; Hurley, A.; Barzi, M.; Doerfler, A.M.; Cherayil, N.A.; Smith, H.E.; Brown, J.D.; Lin, C.Y.; Bissig, K.-D.; et al. Targeting the Apoa1 locus for liver-directed gene therapy. Mol. Ther. Methods Clin. Dev. 2021, 21, 656–669. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, S.; Pisciotta, L.; Rabacchi, C.; Cefalù, A.B.; Noto, D.; Fasano, T.; Signori, A.; Fresa, R.; Averna, M.; Calandra, S. Spectrum of mutations and phenotypic expression in patients with autosomal dominant hypercholesterolemia identified in Italy. Atherosclerosis 2013, 227, 342–348. [Google Scholar] [CrossRef]
- Young, S.G.; Northey, S.T.; McCarthy, B.J. Low plasma cholesterol levels caused by a short deletion in the apolipoprotein B gene. Science 1988, 241, 591–593. [Google Scholar] [CrossRef]
- Cohn, J.S.; Tremblay, M.; Batal, R.; Jacques, H.; Rodriguez, C.; Steiner, G.; Mamer, O.; Davignon, J. Increased apoC-III production is a characteristic feature of patients with hypertriglyceridemia. Atherosclerosis 2004, 177, 137–145. [Google Scholar] [CrossRef]
- Sacks, F.M.; Alaupovic, P.; Moye, L.A.; Cole, T.G.; Sussex, B.; Stampfer, M.J.; Pfeffer, M.A.; Braunwald, E. VLDL, apolipoproteins B, CIII, and E, and risk of recurrent coronary events in the Cholesterol and Recurrent Events (CARE) trial. Circulation 2000, 102, 1886–1892. [Google Scholar] [CrossRef]
- Li, H.; Han, Y.; Qi, R.; Wang, Y.; Zhang, X.; Yu, M.; Tang, Y.; Wang, M.; Shu, Y.-N.; Huang, W.; et al. Aggravated restenosis and atherogenesis in ApoCIII transgenic mice but lack of protection in ApoCIII knockouts: The effect of authentic triglyceride-rich lipoproteins with and without ApoCIII. Cardiovasc. Res. 2015, 107, 579–589. [Google Scholar] [CrossRef]
- Guo, M.; Xu, Y.; Dong, Z.; Zhou, Z.; Cong, N.X.; Gao, M.; Huang, W.; Wang, Y.; Liu, G.; Xian, X. Inactivation of ApoC3 by CRISPR/Cas9 Protects Against Atherosclerosis in Hamsters. Circ. Res. 2020, 127, 1456–1458. [Google Scholar] [CrossRef]
- Zha, Y.; Lu, Y.; Zhang, T.; Yan, K.; Zhuang, W.; Liang, J.; Cheng, Y.; Wang, Y. CRISPR/Cas9-mediated knockout of APOC3 stabilizes plasma lipids and inhibits atherosclerosis in rabbits. Lipids Health Dis. 2021, 20, 180. [Google Scholar] [CrossRef]
- Modrzejewski, D.; Hartung, F.; Lehnert, H.; Sprink, T.; Kohl, C.; Keilwagen, J.; Wilhelm, R. Which Factors Affect the Occurrence of Off-Target Effects Caused by the Use of CRISPR/Cas: A Systematic Review in Plants. Front. Plant Sci. 2020, 11, 574959. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Alkan, F.; Wenzel, A.; Anthon, C.; Havgaard, J.H.; Gorodkin, J. CRISPR-Cas9 off-targeting assessment with nucleic acid duplex energy parameters. Genome Biol. 2018, 19, 177. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Zuo, E.; Sun, Y.; Wei, W.; Yuan, T.; Ying, W.; Sun, H.; Yuan, L.; Steinmetz, L.M.; Li, Y.; Yang, H. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science 2019, 364, 289–292. [Google Scholar] [CrossRef]
- Grünewald, J.; Zhou, R.; Garcia, S.P.; Iyer, S.; Lareau, C.A.; Aryee, M.J.; Joung, J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 2019, 569, 433–437. [Google Scholar] [CrossRef]
- Yin, J.; Liu, M.; Liu, Y.; Wu, J.; Gan, T.; Zhang, W.; Li, Y.; Zhou, Y.; Hu, J. Optimizing genome editing strategy by primer-extension-mediated sequencing. Cell Discov. 2019, 5, 18. [Google Scholar] [CrossRef]
- Ayabe, S.; Nakashima, K.; Yoshiki, A. Off- and on-target effects of genome editing in mouse embryos. J. Reprod. Dev. 2019, 65, 1–5. [Google Scholar] [CrossRef]
- Lee, J.K.; Jeong, E.; Lee, J.; Jung, M.; Shin, E.; Kim, Y.-H.; Lee, K.; Jung, I.; Kim, D.; Kim, S.; et al. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat. Commun. 2018, 9, 3048. [Google Scholar] [CrossRef]
- Chen, J.S.; Dagdas, Y.S.; Kleinstiver, B.P.; Welch, M.M.; Sousa, A.A.; Harrington, L.B.; Sternberg, S.H.; Joung, J.K.; Yildiz, A.; Doudna, J.A. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 2017, 550, 407–410. [Google Scholar] [CrossRef]
- Frock, R.L.; Hu, J.; Meyers, R.M.; Ho, Y.-J.; Kii, E.; Alt, F.W. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 2015, 33, 179–186. [Google Scholar] [CrossRef]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef]
- Kim, D.; Kim, S.; Kim, S.; Park, J.; Kim, J.-S. Genome-wide target specificities of CRISPR-Cas9 nucleases revealed by multiplex Digenome-seq. Genome Res. 2016, 26, 406–415. [Google Scholar] [CrossRef]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.-S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef]
- Taha, E.A.; Lee, J.; Hotta, A. Delivery of CRISPR-Cas tools for in vivo genome editing therapy: Trends and challenges. J. Control. Release 2022, 342, 345–361. [Google Scholar] [CrossRef]
- Lu, B.; Javidi-Parsijani, P.; Makani, V.; Mehraein-Ghomi, F.; Sarhan, W.M.; Sun, D.; Yoo, K.W.; Atala, Z.P.; Lyu, P.; Atala, A. Delivering SaCas9 mRNA by lentivirus-like bionanoparticles for transient expression and efficient genome editing. Nucleic Acids Res. 2019, 47, e44. [Google Scholar] [CrossRef]
- Hanlon, K.S.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Volak, A.; Spirig, S.E.; Muller, A.; Sousa, A.A.; Tsai, S.Q.; Bengtsson, N.E.; et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019, 10, 4439. [Google Scholar] [CrossRef]
- Arjomandnejad, M.; Dasgupta, I.; Flotte, T.R.; Keeler, A.M. Immunogenicity of Recombinant Adeno-Associated Virus (AAV) Vectors for Gene Transfer. BioDrugs 2023, 37, 311–329. [Google Scholar] [CrossRef]
- Ghani, M.W.; Iqbal, A.; Ghani, H.; Bibi, S.; Wang, Z.; Pei, R. Recent advances in nanocomposite-based delivery systems for targeted CRISPR/Cas delivery and therapeutic genetic manipulation. J. Mater. Chem. B 2023, 11, 5251–5271. [Google Scholar] [CrossRef]
- Martin, F.; Sánchez-Hernández, S.; Gutiérrez-Guerrero, A.; Pinedo-Gomez, J.; Benabdellah, K. Biased and Unbiased Methods for the Detection of Off-Target Cleavage by CRISPR/Cas9: An Overview. Int. J. Mol. Sci. 2016, 17, 1507. [Google Scholar] [CrossRef]
- Zhang, X.-H.; Tee, L.Y.; Wang, X.-G.; Huang, Q.-S.; Yang, S.-H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Chew, W.L.; Tabebordbar, M.; Cheng, J.K.; Mali, P.; Wu, E.Y.; Ng, A.H.; Zhu, K.; Wagers, A.J.; Church, G.M. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat. Methods 2016, 13, 868–874. [Google Scholar] [CrossRef]
- Charlesworth, C.T.; Deshpande, P.S.; Dever, D.P.; Camarena, J.; Lemgart, V.T.; Cromer, M.K.; Vakulskas, C.A.; Collingwood, M.A.; Zhang, L.; Bode, N.M.; et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019, 25, 249–254. [Google Scholar] [CrossRef]
- Zhao, Z.; Tuakli-Wosornu, Y.; Lagace, T.A.; Kinch, L.; Grishin, N.V.; Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am. J. Hum. Genet. 2006, 79, 514–523. [Google Scholar] [CrossRef]
- Hooper, A.J.; Marais, A.D.; Tanyanyiwa, D.M.; Burnett, J.R. The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis 2007, 193, 445–448. [Google Scholar] [CrossRef]
- Romeo, S.; Yin, W.; Kozlitina, J.; Pennacchio, L.A.; Boerwinkle, E.; Hobbs, H.H.; Cohen, J.C. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J. Clin. Investig. 2009, 119, 70–79. [Google Scholar] [CrossRef]
Figure 1.
Main therapies used for the treatment of hyperlipidemia with the associated mechanism of action. Different drugs are indicated in red, and the arrows point to the specific molecular targets. A few compounds can act at different levels. Figure modified from [9], licensed under CC-BY 4.0.
Figure 1.
Main therapies used for the treatment of hyperlipidemia with the associated mechanism of action. Different drugs are indicated in red, and the arrows point to the specific molecular targets. A few compounds can act at different levels. Figure modified from [9], licensed under CC-BY 4.0.
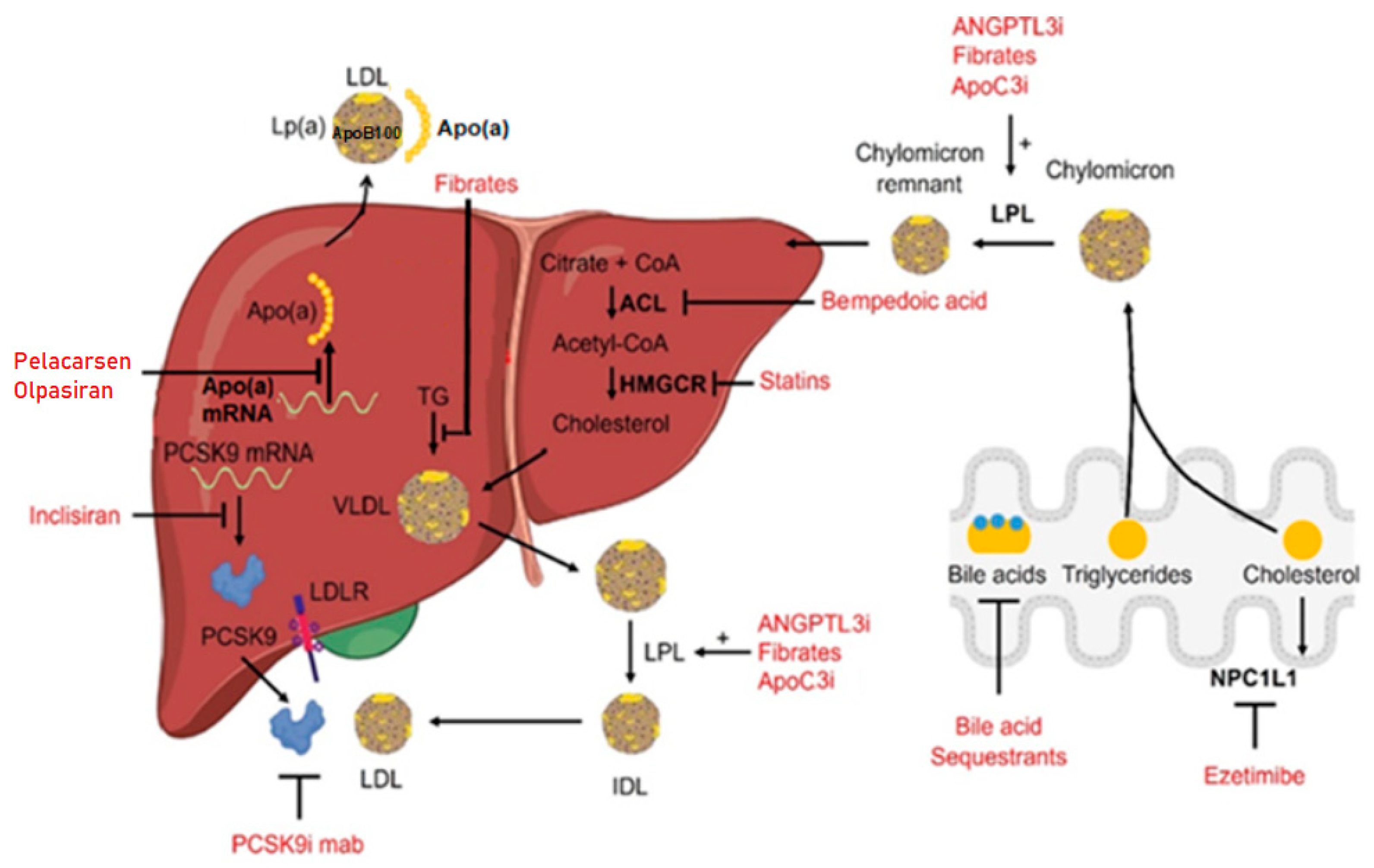
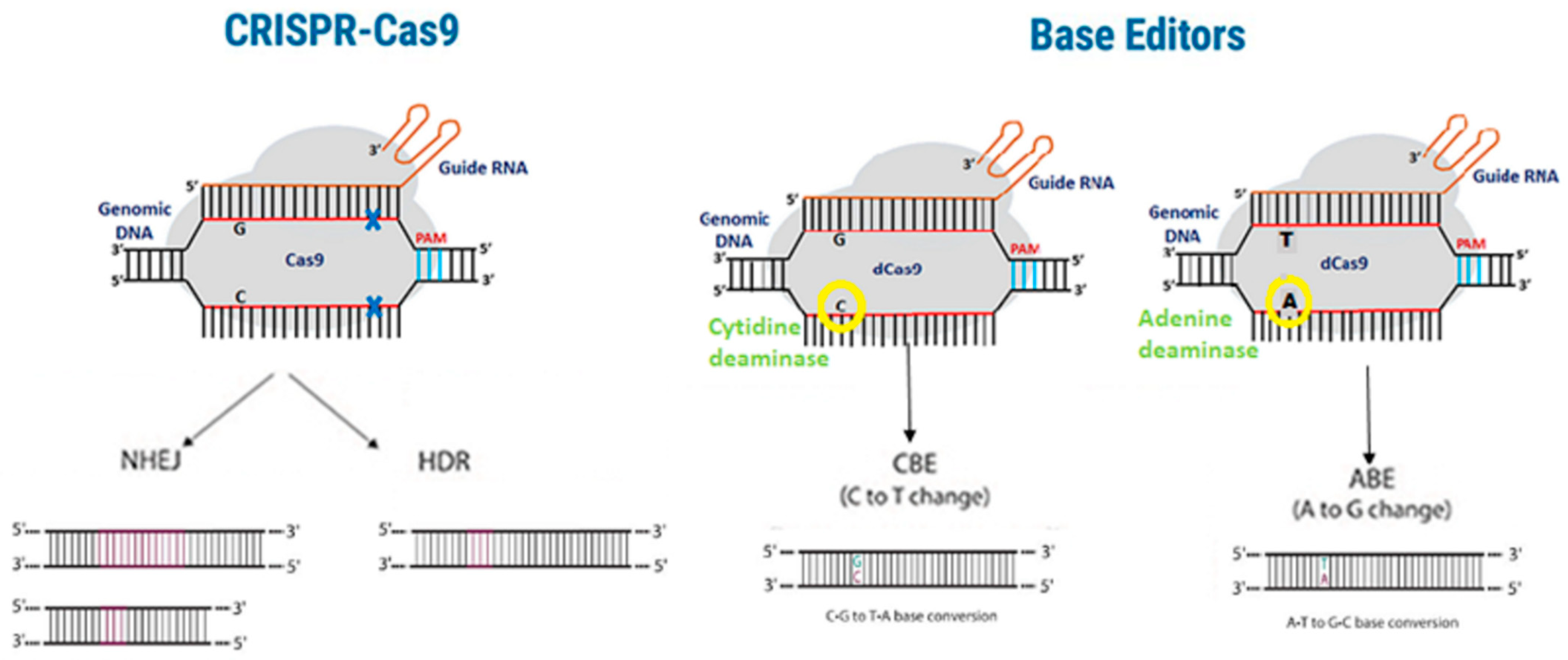
Figure 2.
Differences between the CRISPR-Cas9 and base editors methodologies. Traditional CRISPR-Cas9 gene editing (left panel) introduces double-strand breaks, which can lead to off-target effects. Base editing (right panel) avoids double-strand breaks due to catalytically inactive Cas9 (dCas9), thereby limiting the occurrence of off-target effects.
Figure 2.
Differences between the CRISPR-Cas9 and base editors methodologies. Traditional CRISPR-Cas9 gene editing (left panel) introduces double-strand breaks, which can lead to off-target effects. Base editing (right panel) avoids double-strand breaks due to catalytically inactive Cas9 (dCas9), thereby limiting the occurrence of off-target effects.

{kind=link}
{kind=link}
Table 1.
Main animal model studies targeting genes associated with dyslipidemia.
| Species | Delivery Vehicle | Editing Method | References | ||
|---|---|---|---|---|---|
| PCSK9 | Mouse | Adenovirus | CRISPR | [57] | |
| Mouse | AAV | CRISPR | [58] | ||
| Mouse | AAV | CRISPR | [63] | ||
| Mouse | AAV | CRISPR | [64] | ||
| Mouse | LLN | CRISPR | [65] | ||
| Mouse | LNP | CRISPR | [67] | ||
| Mouse | LNP | CRISPR | [68] | ||
| Mouse | Adenovirus | Base editor | [69] | ||
| NHP | AAV | Meganuclease | [70] | ||
| NHP | LNP | Base editor | [73] | ||
| NHP | LNP | Base editor | [74] | ||
| ANGPTL3 | Mouse | Adenovirus | Base editor | [85] | |
| Mouse | LNP | CRISPR | [88] | ||
| Mouse | AAV | Base editor | [89] | ||
| Mouse | AAV | Base editor | [90] | ||
| NHP | GalNAc-LNP | Base editor | [93] | ||
| LDLR | Mouse | AAV | CRISPR | [97] | |
| Mouse | AAV | CRISPR | [99] | ||
| Rabbit | * | CRISPR | [105] | ||
| Hamster | * | CRISPR | [106] | ||
| APO | A1 | Mouse | AAV | CRISPR | [113] |
| B | Mouse | AAV | CRISPR | [3] | |
| B | Mouse | AAV | CRISPR | [63] | |
| C3 | Hamster | * | CRISPR | [119] | |
| C3 | Rabbit | * | CRISPR | [120] | |
* = microinjections in embryos.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Preta, G. Development of New Genome Editing Tools for the Treatment of Hyperlipidemia. Cells 2023, 12, 2466. https://doi.org/10.3390/cells12202466
AMA Style
Preta G. Development of New Genome Editing Tools for the Treatment of Hyperlipidemia. Cells. 2023; 12(20):2466. https://doi.org/10.3390/cells12202466
Chicago/Turabian StylePreta, Giulio. 2023. "Development of New Genome Editing Tools for the Treatment of Hyperlipidemia" Cells 12, no. 20: 2466. https://doi.org/10.3390/cells12202466
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.