
Liquid Biopsy in Alzheimer’s Disease Patients Reveals Epigenetic Changes in the PRLHR Gene

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Hippocampal Samples
2.2. Validation of PRLHR Methylation Changes in Hippocampal Samples Using Bisulfite Cloning Sequencing
2.3. PRLHR mRNA Expression Analysis Using Real-Time Quantitative PCR (RT-qPCR)
2.4. Plasma Samples
2.5. PRLHR Methylation Measurement in Plasma cfDNA Using Pyrosequencing
2.6. Statistical Analysis
3. Results
3.1. Samples Characteristics
3.2. DNA Methylation Levels in PRLHR Are Increased in Hippocampus of AD Patients Compared to Controls
3.3. PRLHR Gene Expression Is Decreased in AD Hippocampus Compared to Controls
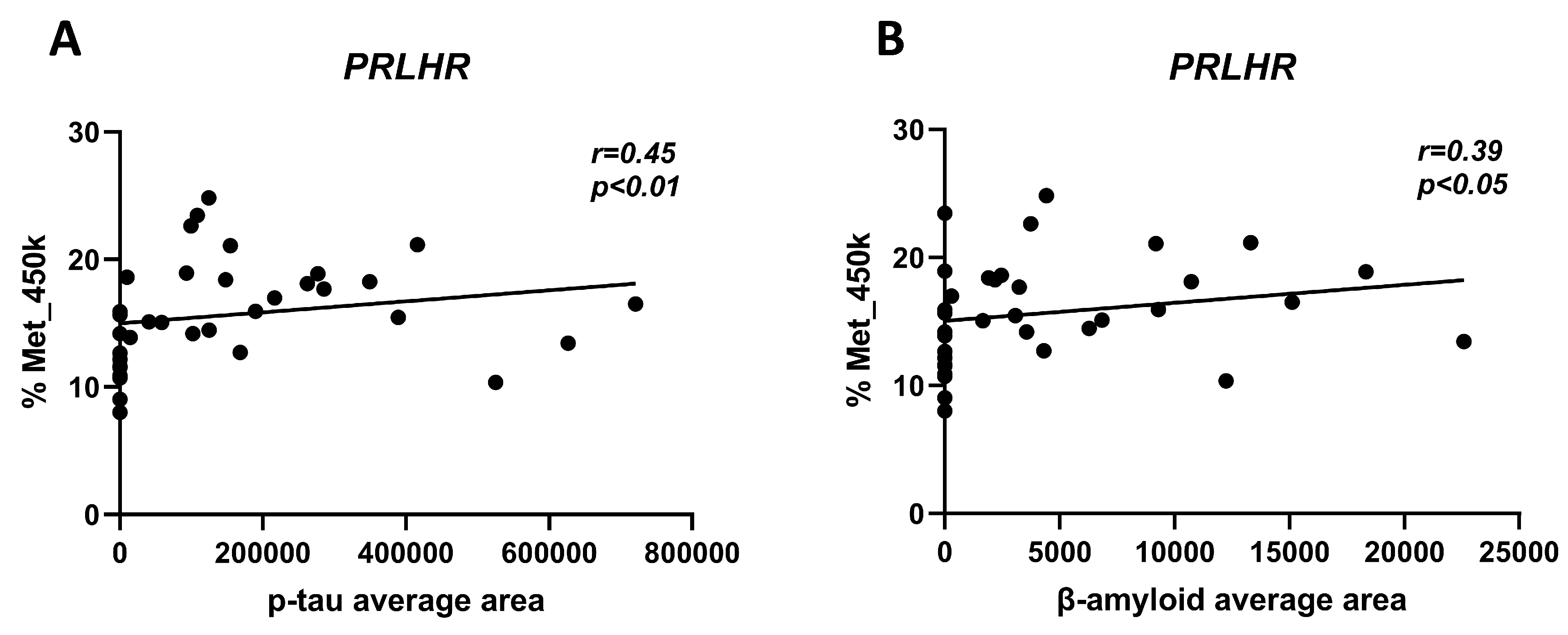
3.4. Correlation of PRLHR Methylation Levels in Hippocampal Samples with β-Amyloid and p-tau Deposits
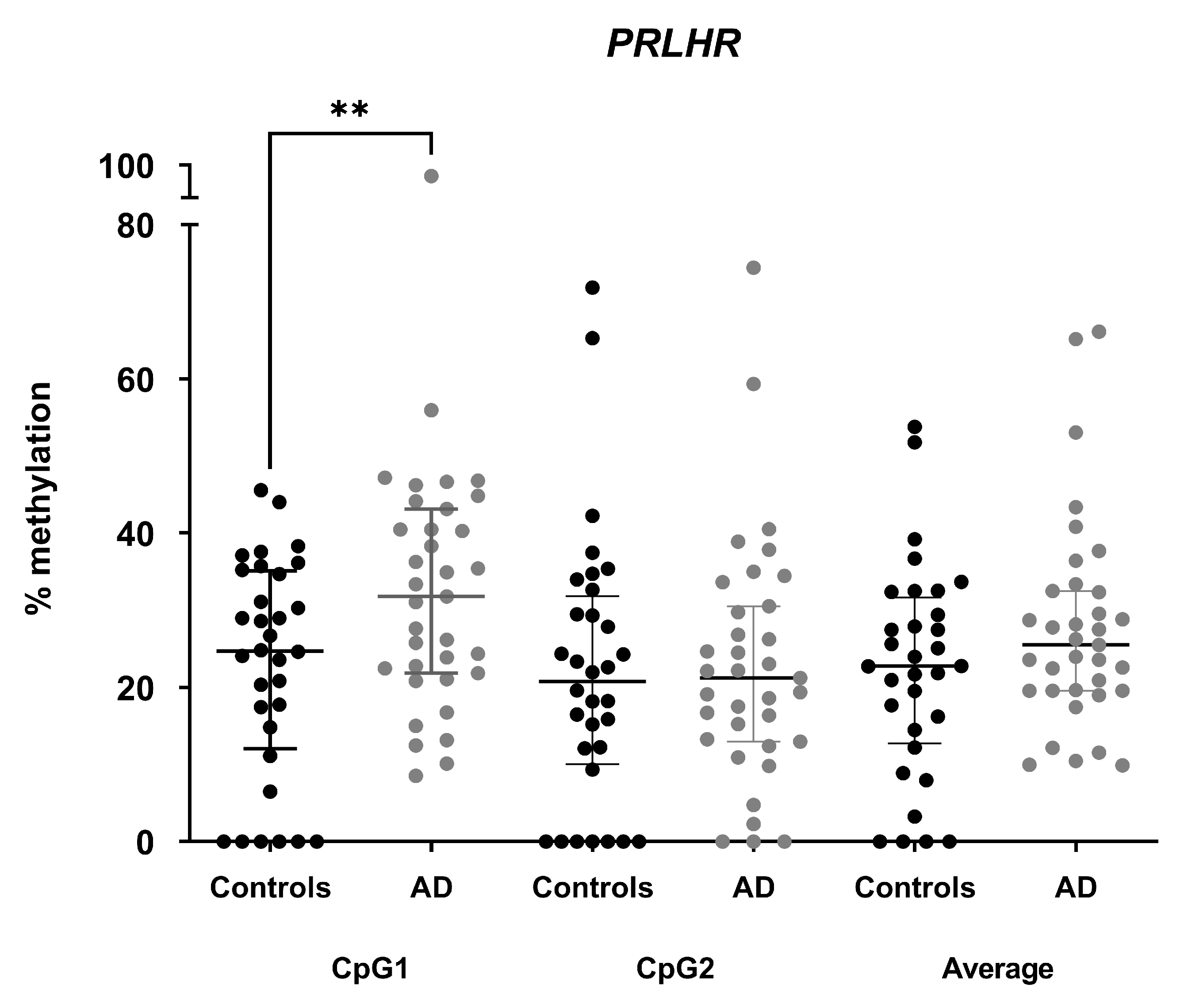
3.5. Methylation Differences in PRLHR Can Be Detected in Plasma cfDNA of AD Patients Compared to Controls
3.6. Correlation of PRLHR Methylation Levels in cfDNA with Clinical Parameters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef]
- Pražienková, V.; Popelová, A.; Kuneš, J.; Maletínská, L. Prolactin-Releasing Peptide: Physiological and Pharmacological Properties. Int. J. Mol. Sci. 2019, 20, 5297. [Google Scholar] [CrossRef]
- Sanchez-Mut, J.V.; Gräff, J. Epigenetic Alterations in Alzheime’s Disease. Front. Behav. Neurosci. 2015, 9, 347. [Google Scholar] [CrossRef]
- Lunnon, K.; Smith, R.; Hannon, E.; De Jager, P.L.; Srivastava, G.; Volta, M.; Troakes, C.; Al-Sarraj, S.; Burrage, J.; Macdonald, R.; et al. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat. Neurosci. 2014, 17, 1164–1170. [Google Scholar] [CrossRef]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef]
- Smith, R.G.; Hannon, E.; De Jager, P.L.; Chibnik, L.; Lott, S.J.; Condliffe, D.; Smith, A.R.; Haroutunian, V.; Troakes, C.; Al-Sarraj, S.; et al. Elevated DNA methylation across a 48-kb region spanning the HOXA gene cluster is associated with Alzheimer’s disease neuropathology. Alzheimer’s Dement. 2018, 14, 1580–1588. [Google Scholar] [CrossRef]
- Li, Q.S.; Sun, Y.; Wang, T. Epigenome-wide association study of Alzheimer’s disease replicates 22 differentially methylated positions and 30 differentially methylated regions. Clin. Epigenet. 2020, 12, 149. [Google Scholar] [CrossRef]
- Smith, A.R.; Smith, R.G.; Condliffe, D.; Hannon, E.; Schalkwyk, L.; Mill, J.; Lunnon, K. Increased DNA methylation near TREM2 is consistently seen in the superior temporal gyrus in Alzheimer’s disease brain. Neurobiol. Aging 2016, 47, 35–40. [Google Scholar] [CrossRef]
- Zhang, L.; Silva, T.C.; Young, J.I.; Gomez, L.; Schmidt, M.A.; Hamilton-Nelson, K.L.; Kunkle, B.W.; Chen, X.; Martin, E.R.; Wang, L. Epigenome-wide meta-analysis of DNA methylation differences in prefrontal cortex implicates the immune processes in Alzheimer’s disease. Nat. Commun. 2020, 11, 6114. [Google Scholar] [CrossRef]
- Wenk, G.L. Neuropathologic changes in Alzheimer’s disease. J. Clin. Psychiatry 2003, 64 (Suppl. 9), 7–10. [Google Scholar]
- Altuna, M.; Urdanoz-Casado, A.; Sanchez-Ruiz de Gordoa, J.; Zelaya, M.V.; Labarga, A.; Lepesant, J.M.J.; Roldan, M.; Blanco-Luquin, I.; Perdones, A.; Larumbe, R.; et al. DNA methylation signature of human hippocampus in Alzheimer’s disease is linked to neurogenesis. Clin. Epigenet. 2019, 11, 91. [Google Scholar] [CrossRef]
- Gaitsch, H.; Franklin, R.J.M.; Reich, D.S. Cell-free DNA-based liquid biopsies in neurology. Brain 2022, 146, 1758–1774. [Google Scholar] [CrossRef]
- Southwood, D.; Singh, S.; Chatterton, Z. Brain-derived cell-free DNA. Neural Regen. Res. 2022, 17, 2213–2214. [Google Scholar] [CrossRef]
- Macías, M.; Alegre, E.; Díaz-Lagares, A.; Patiño, A.; Pérez-Gracia, J.L.; Sanmamed, M.; López-López, R.; Varo, N.; González, A. Liquid Biopsy: From Basic Research to Clinical Practice. Adv. Clin. Chem. 2018, 83, 73–119. [Google Scholar] [CrossRef]
- Crowley, E.; Di Nicolantonio, F.; Loupakis, F.; Bardelli, A. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013, 10, 472–484. [Google Scholar] [CrossRef]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. USA 2008, 105, 16266–16271. [Google Scholar] [CrossRef]
- Lehmann-Werman, R.; Neiman, D.; Zemmour, H.; Moss, J.; Magenheim, J.; Vaknin-Dembinsky, A.; Rubertsson, S.; Nellgård, B.; Blennow, K.; Zetterberg, H.; et al. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc. Natl. Acad. Sci. USA 2016, 113, E1826–E1834. [Google Scholar] [CrossRef]
- Noe, C.R.; Noe-Letschnig, M.; Handschuh, P.; Noe, C.A.; Lanzenberger, R. Dysfunction of the Blood-Brain Barrier-A Key Step in Neurodegeneration and Dementia. Front. Aging Neurosci. 2020, 12, 185. [Google Scholar] [CrossRef]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef]
- Chen, L.; Shen, Q.; Xu, S.; Yu, H.; Pei, S.; Zhang, Y.; He, X.; Wang, Q.; Li, D. 5-Hydroxymethylcytosine Signatures in Circulating Cell-Free DNA as Diagnostic Biomarkers for Late-Onset Alzheimer’s Disease. J. Alzheimer’s Dis. 2022, 85, 573–585. [Google Scholar] [CrossRef]
- Pai, M.C.; Kuo, Y.M.; Wang, I.F.; Chiang, P.M.; Tsai, K.J. The Role of Methylated Circulating Nucleic Acids as a Potential Biomarker in Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 2440–2449. [Google Scholar] [CrossRef]
- Guemri, J.; Pierre-Jean, M.; Brohard, S.; Oussada, N.; Horgues, C.; Bonnet, E.; Mauger, F.; Deleuze, J.F. Methylated ccfDNA from plasma biomarkers of Alzheimer’s disease using targeted bisulfite sequencing. Epigenomics 2022, 14, 451–468. [Google Scholar] [CrossRef]
- Mendioroz, M.; Martínez-Merino, L.; Blanco-Luquin, I.; Urdánoz, A.; Roldán, M.; Jericó, I. Liquid biopsy: A new source of candidate biomarkers in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 763–768. [Google Scholar] [CrossRef]
- Hinuma, S.; Habata, Y.; Fujii, R.; Kawamata, Y.; Hosoya, M.; Fukusumi, S.; Kitada, C.; Masuo, Y.; Asano, T.; Matsumoto, H.; et al. A prolactin-releasing peptide in the brain. Nature 1998, 393, 272–276. [Google Scholar] [CrossRef]
- Špolcová, A.; Mikulášková, B.; Holubová, M.; Nagelová, V.; Pirnik, Z.; Zemenová, J.; Haluzík, M.; Železná, B.; Galas, M.C.; Maletínská, L. Anorexigenic lipopeptides ameliorate central insulin signaling and attenuate tau phosphorylation in hippocampi of mice with monosodium glutamate-induced obesity. J. Alzheimer’s Dis. 2015, 45, 823–835. [Google Scholar] [CrossRef]
- Bell, J.E.; Alafuzoff, I.; Al-Sarraj, S.; Arzberger, T.; Bogdanovic, N.; Budka, H.; Dexter, D.T.; Falkai, P.; Ferrer, I.; Gelpi, E.; et al. Management of a twenty-first century brain bank: Experience in the BrainNet Europe consortium. Acta Neuropathol. 2008, 115, 497–507. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef]
- Celarain, N.; Sánchez-Ruiz de Gordoa, J.; Zelaya, M.V.; Roldán, M.; Larumbe, R.; Pulido, L.; Echavarri, C.; Mendioroz, M. TREM2 upregulation correlates with 5-hydroxymethycytosine enrichment in Alzheimer’s disease hippocampus. Clin. Epigenet. 2016, 8, 37. [Google Scholar] [CrossRef]
- Kurdyukov, S.; Bullock, M. DNA Methylation Analysis: Choosing the Right Method. Biology 2016, 5, 3. [Google Scholar] [CrossRef]
- The Blueprint consortium, Quantitative comparison of DNA methylation assays for biomarker development and clinical applications. Nat. Biotechnol. 2016, 34, 726–737. [CrossRef]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef]
- Li, L.C.; Dahiya, R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef]
- Kumaki, Y.; Oda, M.; Okano, M. QUMA: Quantification tool for methylation analysis. Nucleic Acids Res. 2008, 36, W170–W175. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Reisberg, B.; Ferris, S.H.; de Leon, M.J.; Crook, T. The Global Deterioration Scale for assessment of primary degenerative dementia. Am. J. Psychiatry 1982, 139, 1136–1139. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Affinito, O.; Palumbo, D.; Fierro, A.; Cuomo, M.; De Riso, G.; Monticelli, A.; Miele, G.; Chiariotti, L.; Cocozza, S. Nucleotide distance influences co-methylation between nearby CpG sites. Genomics 2020, 112, 144–150. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Aromolaran, K.A.; Zukin, R.S. The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat. Rev. Neurosci. 2017, 18, 347–361. [Google Scholar] [CrossRef]
- Bacalini, M.G.; Boattini, A.; Gentilini, D.; Giampieri, E.; Pirazzini, C.; Giuliani, C.; Fontanesi, E.; Remondini, D.; Capri, M.; Del Rio, A.; et al. A meta-analysis on age-associated changes in blood DNA methylation: Results from an original analysis pipeline for Infinium 450k data. Aging 2015, 7, 97–109. [Google Scholar] [CrossRef]
- Mansego, M.L.; Milagro, F.I.; Zulet, M.Á.; Moreno-Aliaga, M.J.; Martínez, J.A. Differential DNA Methylation in Relation to Age and Health Risks of Obesity. Int. J. Mol. Sci. 2015, 16, 16816–16832. [Google Scholar] [CrossRef]
- Tan, Q.; Heijmans, B.T.; Hjelmborg, J.V.; Soerensen, M.; Christensen, K.; Christiansen, L. Epigenetic drift in the aging genome: A ten-year follow-up in an elderly twin cohort. Int. J. Epidemiol. 2016, 45, 1146–1158. [Google Scholar] [CrossRef]
- Moreno-Jimenez, E.P.; Flor-Garcia, M.; Terreros-Roncal, J.; Rabano, A.; Cafini, F.; Pallas-Bazarra, N.; Avila, J.; Llorens-Martin, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef]
- Zmeškalová, A.; Popelová, A.; Exnerová, A.; Železná, B.; Kuneš, J.; Maletínská, L. Cellular Signaling and Anti-Apoptotic Effects of Prolactin-Releasing Peptide and Its Analog on SH-SY5Y Cells. Int. J. Mol. Sci. 2020, 21, 6343. [Google Scholar] [CrossRef]
- Mrazikova, L.; Hojna, S.; Pacesova, A.; Hruba, L.; Strnadova, V.; Neprasova, B.; Zelezna, B.; Kunes, J.; Maletinska, L. Palmitoylated prolactin-releasing peptide treatment had neuroprotective but not anti-obesity effect in fa/fa rats with leptin signaling disturbances. Nutr. Diabetes 2022, 12, 26. [Google Scholar] [CrossRef]
- Bjursell, M.; Lennerås, M.; Göransson, M.; Elmgren, A.; Bohlooly, Y.M. GPR10 deficiency in mice results in altered energy expenditure and obesity. Biochem. Biophys. Res. Commun. 2007, 363, 633–638. [Google Scholar] [CrossRef]
- Parhar, I.; Ogawa, S.; Kitahashi, T. RFamide peptides as mediators in environmental control of GnRH neurons. Prog. Neurobiol. 2012, 98, 176–196. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Ta, Q.T.H.; Nguyen, T.K.O.; Nguyen, T.T.D.; Giau, V.V. Type 3 Diabetes and Its Role Implications in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 3165. [Google Scholar] [CrossRef]
- Selman, A.; Burns, S.; Reddy, A.P.; Culberson, J.; Reddy, P.H. The Role of Obesity and Diabetes in Dementia. Int. J. Mol. Sci. 2022, 23, 9267. [Google Scholar] [CrossRef]
- Vargas-Soria, M.; Carranza-Naval, M.J.; Del Marco, A.; Garcia-Alloza, M. Role of liraglutide in Alzheimer’s disease pathology. Alzheimers Res. Ther. 2021, 13, 112. [Google Scholar] [CrossRef]
- Holubova, M.; Hruba, L.; Popelova, A.; Bencze, M.; Prazienkova, V.; Gengler, S.; Kratochvilova, H.; Haluzik, M.; Zelezna, B.; Kunes, J.; et al. Liraglutide and a lipidized analog of prolactin-releasing peptide show neuroprotective effects in a mouse model of beta-amyloid pathology. Neuropharmacology 2019, 144, 377–387. [Google Scholar] [CrossRef]
- Maletínská, L.; Popelová, A.; Železná, B.; Bencze, M.; Kuneš, J. The impact of anorexigenic peptides in experimental models of Alzheimer’s disease pathology. J. Endocrinol. 2019, 240, R47–R72. [Google Scholar] [CrossRef]
- Santamaria, B.A.; Corroza, J.; Gordoa, J.S.-R.d.; Cabello, C.; Robles, M.; Méndez-López, I.; Conde, M.M.; Zueco, S.; Roldan, M.; Urdánoz-Casado, A.; et al. Association of Blood-Based DNA Methylation Markers with Late-Onset Alzheimer Disease: A Potential Diagnostic Approach. Neurology 2023. Online ahead of print. [Google Scholar] [CrossRef]
- Nazarian, A.; Yashin, A.I.; Kulminski, A.M. Summary-Based Methylome-Wide Association Analyses Suggest Potential Genetically Driven Epigenetic Heterogeneity of Alzheimer’s Disease. J. Clin. Med. 2020, 9, 1489. [Google Scholar] [CrossRef]
- Monti, N.; Cavallaro, R.A.; Stoccoro, A.; Nicolia, V.; Scarpa, S.; Kovacs, G.G.; Fiorenza, M.T.; Lucarelli, M.; Aronica, E.; Ferrer, I.; et al. CpG and non-CpG Presenilin1 methylation pattern in course of neurodevelopment and neurodegeneration is associated with gene expression in human and murine brain. Epigenetics 2020, 15, 781–799. [Google Scholar] [CrossRef]
- Konki, M.; Lindgren, N.; Kylaniemi, M.; Venho, R.; Laajala, E.; Ghimire, B.; Lahesmaa, R.; Kaprio, J.; Rinne, J.O.; Lund, R.J. Plasma cell-free DNA methylation marks for episodic memory impairment: A pilot twin study. Sci. Rep. 2020, 10, 14192. [Google Scholar] [CrossRef]
- El Messaoudi, S.; Rolet, F.; Mouliere, F.; Thierry, A.R. Circulating cell free DNA: Preanalytical considerations. Clin. Chim. Acta 2013, 424, 222–230. [Google Scholar] [CrossRef]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef]
- de Mendoza, A.; Poppe, D.; Buckberry, S.; Pflueger, J.; Albertin, C.B.; Daish, T.; Bertrand, S.; de la Calle-Mustienes, E.; Gómez-Skarmeta, J.L.; Nery, J.R.; et al. The emergence of the brain non-CpG methylation system in vertebrates. Nat. Ecol. Evol. 2021, 5, 369–378. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotypical Features | Controls (n = 35) | AD Patients (n = 35) | p-Value |
|---|---|---|---|
| Median (IQR) | |||
| Age (years) | 77 (72–79) | 78 (773–783) | 0.154 |
| MMSE | 30 (29–30) | 22 (19–26) | <0.001 |
| GDS | 1 (1–1) | 4 (4–4) | <0.001 |
| cfDNA conc (ng/µL) | 2.1 (1.0–5.1) | 1.8 (0.7–5.5) | 0.445 |
| N (%) | |||
| Sex | 0.811 | ||
| Female | 17 (49) | 18 (51) | |
| Male | 18 (51) | 17 (49) |
| Variable | B | Wald | p-Value | OR | 95% C.I. for OR | |
|---|---|---|---|---|---|---|
| Hippocampus | MET_PRLHR_450K | 0.50 | 4.37 | 0.04 * | 1.65 | 1.03–2.63 |
| Age | −2.28 | 3.79 | 0.05 | 0.10 | 0.01–1.02 | |
| Sex (female) | −0.65 | 0.28 | 0.60 | 0.52 | 0.05–5.92 | |
| Constant | −4.89 | 2.05 | 0.15 | 0.01 | ||
| cfDNA | MET_PRLHR_CpG1 | 0.05 | 5.69 | 0.02 * | 1.05 | 1.01–1.09 |
| Age | 0.06 | 1.31 | 0.25 | 1.06 | 0.96–1.16 | |
| Sex (female) | 0.39 | 0.53 | 0.47 | 1.47 | 0.52–4.17 | |
| Constant | −5.64 | 2.20 | 0.14 | 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macías, M.; Acha, B.; Corroza, J.; Urdánoz-Casado, A.; Roldan, M.; Robles, M.; Sánchez-Ruiz de Gordoa, J.; Erro, M.E.; Jericó, I.; Blanco-Luquin, I.; et al. Liquid Biopsy in Alzheimer’s Disease Patients Reveals Epigenetic Changes in the PRLHR Gene. Cells 2023, 12, 2679. https://doi.org/10.3390/cells12232679
Macías M, Acha B, Corroza J, Urdánoz-Casado A, Roldan M, Robles M, Sánchez-Ruiz de Gordoa J, Erro ME, Jericó I, Blanco-Luquin I, et al. Liquid Biopsy in Alzheimer’s Disease Patients Reveals Epigenetic Changes in the PRLHR Gene. Cells. 2023; 12(23):2679. https://doi.org/10.3390/cells12232679
Chicago/Turabian StyleMacías, Mónica, Blanca Acha, Jon Corroza, Amaya Urdánoz-Casado, Miren Roldan, Maitane Robles, Javier Sánchez-Ruiz de Gordoa, María Elena Erro, Ivonne Jericó, Idoia Blanco-Luquin, and et al. 2023. "Liquid Biopsy in Alzheimer’s Disease Patients Reveals Epigenetic Changes in the PRLHR Gene" Cells 12, no. 23: 2679. https://doi.org/10.3390/cells12232679