
A Novel Co-Culture Model Reveals Enhanced CFTR Rescue in Primary Cystic Fibrosis Airway Epithelial Cultures with Persistent Pseudomonas aeruginosa Infection

 , , , ,
, , , ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Epithelial and Bacterial Co-Culture
2.2. Drug Treatments
2.3. Antimicrobial Activity Assays
2.4. Western Blot Analysis
2.5. Ussing Chamber Measurements
2.6. Microscopy
2.7. mRNA Analysis
2.8. Cytokine Analysis
2.9. Statistics
3. Results
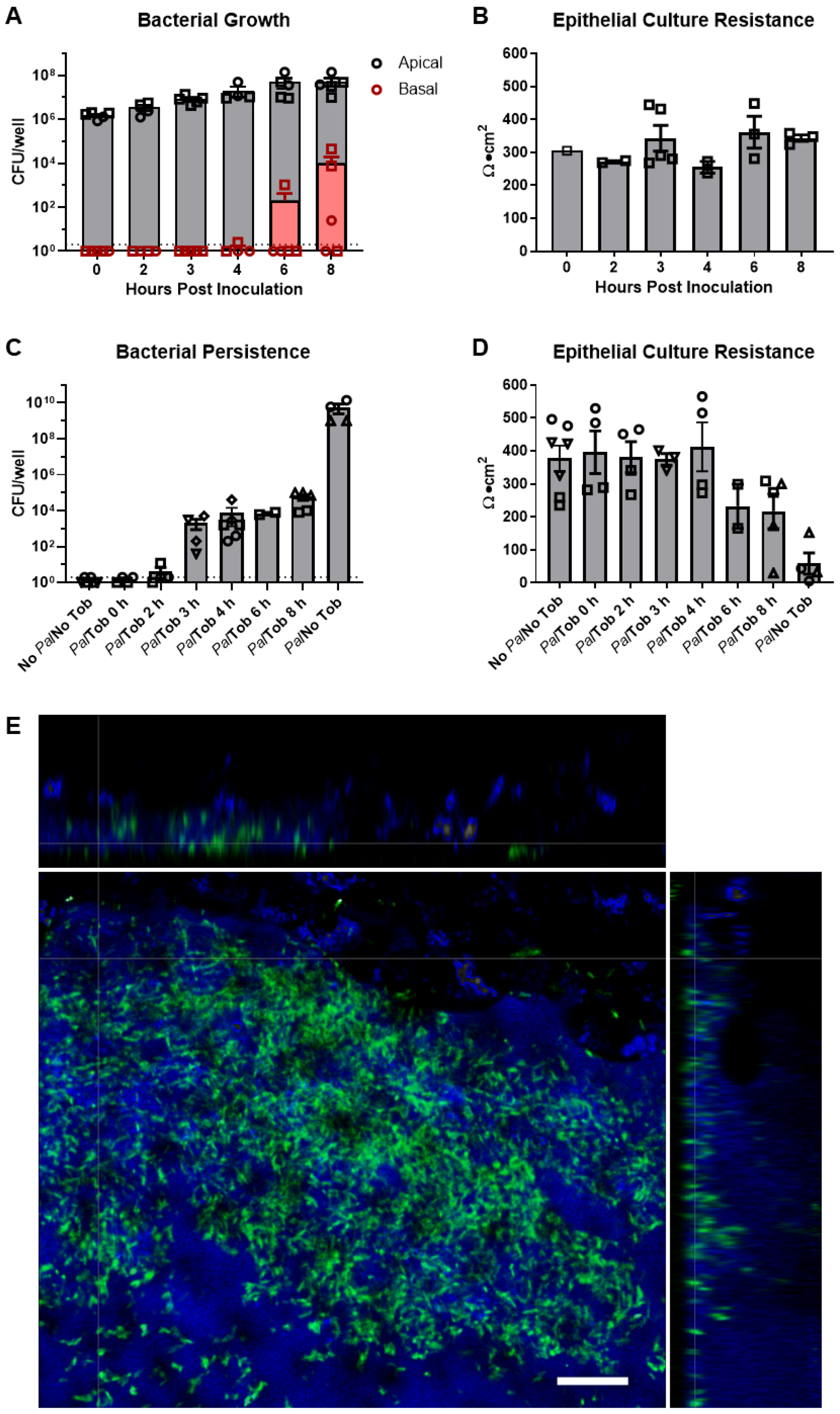
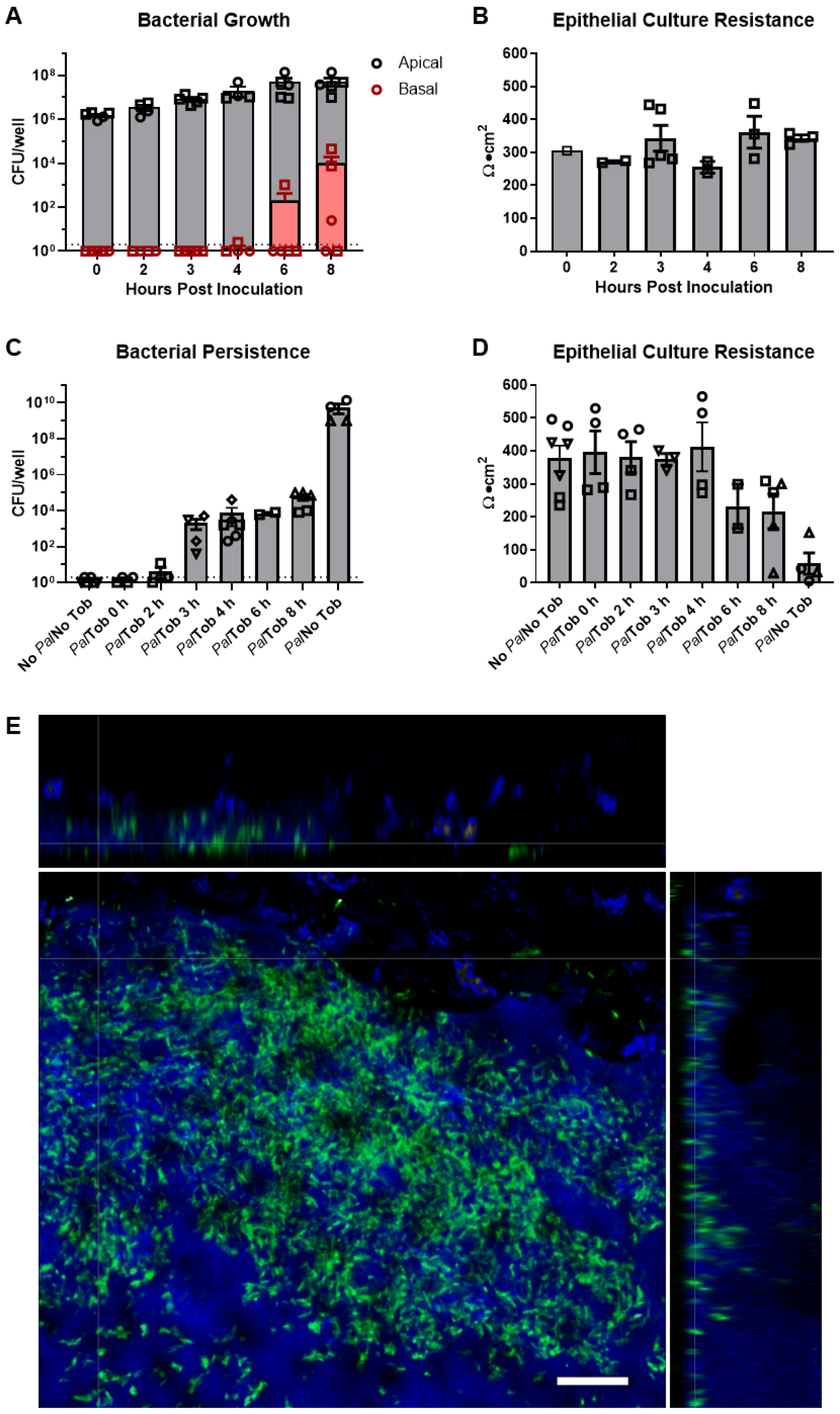
3.1. Model of Persistent P. aeruginosa Airway Infection Using Well-Differentiated Primary HBE Cultures
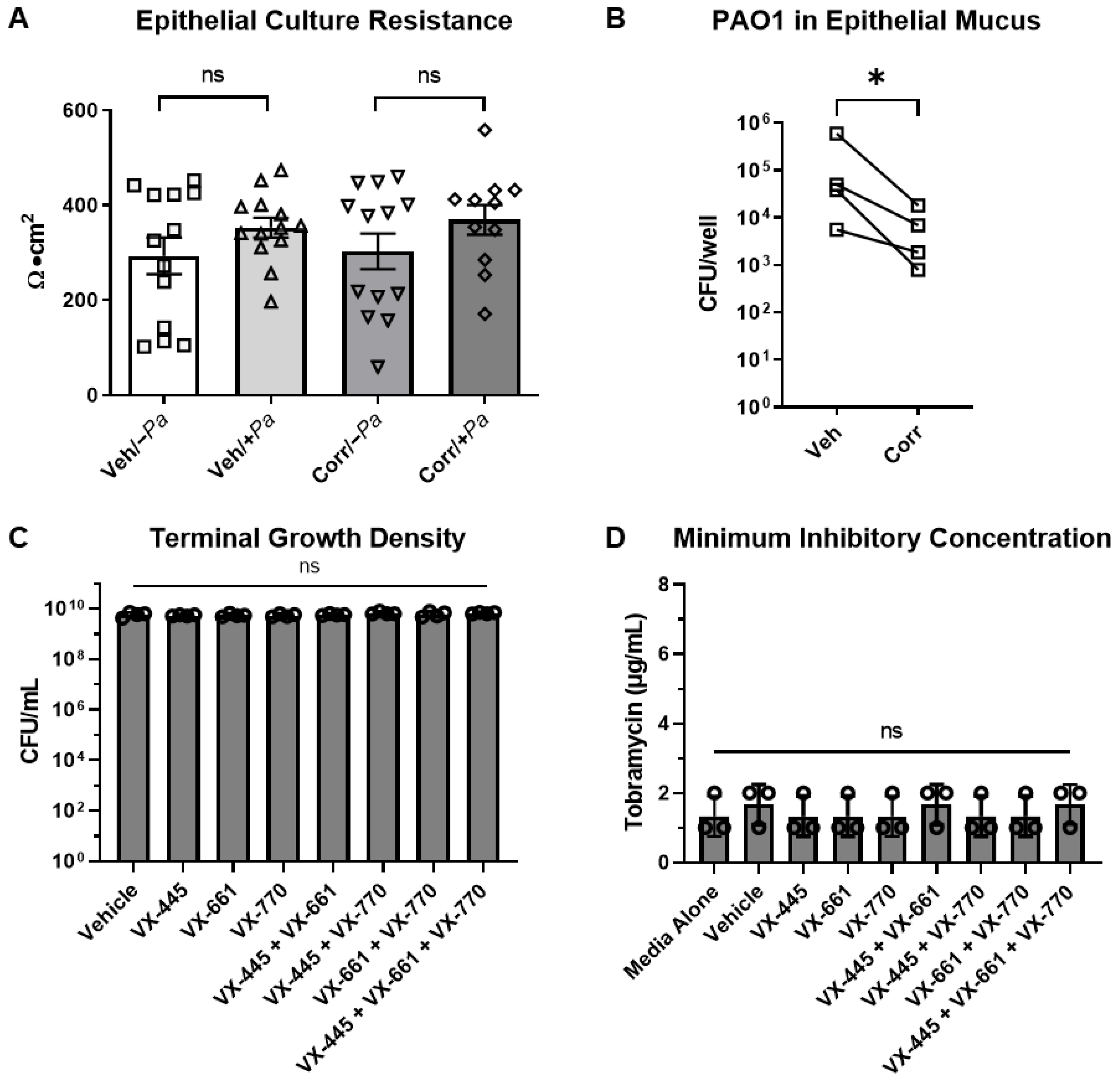
3.2. CFTR Correctors Caused a Decrease in P. aeruginosa Burden When Co-Cultured with HBE Cells
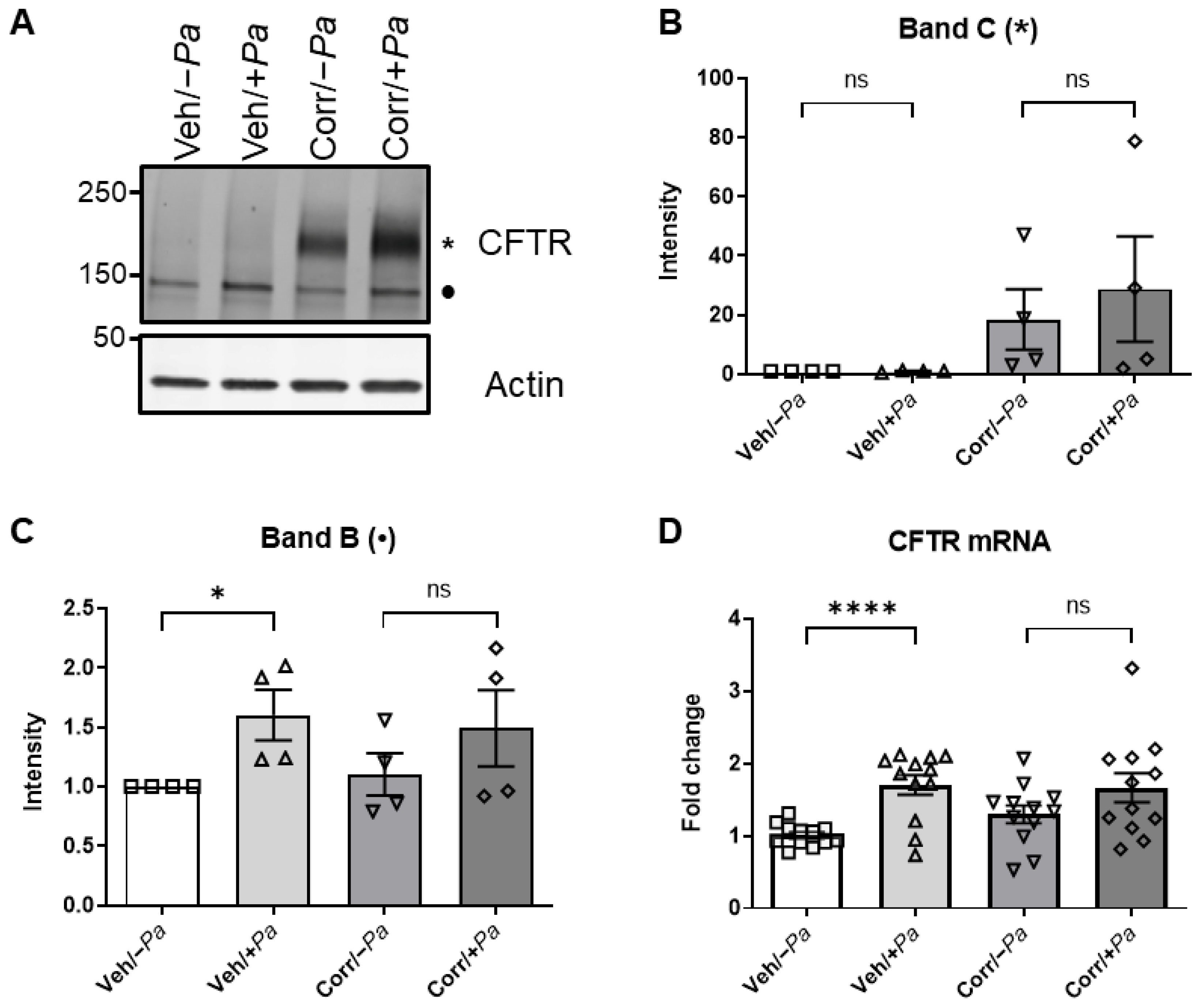
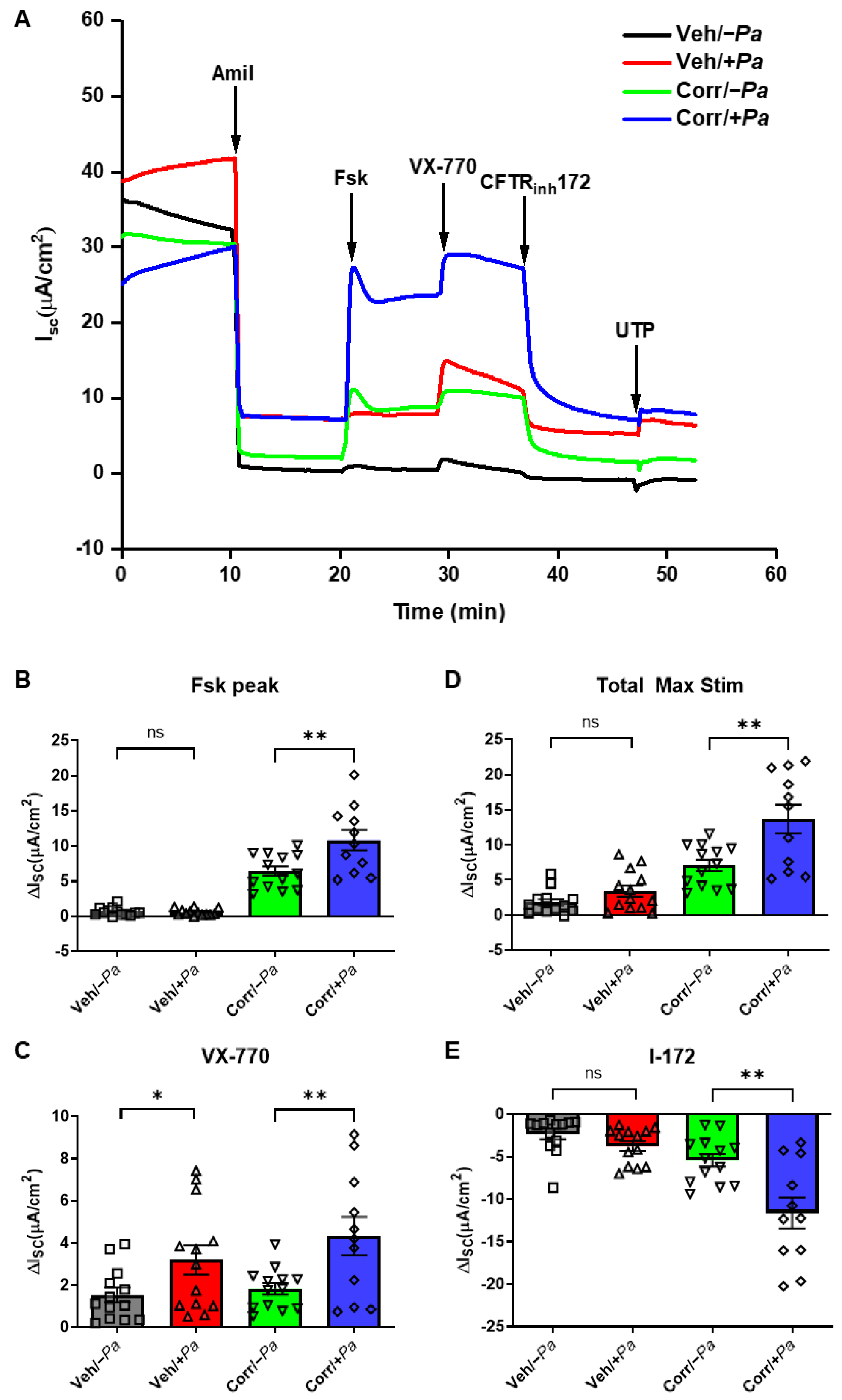
3.3. The Presence of P. aeruginosa Enhanced CFTR Protein Maturation, mRNA, and Function
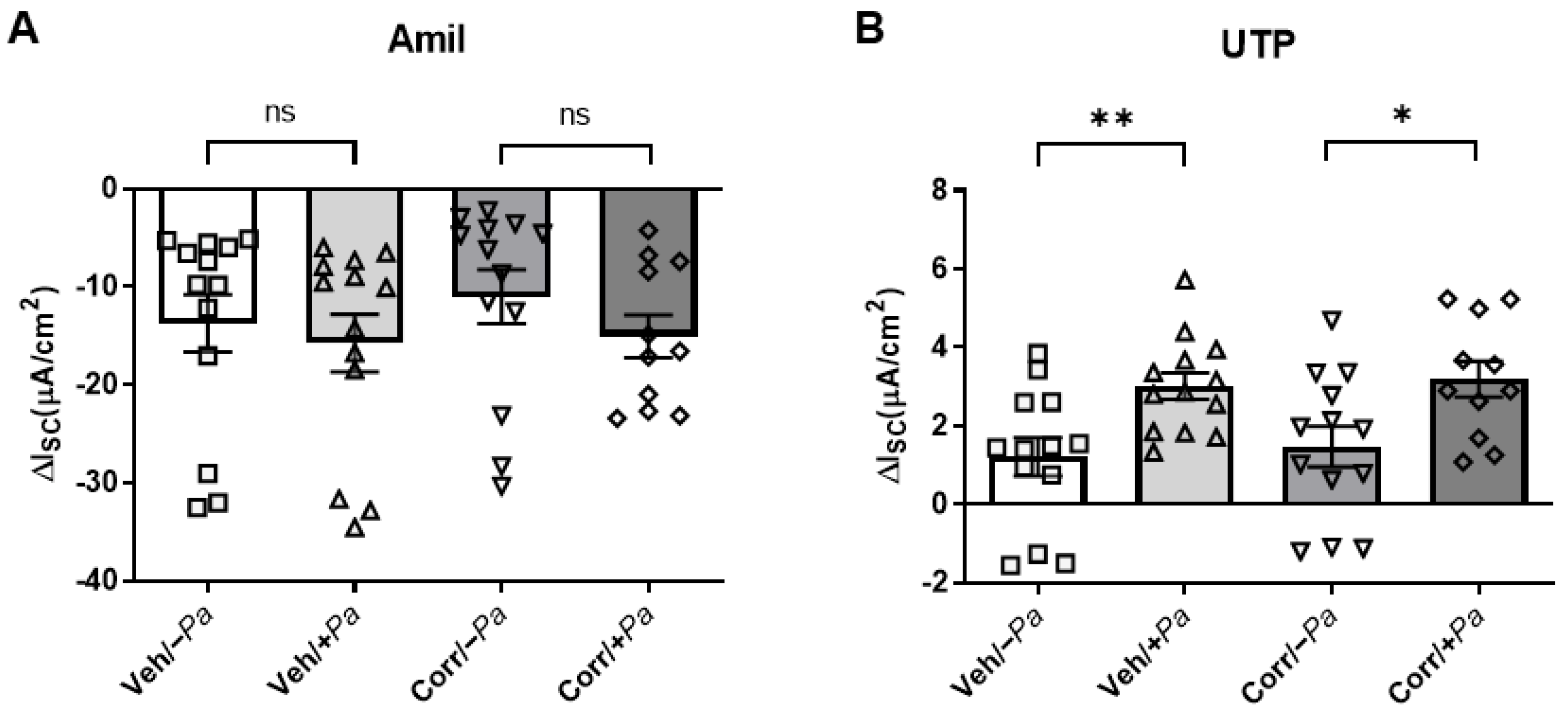
3.4. P. aeruginosa Infection Increased Activity of CaCC in HBE Cultures
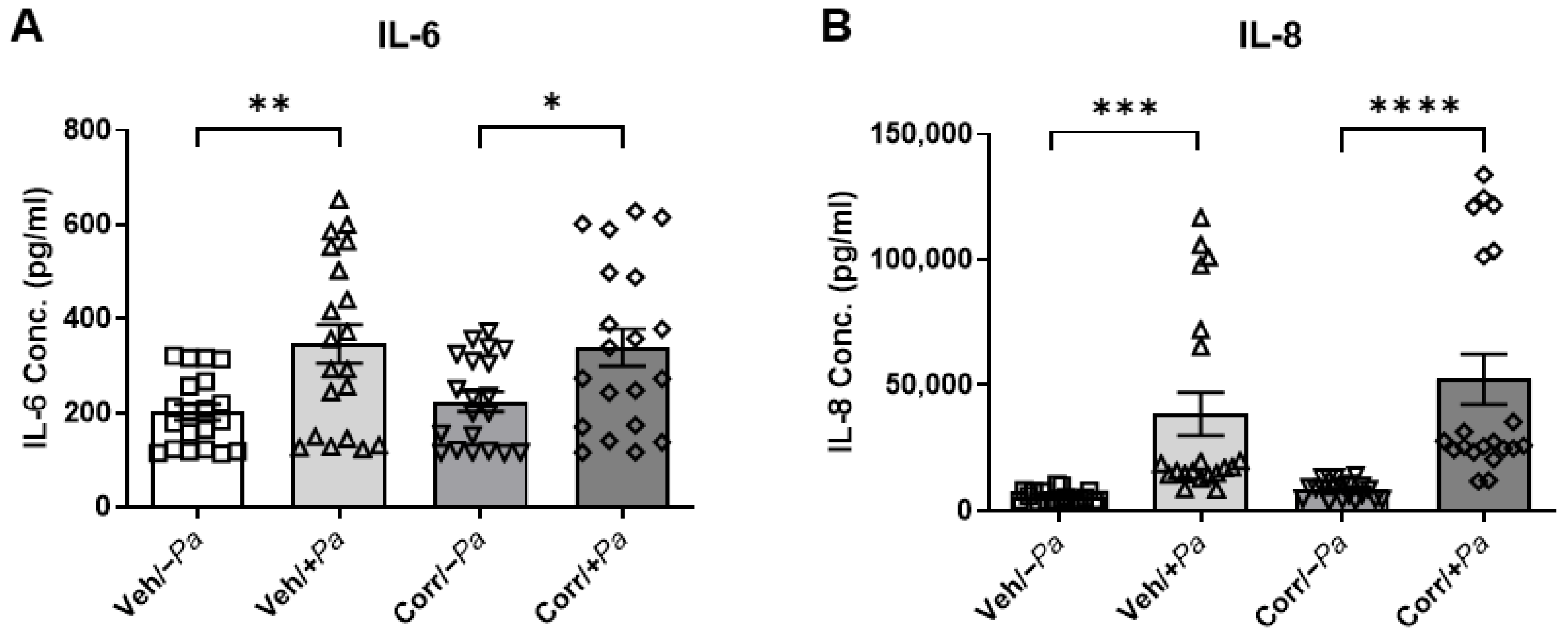
3.5. Cytokine Secretion Is Increased upon Bacterial Infection
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boucher, R.C. Airway surface dehydration in cystic fibrosis: Pathogenesis and therapy. Annu. Rev. Med. 2007, 58, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Chmiel, J.F.; Davis, P.B. State of the art: Why do the lungs of patients with cystic fibrosis become infected and why can’t they clear the infection? Respir. Res. 2003, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Kwilas, A.R.; Yednak, M.A.; Zhang, L.; Liesman, R.; Collins, P.L.; Pickles, R.J.; Peeples, M.E. Respiratory syncytial virus engineered to express the cystic fibrosis transmembrane conductance regulator corrects the bioelectric phenotype of human cystic fibrosis airway epithelium in vitro. J. Virol. 2010, 84, 7770–7781. [Google Scholar] [CrossRef] [PubMed]
- Esther, C.R., Jr.; Muhlebach, M.S.; Ehre, C.; Hill, D.B.; Wolfgang, M.C.; Kesimer, M.; Ramsey, K.A.; Markovetz, M.R.; Garbarine, I.C.; Forest, M.G.; et al. Mucus accumulation in the lungs precedes structural changes and infection in children with cystic fibrosis. Sci. Transl. Med. 2019, 11, eaav3488. [Google Scholar] [CrossRef]
- Zemanick, E.T.; Hoffman, L.R. Cystic Fibrosis: Microbiology and Host Response. Pediatr. Clin. N. Am. 2016, 63, 617–636. [Google Scholar] [CrossRef]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Lung infections associated with cystic fibrosis. Clin. Microbiol. Rev. 2002, 15, 194–222. [Google Scholar] [CrossRef]
- Li, Z.; Kosorok, M.R.; Farrell, P.M.; Laxova, A.; West, S.E.; Green, C.G.; Collins, J.; Rock, M.J.; Splaingard, M.L. Longitudinal development of mucoid Pseudomonas aeruginosa infection and lung disease progression in children with cystic fibrosis. JAMA 2005, 293, 581–588. [Google Scholar] [CrossRef]
- Mayer-Hamblett, N.; Ramsey, B.W.; Kulasekara, H.D.; Wolter, D.J.; Houston, L.S.; Pope, C.E.; Kulasekara, B.R.; Armbruster, C.R.; Burns, J.L.; Retsch-Bogart, G.; et al. Pseudomonas aeruginosa phenotypes associated with eradication failure in children with cystic fibrosis. Clin. Infect. Dis. 2014, 59, 624–631. [Google Scholar] [CrossRef]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef]
- Davies, J.C.; Wainwright, C.E.; Canny, G.J.; Chilvers, M.A.; Howenstine, M.S.; Munck, A.; Mainz, J.G.; Rodriguez, S.; Li, H.; Yen, K.; et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 2013, 187, 1219–1225. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Daines, C.; Ringshausen, F.C.; Kerem, E.; Wilson, J.; Tullis, E.; Nair, N.; Simard, C.; Han, L.; Ingenito, E.P.; et al. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035. [Google Scholar] [CrossRef] [PubMed]
- Cholon, D.M.; Quinney, N.L.; Fulcher, M.L.; Esther, C.R., Jr.; Das, J.; Dokholyan, N.V.; Randell, S.H.; Boucher, R.C.; Gentzsch, M. Potentiator ivacaftor abrogates pharmacological correction of DeltaF508 CFTR in cystic fibrosis. Sci. Transl. Med. 2014, 6, 246ra296. [Google Scholar] [CrossRef]
- Veit, G.; Avramescu, R.G.; Perdomo, D.; Phuan, P.W.; Bagdany, M.; Apaja, P.M.; Borot, F.; Szollosi, D.; Wu, Y.S.; Finkbeiner, W.E.; et al. Some gating potentiators, including VX-770, diminish DeltaF508-CFTR functional expression. Sci. Transl. Med. 2014, 6, 246ra297. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Zemanick, E.T.; Taylor-Cousar, J.L.; Davies, J.; Gibson, R.L.; Mall, M.A.; McKone, E.F.; McNally, P.; Ramsey, B.W.; Rayment, J.H.; Rowe, S.M.; et al. A Phase 3 Open-Label Study of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 through 11 Years of Age with Cystic Fibrosis and at Least One F508del Allele. Am. J. Respir. Crit. Care Med. 2021, 203, 1522–1532. [Google Scholar] [CrossRef]
- Mall, M.A.; Mayer-Hamblett, N.; Rowe, S.M. Cystic Fibrosis: Emergence of Highly Effective Targeted Therapeutics and Potential Clinical Implications. Am. J. Respir. Crit. Care Med. 2020, 201, 1193–1208. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Hug, M.J.; Sommerburg, O.; Hirtz, S.; Hentschel, J.; Heinzmann, A.; Dopfer, C.; Schulz, A.; Mainz, J.G.; Tummler, B.; et al. Intestinal Current Measurements Detect Activation of Mutant CFTR in Patients with Cystic Fibrosis with the G551D Mutation Treated with Ivacaftor. Am. J. Respir. Crit. Care Med. 2015, 192, 1252–1255. [Google Scholar] [CrossRef] [PubMed]
- Accurso, F.J.; Rowe, S.M.; Clancy, J.P.; Boyle, M.P.; Dunitz, J.M.; Durie, P.R.; Sagel, S.D.; Hornick, D.B.; Konstan, M.W.; Donaldson, S.H.; et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N. Engl. J. Med. 2010, 363, 1991–2003. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; McCann, J.D.; Anderson, M.P.; Clancy, J.P.; Liedtke, C.M.; Nairn, A.C.; Greengard, P.; Welsch, M.J. Regulation of chloride channels by protein kinase C in normal and cystic fibrosis airway epithelia. Science 1989, 244, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, S.H.; Laube, B.L.; Corcoran, T.E.; Bhambhvani, P.; Zeman, K.; Ceppe, A.; Zeitlin, P.L.; Mogayzel, P.J., Jr.; Boyle, M.; Locke, L.W.; et al. Effect of ivacaftor on mucociliary clearance and clinical outcomes in cystic fibrosis patients with G551D-CFTR. JCI Insight 2018, 3, e122695. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Heltshe, S.L.; Gonska, T.; Donaldson, S.H.; Borowitz, D.; Gelfond, D.; Sagel, S.D.; Khan, U.; Mayer-Hamblett, N.; Van Dalfsen, J.M.; et al. Clinical mechanism of the cystic fibrosis transmembrane conductance regulator potentiator ivacaftor in G551D-mediated cystic fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 175–184. [Google Scholar] [CrossRef]
- Hayes, D., Jr.; Long, F.R.; McCoy, K.S.; Sheikh, S.I. Improvement in bronchiectasis on CT imaging in a pediatric patient with cystic fibrosis on ivacaftor therapy. Respiration 2014, 88, 345. [Google Scholar] [CrossRef]
- Hayes, D., Jr.; McCoy, K.S.; Sheikh, S.I. Improvement of sinus disease in cystic fibrosis with ivacaftor therapy. Am. J. Respir. Crit. Care Med. 2014, 190, 468. [Google Scholar] [CrossRef]
- Sheikh, S.I.; Long, F.R.; McCoy, K.S.; Johnson, T.; Ryan-Wenger, N.A.; Hayes, D., Jr. Computed tomography correlates with improvement with ivacaftor in cystic fibrosis patients with G551D mutation. J. Cyst. Fibros. 2015, 14, 84–89. [Google Scholar] [CrossRef]
- Hayes, D., Jr.; Long, F.R.; McCoy, K.S.; Sheikh, S.I. CT imaging of pediatric patients with cystic fibrosis on ivacaftor therapy. Lung 2014, 192, 823–824. [Google Scholar] [CrossRef]
- Hoare, S.; McEvoy, S.; McCarthy, C.J.; Kilcoyne, A.; Brady, D.; Gibney, B.; Gallagher, C.G.; McKone, E.F.; Dodd, J.D. Ivacaftor imaging response in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2014, 189, 484. [Google Scholar] [CrossRef]
- Chang, E.H.; Tang, X.X.; Shah, V.S.; Launspach, J.L.; Ernst, S.E.; Hilkin, B.; Karp, P.H.; Abou Alaiwa, M.H.; Graham, S.M.; Hornick, D.B.; et al. Medical reversal of chronic sinusitis in a cystic fibrosis patient with ivacaftor. Int. Forum Allergy Rhinol. 2015, 5, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, S.I.; Long, F.R.; McCoy, K.S.; Johnson, T.; Ryan-Wenger, N.A.; Hayes, D., Jr. Ivacaftor improves appearance of sinus disease on computerised tomography in cystic fibrosis patients with G551D mutation. Clin. Otolaryngol. 2015, 40, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Gelfond, D.; Heltshe, S.; Ma, C.; Rowe, S.M.; Frederick, C.; Uluer, A.; Sicilian, L.; Konstan, M.; Tullis, E.; Roach, R.N.; et al. Impact of CFTR Modulation on Intestinal pH, Motility, and Clinical Outcomes in Patients With Cystic Fibrosis and the G551D Mutation. Clin. Transl. Gastroenterol. 2017, 8, e81. [Google Scholar] [CrossRef] [PubMed]
- Bellin, M.D.; Laguna, T.; Leschyshyn, J.; Regelmann, W.; Dunitz, J.; Billings, J.; Moran, A. Insulin secretion improves in cystic fibrosis following ivacaftor correction of CFTR: A small pilot study. Pediatr. Diabetes 2013, 14, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Tsabari, R.; Elyashar, H.I.; Cymberknowh, M.C.; Breuer, O.; Armoni, S.; Livnat, G.; Kerem, E.; Zangen, D.H. CFTR potentiator therapy ameliorates impaired insulin secretion in CF patients with a gating mutation. J. Cyst. Fibros. 2016, 15, e25–e27. [Google Scholar] [CrossRef]
- Hayes, D., Jr.; McCoy, K.S.; Sheikh, S.I. Resolution of cystic fibrosis-related diabetes with ivacaftor therapy. Am. J. Respir. Crit. Care Med. 2014, 190, 590–591. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Hisert, K.B.; Heltshe, S.L.; Pope, C.; Jorth, P.; Wu, X.; Edwards, R.M.; Radey, M.; Accurso, F.J.; Wolter, D.J.; Cooke, G.; et al. Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function Reduces Airway Bacteria and Inflammation in People with Cystic Fibrosis and Chronic Lung Infections. Am. J. Respir. Crit. Care Med. 2017, 195, 1617–1628. [Google Scholar] [CrossRef]
- Heltshe, S.L.; Mayer-Hamblett, N.; Burns, J.L.; Khan, U.; Baines, A.; Ramsey, B.W.; Rowe, S.M.; GOAL (The G551D Observation-AL) Investigators of the Cystic Fibrosis Foundation Therapeutics Development Network. Pseudomonas aeruginosa in cystic fibrosis patients with G551D-CFTR treated with ivacaftor. Clin. Infect. Dis. 2015, 60, 703–712. [Google Scholar] [CrossRef]
- Nichols, D.P.; Morgan, S.J.; Skalland, M.; Vo, A.T.; Van Dalfsen, J.M.; Singh, S.B.; Ni, W.; Hoffman, L.R.; McGeer, K.; Heltshe, S.L.; et al. Pharmacologic improvement of CFTR function rapidly decreases sputum pathogen density, but lung infections generally persist. J. Clin. Investig. 2023, 133, e167957. [Google Scholar] [CrossRef]
- Schaupp, L.; Addante, A.; Voller, M.; Fentker, K.; Kuppe, A.; Bardua, M.; Duerr, J.; Piehler, L.; Rohmel, J.; Thee, S.; et al. Longitudinal Effects of Elexacaftor/Tezacaftor/Ivacaftor on Sputum Viscoelastic Properties, Airway Infection and Inflammation in Patients with Cystic Fibrosis. Eur. Respir. J. 2023, 62, 2202153. [Google Scholar] [CrossRef] [PubMed]
- Caverly, L.J.; Riquelme, S.A.; Hisert, K.B. The Impact of Highly Effective Modulator Therapy on Cystic Fibrosis Microbiology and Inflammation. Clin. Chest Med. 2022, 43, 647–665. [Google Scholar] [CrossRef] [PubMed]
- Stanton, B.A.; Coutermarsh, B.; Barnaby, R.; Hogan, D. Pseudomonas aeruginosa Reduces VX-809 Stimulated F508del-CFTR Chloride Secretion by Airway Epithelial Cells. PLoS ONE 2015, 10, e0127742. [Google Scholar] [CrossRef] [PubMed]
- Rubino, R.; Bezzerri, V.; Favia, M.; Facchini, M.; Tebon, M.; Singh, A.K.; Riederer, B.; Seidler, U.; Iannucci, A.; Bragonzi, A.; et al. Pseudomonas aeruginosa reduces the expression of CFTR via post-translational modification of NHERF1. Pflug. Arch. 2014, 466, 2269–2278. [Google Scholar] [CrossRef]
- Swiatecka-Urban, A.; Moreau-Marquis, S.; Maceachran, D.P.; Connolly, J.P.; Stanton, C.R.; Su, J.R.; Barnaby, R.; O’Toole, G.A.; Stanton, B.A. Pseudomonas aeruginosa inhibits endocytic recycling of CFTR in polarized human airway epithelial cells. Am. J. Physiol. Cell Physiol. 2006, 290, C862–C872. [Google Scholar] [CrossRef]
- Hirsch, M.J.; Hughes, E.M.; Easter, M.M.; Bollenbecker, S.E.; Howze Iv, P.H.; Birket, S.E.; Barnes, J.W.; Kiedrowski, M.R.; Krick, S. A novel in vitro model to study prolonged Pseudomonas aeruginosa infection in the cystic fibrosis bronchial epithelium. PLoS ONE 2023, 18, e0288002. [Google Scholar] [CrossRef]
- Malet, J.K.; Hennemann, L.C.; Hua, E.M.L.; Faure, E.; Waters, V.; Rousseau, S.; Nguyen, D. A Model of Intracellular Persistence of Pseudomonas aeruginosa in Airway Epithelial Cells. Cell. Microbiol. 2022, 2022, 5431666. [Google Scholar] [CrossRef]
- Hild, M.; Jaffe, A.B. Production of 3-D Airway Organoids From Primary Human Airway Basal Cells and Their Use in High-Throughput Screening. Curr. Protoc. Stem Cell Biol. 2016, 37, IE.9.1–IE.9.15. [Google Scholar] [CrossRef]
- Fulcher, M.L.; Gabriel, S.; Burns, K.A.; Yankaskas, J.R.; Randell, S.H. Well-differentiated human airway epithelial cell cultures. Methods Mol. Med. 2005, 107, 183–206. [Google Scholar] [CrossRef]
- Fulcher, M.L.; Randell, S.H. Human nasal and tracheo-bronchial respiratory epithelial cell culture. Methods Mol. Biol. 2013, 945, 109–121. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2015; Volume 35, p. M07-A11. [Google Scholar]
- Hull-Ryde, E.A.; Minges, J.T.; Martino, M.E.B.; Kato, T.; Norris-Drouin, J.L.; Ribeiro, C.M.P. IRE1alpha Is a Therapeutic Target for Cystic Fibrosis Airway Inflammation. Int. J. Mol. Sci. 2021, 22, 3063. [Google Scholar] [CrossRef] [PubMed]
- Filkins, L.M.; Graber, J.A.; Olson, D.G.; Dolben, E.L.; Lynd, L.R.; Bhuju, S.; O’Toole, G.A. Coculture of Staphylococcus aureus with Pseudomonas aeruginosa Drives S. aureus towards Fermentative Metabolism and Reduced Viability in a Cystic Fibrosis Model. J. Bacteriol. 2015, 197, 2252–2264. [Google Scholar] [CrossRef]
- Anderson, G.G.; Moreau-Marquis, S.; Stanton, B.A.; O’Toole, G.A. In vitro analysis of tobramycin-treated Pseudomonas aeruginosa biofilms on cystic fibrosis-derived airway epithelial cells. Infect. Immun. 2008, 76, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Garnett, J.P.; Kalsi, K.K.; Sobotta, M.; Bearham, J.; Carr, G.; Powell, J.; Brodlie, M.; Ward, C.; Tarran, R.; Baines, D.L. Hyperglycaemia and Pseudomonas aeruginosa acidify cystic fibrosis airway surface liquid by elevating epithelial monocarboxylate transporter 2 dependent lactate-H(+) secretion. Sci. Rep. 2016, 6, 37955. [Google Scholar] [CrossRef]
- Heiniger, R.W.; Winther-Larsen, H.C.; Pickles, R.J.; Koomey, M.; Wolfgang, M.C. Infection of human mucosal tissue by Pseudomonas aeruginosa requires sequential and mutually dependent virulence factors and a novel pilus-associated adhesin. Cell. Microbiol. 2010, 12, 1158–1173. [Google Scholar] [CrossRef] [PubMed]
- Worlitzsch, D.; Tarran, R.; Ulrich, M.; Schwab, U.; Cekici, A.; Meyer, K.C.; Birrer, P.; Bellon, G.; Berger, J.; Weiss, T.; et al. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J. Clin. Investig. 2002, 109, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Baltimore, R.S.; Christie, C.D.; Smith, G.J. Immunohistopathologic localization of Pseudomonas aeruginosa in lungs from patients with cystic fibrosis. Implications for the pathogenesis of progressive lung deterioration. Am. Rev. Respir. Dis. 1989, 140, 1650–1661. [Google Scholar] [CrossRef]
- Halldorsson, S.; Gudjonsson, T.; Gottfredsson, M.; Singh, P.K.; Gudmundsson, G.H.; Baldursson, O. Azithromycin maintains airway epithelial integrity during Pseudomonas aeruginosa infection. Am. J. Respir. Cell Mol. Biol. 2010, 42, 62–68. [Google Scholar] [CrossRef]
- Ruddy, J.; Emerson, J.; Moss, R.; Genatossio, A.; McNamara, S.; Burns, J.L.; Anderson, G.; Rosenfeld, M. Sputum tobramycin concentrations in cystic fibrosis patients with repeated administration of inhaled tobramycin. J. Aerosol Med. Pulm. Drug Deliv. 2013, 26, 69–75. [Google Scholar] [CrossRef]
- Cheer, S.M.; Waugh, J.; Noble, S. Inhaled tobramycin (TOBI): A review of its use in the management of Pseudomonas aeruginosa infections in patients with cystic fibrosis. Drugs 2003, 63, 2501–2520. [Google Scholar] [CrossRef]
- Bjarnsholt, T.; Jensen, P.O.; Fiandaca, M.J.; Pedersen, J.; Hansen, C.R.; Andersen, C.B.; Pressler, T.; Givskov, M.; Hoiby, N. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr. Pulmonol. 2009, 44, 547–558. [Google Scholar] [CrossRef] [PubMed]
- DePas, W.H.; Starwalt-Lee, R.; Van Sambeek, L.; Ravindra Kumar, S.; Gradinaru, V.; Newman, D.K. Exposing the Three-Dimensional Biogeography and Metabolic States of Pathogens in Cystic Fibrosis Sputum via Hydrogel Embedding, Clearing, and rRNA Labeling. mBio 2016, 7, e00796-16. [Google Scholar] [CrossRef] [PubMed]
- Reznikov, L.R.; Abou Alaiwa, M.H.; Dohrn, C.L.; Gansemer, N.D.; Diekema, D.J.; Stoltz, D.A.; Welsh, M.J. Antibacterial properties of the CFTR potentiator ivacaftor. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2014, 13, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Saber, M.M.; Donner, J.; Levade, I.; Acosta, N.; Parkins, M.D.; Boyle, B.; Levesque, R.C.; Nguyen, D.; Shapiro, B.J. Single nucleotide variants in Pseudomonas aeruginosa populations from sputum correlate with baseline lung function and predict disease progression in individuals with cystic fibrosis. Microb. Genom. 2023, 9, mgen000981. [Google Scholar] [CrossRef]
- Iwanska, A.; Trafny, E.A.; Czopowicz, M.; Augustynowicz-Kopec, E. Phenotypic and genotypic characteristics of Pseudomonas aeruginosa isolated from cystic fibrosis patients with chronic infections. Sci. Rep. 2023, 13, 11741. [Google Scholar] [CrossRef]
- Stanton, B.A. Effects of Pseudomonas aeruginosa on CFTR chloride secretion and the host immune response. Am. J. Physiol. Cell Physiol. 2017, 312, C357–C366. [Google Scholar] [CrossRef]
- Abou Alaiwa, M.H.; Reznikov, L.R.; Gansemer, N.D.; Sheets, K.A.; Horswill, A.R.; Stoltz, D.A.; Zabner, J.; Welsh, M.J. pH modulates the activity and synergism of the airway surface liquid antimicrobials beta-defensin-3 and LL-37. Proc. Natl. Acad. Sci. USA 2014, 111, 18703–18708. [Google Scholar] [CrossRef]
- Guimbellot, J.S.; Acosta, E.P.; Rowe, S.M. Sensitivity of ivacaftor to drug-drug interactions with rifampin, a cytochrome P450 3A4 inducer. Pediatr. Pulmonol. 2018, 53, E6–E8. [Google Scholar] [CrossRef]
- Schneider, E.K.; Azad, M.A.; Han, M.L.; Tony Zhou, Q.; Wang, J.; Huang, J.X.; Cooper, M.A.; Doi, Y.; Baker, M.A.; Bergen, P.J.; et al. An “Unlikely” Pair: The Antimicrobial Synergy of Polymyxin B in Combination with the Cystic Fibrosis Transmembrane Conductance Regulator Drugs KALYDECO and ORKAMBI. ACS Infect. Dis. 2016, 2, 478–488. [Google Scholar] [CrossRef]
- Cho, D.Y.; Lim, D.J.; Mackey, C.; Skinner, D.; Zhang, S.; McCormick, J.; Woodworth, B.A. Ivacaftor, a Cystic Fibrosis Transmembrane Conductance Regulator Potentiator, Enhances Ciprofloxacin Activity Against Pseudomonas aeruginosa. Am. J. Rhinol. Allergy 2019, 33, 129–136. [Google Scholar] [CrossRef]
- Payne, J.E.; Dubois, A.V.; Ingram, R.J.; Weldon, S.; Taggart, C.C.; Elborn, J.S.; Tunney, M.M. Activity of innate antimicrobial peptides and ivacaftor against clinical cystic fibrosis respiratory pathogens. Int. J. Antimicrob. Agents 2017, 50, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Cigana, C.; Giannella, R.; Colavolpe, A.; Alcala-Franco, B.; Mancini, G.; Colombi, F.; Bigogno, C.; Bastrup, U.; Bertoni, G.; Bragonzi, A. Mutual Effects of Single and Combined CFTR Modulators and Bacterial Infection in Cystic Fibrosis. Microbiol. Spectr. 2023, 11, e0408322. [Google Scholar] [CrossRef]
- Henderson, A.G.; Ehre, C.; Button, B.; Abdullah, L.H.; Cai, L.H.; Leigh, M.W.; DeMaria, G.C.; Matsui, H.; Donaldson, S.H.; Davis, C.W.; et al. Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J. Clin. Investig. 2014, 124, 3047–3060. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Wagner, V.E.; Hill, D.B.; Schwab, U.E.; Rogers, T.D.; Button, B.; Taylor, R.M., 2nd; Superfine, R.; Rubinstein, M.; Iglewski, B.H.; et al. A physical linkage between cystic fibrosis airway surface dehydration and Pseudomonas aeruginosa biofilms. Proc. Natl. Acad. Sci. USA 2006, 103, 18131–18136. [Google Scholar] [CrossRef]
- Tarran, R.; Grubb, B.R.; Parsons, D.; Picher, M.; Hirsh, A.J.; Davis, C.W.; Boucher, R.C. The CF salt controversy: In vivo observations and therapeutic approaches. Mol. Cell 2001, 8, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.B.; Shaffer, K.M.; Araba, K.C.; Markovetz, M.R.; Wykoff, J.A.; Quinney, N.L.; Hao, S.; Delion, M.F.; Flen, A.L.; Morton, L.C.; et al. Treatment of cystic fibrosis airway cells with CFTR modulators reverses aberrant mucus properties via hydration. Eur. Respir. J. 2022, 59, 2100185. [Google Scholar] [CrossRef]
- Okuda, K.; Shaffer, K.M.; Ehre, C. Mucins and CFTR: Their Close Relationship. Int. J. Mol. Sci. 2022, 23, 10232. [Google Scholar] [CrossRef]
- LeSimple, P.; Liao, J.; Robert, R.; Gruenert, D.C.; Hanrahan, J.W. Cystic fibrosis transmembrane conductance regulator trafficking modulates the barrier function of airway epithelial cell monolayers. J. Physiol. 2010, 588, 1195–1209. [Google Scholar] [CrossRef]
- Simonin, J.L.; Luscher, A.; Losa, D.; Badaoui, M.; van Delden, C.; Kohler, T.; Chanson, M. Surface Hydration Protects Cystic Fibrosis Airways from Infection by Restoring Junctional Networks. Cells 2022, 11, 1587. [Google Scholar] [CrossRef]
- Boucher, R.C.; Stutts, M.J.; Knowles, M.R.; Cantley, L.; Gatzy, J.T. Na+ transport in cystic fibrosis respiratory epithelia. Abnormal basal rate and response to adenylate cyclase activation. J. Clin. Investig. 1986, 78, 1245–1252. [Google Scholar] [CrossRef]
- Zhang, L.; Button, B.; Gabriel, S.E.; Burkett, S.; Yan, Y.; Skiadopoulos, M.H.; Dang, Y.L.; Vogel, L.N.; McKay, T.; Mengos, A.; et al. CFTR delivery to 25% of surface epithelial cells restores normal rates of mucus transport to human cystic fibrosis airway epithelium. PLoS Biol. 2009, 7, e1000155. [Google Scholar] [CrossRef]
- Tarran, R.; Button, B.; Picher, M.; Paradiso, A.M.; Ribeiro, C.M.; Lazarowski, E.R.; Zhang, L.; Collins, P.L.; Pickles, R.J.; Fredberg, J.J.; et al. Normal and cystic fibrosis airway surface liquid homeostasis. The effects of phasic shear stress and viral infections. J. Biol. Chem. 2005, 280, 35751–35759. [Google Scholar] [CrossRef] [PubMed]
- Solomon, G.M.; Fu, L.; Rowe, S.M.; Collawn, J.F. The therapeutic potential of CFTR modulators for COPD and other airway diseases. Curr. Opin. Pharmacol. 2017, 34, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.D.; Bono, T.R.; Rowe, S.M.; Solomon, G.M. CFTR targeted therapies: Recent advances in cystic fibrosis and possibilities in other diseases of the airways. Eur. Respir. Rev. 2020, 29, 190068. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.M.; Paradiso, A.M.; Schwab, U.; Perez-Vilar, J.; Jones, L.; O’Neal, W.; Boucher, R.C. Chronic airway infection/inflammation induces a Ca2+i-dependent hyperinflammatory response in human cystic fibrosis airway epithelia. J. Biol. Chem. 2005, 280, 17798–17806. [Google Scholar] [CrossRef] [PubMed]
- Gentzsch, M.; Cholon, D.M.; Quinney, N.L.; Boyles, S.E.; Martino, M.E.B.; Ribeiro, C.M.P. The Cystic Fibrosis Airway Milieu Enhances Rescue of F508del in a Pre-Clinical Model. Eur. Respir. J. 2018, 52, 1801133. [Google Scholar] [CrossRef]
- Gentzsch, M.; Cholon, D.M.; Quinney, N.L.; Martino, M.E.B.; Minges, J.T.; Boyles, S.E.; Guhr Lee, T.N.; Esther, C.R., Jr.; Ribeiro, C.M.P. Airway Epithelial Inflammation In Vitro Augments the Rescue of Mutant CFTR by Current CFTR Modulator Therapies. Front. Pharmacol. 2021, 12, 628722. [Google Scholar] [CrossRef]
- Jaudszus, A.; Arnold, C.; Hentschel, J.; Hunniger, K.; Baier, M.; Mainz, J.G. Increased cytokines in cystic fibrosis patients’ upper airways during a new P. aeruginosa colonization. Pediatr. Pulmonol. 2018, 53, 881–887. [Google Scholar] [CrossRef]
- Saint-Criq, V.; Villeret, B.; Bastaert, F.; Kheir, S.; Hatton, A.; Cazes, A.; Xing, Z.; Sermet-Gaudelus, I.; Garcia-Verdugo, I.; Edelman, A.; et al. Pseudomonas aeruginosa LasB protease impairs innate immunity in mice and humans by targeting a lung epithelial cystic fibrosis transmembrane regulator-IL-6-antimicrobial-repair pathway. Thorax 2018, 73, 49–61. [Google Scholar] [CrossRef]
- Muhlebach, M.S.; Reed, W.; Noah, T.L. Quantitative cytokine gene expression in CF airway. Pediatr. Pulmonol. 2004, 37, 393–399. [Google Scholar] [CrossRef]
- Bonfield, T.L.; Panuska, J.R.; Konstan, M.W.; Hilliard, K.A.; Hilliard, J.B.; Ghnaim, H.; Berger, M. Inflammatory cytokines in cystic fibrosis lungs. Am. J. Respir. Crit. Care Med. 1995, 152, 2111–2118. [Google Scholar] [CrossRef] [PubMed]
- Dean, T.P.; Dai, Y.; Shute, J.K.; Church, M.K.; Warner, J.O. Interleukin-8 concentrations are elevated in bronchoalveolar lavage, sputum, and sera of children with cystic fibrosis. Pediatr. Res. 1993, 34, 159–161. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cholon, D.M.; Greenwald, M.A.; Higgs, M.G.; Quinney, N.L.; Boyles, S.E.; Meinig, S.L.; Minges, J.T.; Chaubal, A.; Tarran, R.; Ribeiro, C.M.P.; et al. A Novel Co-Culture Model Reveals Enhanced CFTR Rescue in Primary Cystic Fibrosis Airway Epithelial Cultures with Persistent Pseudomonas aeruginosa Infection. Cells 2023, 12, 2618. https://doi.org/10.3390/cells12222618
Cholon DM, Greenwald MA, Higgs MG, Quinney NL, Boyles SE, Meinig SL, Minges JT, Chaubal A, Tarran R, Ribeiro CMP, et al. A Novel Co-Culture Model Reveals Enhanced CFTR Rescue in Primary Cystic Fibrosis Airway Epithelial Cultures with Persistent Pseudomonas aeruginosa Infection. Cells. 2023; 12(22):2618. https://doi.org/10.3390/cells12222618
Chicago/Turabian StyleCholon, Deborah M., Matthew A. Greenwald, Matthew G. Higgs, Nancy L. Quinney, Susan E. Boyles, Suzanne L. Meinig, John T. Minges, Ashlesha Chaubal, Robert Tarran, Carla M. P. Ribeiro, and et al. 2023. "A Novel Co-Culture Model Reveals Enhanced CFTR Rescue in Primary Cystic Fibrosis Airway Epithelial Cultures with Persistent Pseudomonas aeruginosa Infection" Cells 12, no. 22: 2618. https://doi.org/10.3390/cells12222618
APA StyleCholon, D. M., Greenwald, M. A., Higgs, M. G., Quinney, N. L., Boyles, S. E., Meinig, S. L., Minges, J. T., Chaubal, A., Tarran, R., Ribeiro, C. M. P., Wolfgang, M. C., & Gentzsch, M. (2023). A Novel Co-Culture Model Reveals Enhanced CFTR Rescue in Primary Cystic Fibrosis Airway Epithelial Cultures with Persistent Pseudomonas aeruginosa Infection. Cells, 12(22), 2618. https://doi.org/10.3390/cells12222618