
Chromosomal Instability-Driven Cancer Progression: Interplay with the Tumour Microenvironment and Therapeutic Strategies


{kind=link}
{kind=link}
Abstract
:1. Chromosomal Instability (CIN) in Cancer
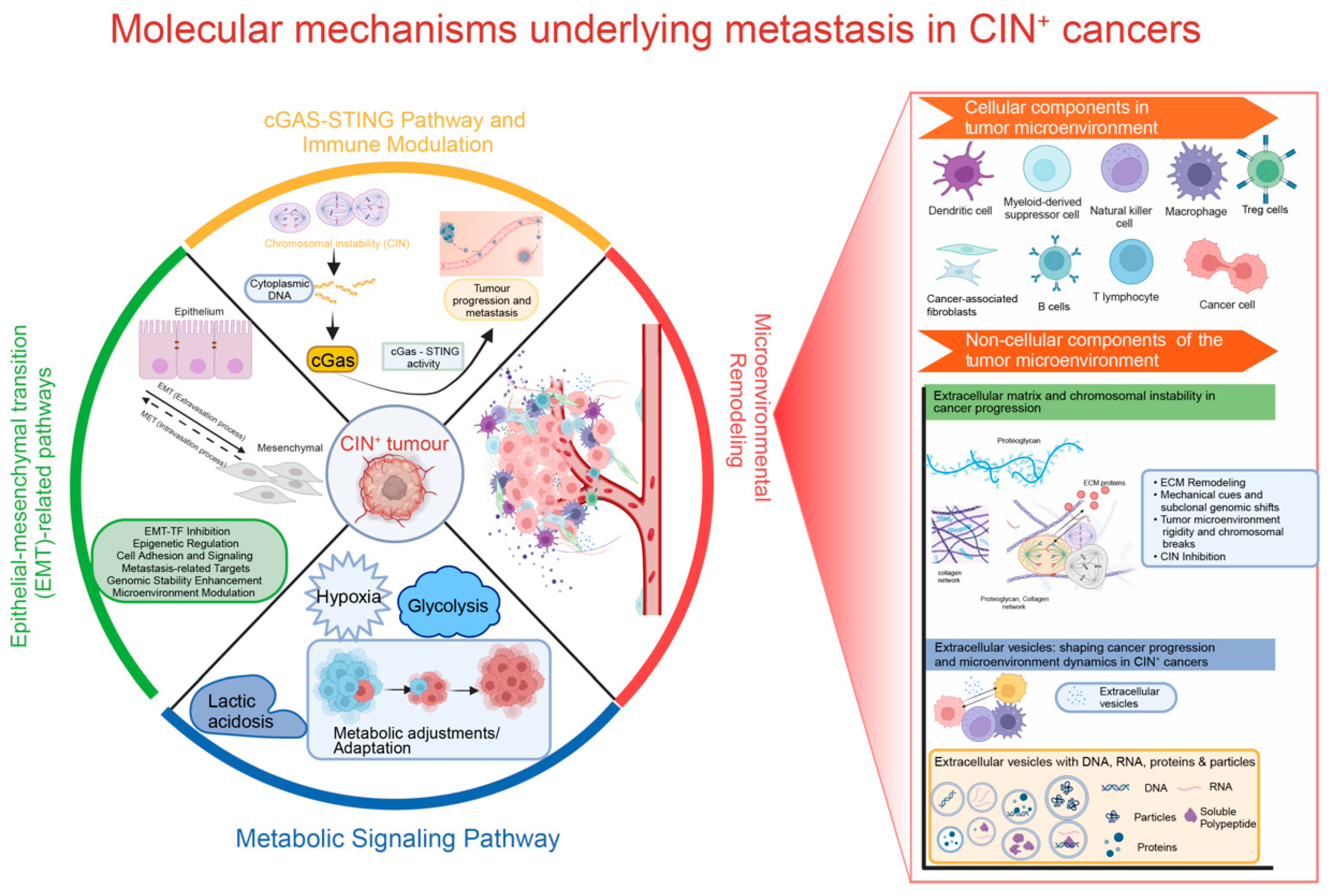
2. Molecular Mechanisms Underlying Metastasis in CIN+ Cancers
2.1. cGAS-STING Signalling and Immune Modulation
2.2. Epithelial-Mesenchymal Transition (EMT) and CIN
2.3. CIN and Metabolic Signalling
2.4. CIN and Remodelling of the Micro-Environment
2.4.1. Cellular Components in the TME
2.4.2. Non-Cellular Components of the Tumour Microenvironment
The Extracellular Matrix (ECM)
Extracellular Vesicles (EVs)
3. Targeting of Mechanisms to Counteract CIN-Driven Cancer
3.1. Targeting the CIN-Altered Immune Landscape
3.2. Exploiting Metabolic Vulnerabilities
3.3. Manipulating CIN and Its Downstream Effects
3.4. Targeting Extracellular Matrix (ECM)
3.5. Targeting Metastasis and STING
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Geigl, J.B.; Obenauf, A.C.; Schwarzbraun, T.; Speicher, M.R. Defining “Chromosomal Instability”. Trends Genet. 2008, 24, 64–69. [Google Scholar] [CrossRef]
- Thompson, S.L.; Bakhoum, S.F.; Compton, D.A. Mechanisms of Chromosomal Instability. Curr. Biol. 2010, 20, R285–R295. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.-A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal Instability Drives Metastasis through a Cytosolic DNA Response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The Causes and Consequences of Genetic Heterogeneity in Cancer Evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A Signature of Chromosomal Instability Inferred from Gene Expression Profiles Predicts Clinical Outcome in Multiple Human Cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef]
- Li, J.; Hubisz, M.J.; Earlie, E.M.; Duran, M.A.; Hong, C.; Varela, A.A.; Lettera, E.; Deyell, M.; Tavora, B.; Havel, J.J.; et al. Non-Cell-Autonomous Cancer Progression from Chromosomal Instability. Nature 2023, 620, 1080–1088. [Google Scholar] [CrossRef]
- Shaikh, N.; Mazzagatti, A.; De Angelis, S.; Johnson, S.C.; Bakker, B.; Spierings, D.C.J.; Wardenaar, R.; Maniati, E.; Wang, J.; Boemo, M.A.; et al. Replication Stress Generates Distinctive Landscapes of DNA Copy Number Alterations and Chromosome Scale Losses. Genome Biol. 2022, 23, 223. [Google Scholar] [CrossRef]
- Santaguida, S.; Richardson, A.; Iyer, D.R.; M’Saad, O.; Zasadil, L.; Knouse, K.A.; Wong, Y.L.; Rhind, N.; Desai, A.; Amon, A. Chromosome Mis-Segregation Generates Cell-Cycle-Arrested Cells with Complex Karyotypes That Are Eliminated by the Immune System. Dev. Cell 2017, 41, 638–651.e5. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The Landscape of Somatic Copy-Number Alteration across Human Cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Drews, R.M.; Hernando, B.; Tarabichi, M.; Haase, K.; Lesluyes, T.; Smith, P.S.; Morrill Gavarró, L.; Couturier, D.L.; Liu, L.; Schneider, M.; et al. A Pan-Cancer Compendium of Chromosomal Instability. Nature 2022, 606, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.D.; Abbasi, A.; Islam, S.M.A.; Bowes, A.L.; Khandekar, A.; Haase, K.; Hames-Fathi, S.; Ajayi, D.; Verfaillie, A.; Dhami, P.; et al. Signatures of Copy Number Alterations in Human Cancer. Nature 2022, 606, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Silkworth, W.T.; Cimini, D. Transient Defects of Mitotic Spindle Geometry and Chromosome Segregation Errors. Cell Div. 2012, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Godek, K.M.; Kabeche, L.; Compton, D.A. Regulation of Kinetochore-Microtubule Attachments through Homeostatic Control during Mitosis. Nat. Rev. Mol. Cell Biol. 2015, 16, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Murnane, J.P. Telomere Loss as a Mechanism for Chromosome Instability in Human Cancer. Cancer Res. 2010, 70, 4255–4259. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.-S.; Wersto, R.P.; Zhou, W.; Chrest, F.J.; Garrett, E.S.; Kwon, T.K.; Gabrielson, E. Variable Levels of Chromosomal Instability and Mitotic Spindle Checkpoint Defects in Breast Cancer. Am. J. Pathol. 2002, 161, 391–397. [Google Scholar] [CrossRef]
- Parine, N.R.; Varsha, R.S.; Alanazi, M.S. Microsatellite Instability in Colorectal Cancer; Abdurakhmonov, I.Y., Ed.; IntechOpen: Rijeka, Croatia, 2016; ISBN 978-953-51-2798-7. [Google Scholar]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer Drug Resistance: An Evolving Paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer Chemotherapy and beyond: Current Status, Drug Candidates, Associated Risks and Progress in Targeted Therapeutics. Genes Dis. 2023, 10, 1367–1401. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Yu, M.; Bardia, A.; Aceto, N.; Bersani, F.; Madden, M.W.; Donaldson, M.C.; Desai, R.; Zhu, H.; Comaills, V.; Zheng, Z.; et al. Ex Vivo Culture of Circulating Breast Tumor Cells for Individualized Testing of Drug Susceptibility. Science 2014, 345, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Wilting, R.H.; Dannenberg, J.-H. Epigenetic Mechanisms in Tumorigenesis, Tumor Cell Heterogeneity and Drug Resistance. Drug Resist. Updates 2012, 15, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Landau, D.A. Chromosomal Instability as a Driver of Tumor Heterogeneity and Evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a029611. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.X.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.M.; Howell, M.; et al. Chromosomal Instability Confers Intrinsic Multidrug Resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Replogle, J.M.; Zhou, W.; Amaro, A.E.; McFarland, J.M.; Villalobos-Ortiz, M.; Ryan, J.; Letai, A.; Yilmaz, O.; Sheltzer, J.; Lippard, S.J.; et al. Aneuploidy Increases Resistance to Chemotherapeutics by Antagonizing Cell Division. Proc. Natl. Acad. Sci. USA 2020, 117, 30566–30576. [Google Scholar] [CrossRef] [PubMed]
- Lasolle, H.; Elsensohn, M.-H.; Wierinckx, A.; Alix, E.; Bonnefille, C.; Vasiljevic, A.; Cortet, C.; Decoudier, B.; Sturm, N.; Gaillard, S.; et al. Chromosomal Instability in the Prediction of Pituitary Neuroendocrine Tumors Prognosis. Acta Neuropathol. Commun. 2020, 8, 190. [Google Scholar] [CrossRef]
- van Riet, J.; van de Werken, H.J.G.; Cuppen, E.; Eskens, F.A.L.M.; Tesselaar, M.; van Veenendaal, L.M.; Klümpen, H.-J.; Dercksen, M.W.; Valk, G.D.; Lolkema, M.P.; et al. The Genomic Landscape of 85 Advanced Neuroendocrine Neoplasms Reveals Subtype-Heterogeneity and Potential Therapeutic Targets. Nat. Commun. 2021, 12, 4612. [Google Scholar] [CrossRef]
- Carloni, V.; Lulli, M.; Madiai, S.; Mello, T.; Hall, A.; Luong, T.V.; Pinzani, M.; Rombouts, K.; Galli, A. CHK2 Overexpression and Mislocalisation within Mitotic Structures Enhances Chromosomal Instability and Hepatocellular Carcinoma Progression. Gut 2018, 67, 348–361. [Google Scholar] [CrossRef]
- Gong, Y.; Zou, S.; Deng, D.; Wang, L.; Hu, H.; Qiu, Z.; Wei, T.; Yang, P.; Zhou, J.; Zhang, Y.; et al. Loss of RanGAP1 Drives Chromosome Instability and Rapid Tumorigenesis of Osteosarcoma. Dev. Cell 2023, 58, 192–210.e11. [Google Scholar] [CrossRef]
- Hong, C.; Schubert, M.; Tijhuis, A.E.; Requesens, M.; Roorda, M.; van den Brink, A.; Ruiz, L.A.; Bakker, P.L.; van der Sluis, T.; Pieters, W.; et al. CGAS-STING Drives the IL-6-Dependent Survival of Chromosomally Instable Cancers. Nature 2022, 607, 366–373. [Google Scholar] [CrossRef]
- Paludan, S.R. Activation and Regulation of DNA-Driven Immune Responses. Microbiol. Mol. Biol. Rev. 2015, 79, 225–241. [Google Scholar] [CrossRef]
- Pan, J.; Fei, C.-J.; Hu, Y.; Wu, X.-Y.; Nie, L.; Chen, J. Current Understanding of the CGAS-STING Signaling Pathway: Structure, Regulatory Mechanisms, and Related Diseases. Zool. Res. 2023, 44, 183–218. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP Is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING Is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Barber, G.N. Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF-ΚB Activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djw199. [Google Scholar] [CrossRef]
- Wörmann, S.M.; Zhang, A.; Thege, F.I.; Cowan, R.W.; Rupani, D.N.; Wang, R.; Manning, S.L.; Gates, C.; Wu, W.; Levin-Klein, R.; et al. APOBEC3A Drives Deaminase Domain-Independent Chromosomal Instability to Promote Pancreatic Cancer Metastasis. Nat. Cancer 2021, 2, 1338–1356. [Google Scholar] [CrossRef]
- Huang, Y.; Li, W.; Yan, W.; Wu, J.; Chen, L.; Yao, X.; Gu, F.; Lv, L.; Zhao, J.; Zhao, M.; et al. Loss of PICH Promotes Chromosome Instability and Cell Death in Triple-Negative Breast Cancer. Cell Death Dis. 2019, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.; McCloy, R.A.; Parker, B.L.; Gallego-Ortega, D.; Law, A.M.K.; Chin, V.T.; Conway, J.R.W.; Fey, D.; Millar, E.K.A.; O’Toole, S.; et al. MASTL Overexpression Promotes Chromosome Instability and Metastasis in Breast Cancer. Oncogene 2018, 37, 4518–4533. [Google Scholar] [CrossRef]
- Bakhoum, M.F.; Francis, J.H.; Agustinus, A.; Earlie, E.M.; Di Bona, M.; Abramson, D.H.; Duran, M.; Masilionis, I.; Molina, E.; Shoushtari, A.N.; et al. Loss of Polycomb Repressive Complex 1 Activity and Chromosomal Instability Drive Uveal Melanoma Progression. Nat. Commun. 2021, 12, 5402. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Dragomir, M.P.; Fabris, L.; Bayraktar, R.; Knutsen, E.; Liu, X.; Tang, C.; Li, Y.; Shimura, T.; Ivkovic, T.C.; et al. The Long Noncoding RNA CCAT2 Induces Chromosomal Instability Through BOP1-AURKB Signaling. Gastroenterology 2020, 159, 2146–2162.e33. [Google Scholar] [CrossRef]
- Li, J.; Duran, M.A.; Dhanota, N.; Chatila, W.K.; Bettigole, S.E.; Kwon, J.; Sriram, R.K.; Humphries, M.P.; Salto-Tellez, M.; James, J.A.; et al. Metastasis and Immune Evasion from Extracellular CGAMP Hydrolysis. Cancer Discov. 2021, 11, 1212–1227. [Google Scholar] [CrossRef]
- Töpfer, K.; Kempe, S.; Müller, N.; Schmitz, M.; Bachmann, M.; Cartellieri, M.; Schackert, G.; Temme, A. Tumor Evasion from T Cell Surveillance. J. Biomed. Biotechnol. 2011, 2011, 918471. [Google Scholar] [CrossRef]
- Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease: Old Views and New Perspectives. Int. J. Dev. Biol. 2009, 53, 1541–1547. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Roschke, A.V.; Glebov, O.K.; Lababidi, S.; Gehlhaus, K.S.; Weinstein, J.N.; Kirsch, I.R. Chromosomal Instability Is Associated with Higher Expression of Genes Implicated in Epithelial-Mesenchymal Transition, Cancer Invasiveness, and Metastasis and with Lower Expression of Genes Involved in Cell Cycle Checkpoints, DNA Repair, and Chromatin Ma. Neoplasia 2008, 10, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Bakir, B.; Chiarella, A.M.; Pitarresi, J.R.; Rustgi, A.K. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020, 30, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Su, Y.; Koeman, J.; Haak, E.; Dykema, K.; Essenberg, C.; Hudson, E.; Petillo, D.; Khoo, S.K.; Vande Woude, G.F. Chromosome Instability Drives Phenotypic Switching to Metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, 14793–14798. [Google Scholar] [CrossRef]
- Celesti, G.; Di Caro, G.; Bianchi, P.; Grizzi, F.; Basso, G.; Marchesi, F.; Doni, A.; Marra, G.; Roncalli, M.; Mantovani, A.; et al. Presence of Twist1-Positive Neoplastic Cells in the Stroma of Chromosome-Unstable Colorectal Tumors. Gastroenterology 2013, 145, 647–657.e15. [Google Scholar] [CrossRef]
- Khot, M.; Sreekumar, D.; Jahagirdar, S.; Kulkarni, A.; Hari, K.; Faseela, E.E.; Sabarinathan, R.; Jolly, M.K.; Sengupta, K. Twist1 Induces Chromosomal Instability (CIN) in Colorectal Cancer Cells. Hum. Mol. Genet. 2020, 29, 1673–1688. [Google Scholar] [CrossRef]
- Jusino, S.; Saavedra, H.I. Role of E2Fs and Mitotic Regulators Controlled by E2Fs in the Epithelial to Mesenchymal Transition. Exp. Biol. Med. 2019, 244, 1419–1429. [Google Scholar] [CrossRef]
- Kumari, A.; Shonibare, Z.; Monavarian, M.; Arend, R.C.; Lee, N.Y.; Inman, G.J.; Mythreye, K. TGFβ Signaling Networks in Ovarian Cancer Progression and Plasticity. Clin. Exp. Metastasis 2021, 38, 139–161. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Sakai, Y. Transforming Growth Factor-β Signaling Pathway in Colorectal Cancer and Its Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 5822. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-E.; Kim, J.-H.; Che, Y.-H.; Kim, Y.-J.; Sung, J.-Y.; Kim, Y.-W.; Choe, B.-G.; Lee, S.; Park, J.-H. Role of the WNT/β-Catenin/ZKSCAN3 Pathway in Regulating Chromosomal Instability in Colon Cancer Cell Lines and Tissues. Int. J. Mol. Sci. 2022, 23, 9302. [Google Scholar] [CrossRef] [PubMed]
- Kumareswaran, R.; Ludkovski, O.; Meng, A.; Sykes, J.; Pintilie, M.; Bristow, R.G. Chronic Hypoxia Compromises Repair of DNA Double-Strand Breaks to Drive Genetic Instability. J. Cell Sci. 2012, 125, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Kanthan, R.; Senger, J.-L.; Kanthan, S.C. Molecular Events in Primary and Metastatic Colorectal Carcinoma: A Review. Patholog. Res. Int. 2012, 2012, 597497. [Google Scholar] [CrossRef]
- Torres, E.M.; Sokolsky, T.; Tucker, C.M.; Chan, L.Y.; Boselli, M.; Dunham, M.J.; Amon, A. Effects of Aneuploidy on Cellular Physiology and Cell Division in Haploid Yeast. Science 2007, 317, 916–924. [Google Scholar] [CrossRef]
- Foijer, F.; DiTommaso, T.; Donati, G.; Hautaviita, K.; Xie, S.Z.; Heath, E.; Smyth, I.; Watt, F.M.; Sorger, P.K.; Bradley, A. Spindle Checkpoint Deficiency Is Tolerated by Murine Epidermal Cells but Not Hair Follicle Stem Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 2928–2933. [Google Scholar] [CrossRef]
- Williams, B.R.; Prabhu, V.R.; Hunter, K.E.; Glazier, C.M.; Whittaker, C.A.; Housman, D.E.; Amon, A. Aneuploidy Affects Proliferation and Spontaneous Immortalization in Mammalian Cells. Science 2008, 322, 703–709. [Google Scholar] [CrossRef]
- Foijer, F.; Xie, S.Z.; Simon, J.E.; Bakker, P.L.; Conte, N.; Davis, S.H.; Kregel, E.; Jonkers, J.; Bradley, A.; Sorger, P.K. Chromosome Instability Induced by Mps1 and P53 Mutation Generates Aggressive Lymphomas Exhibiting Aneuploidy-Induced Stress. Proc. Natl. Acad. Sci. USA 2014, 111, 13427–13432. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Ha, G.; Tseng, Y.-Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-Derived Xenografts Undergo Mouse-Specific Tumor Evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef]
- Pino, M.S.; Chung, D.C. The Chromosomal Instability Pathway in Colon Cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [PubMed]
- Addie, R.D.; Kostidis, S.; Corver, W.E.; Oosting, J.; Aminzadeh-Gohari, S.; Feichtinger, R.G.; Kofler, B.; Aydemirli, M.D.; Giera, M.; Morreau, H. Metabolic Reprogramming Related to Whole-Chromosome Instability in Models for Hürthle Cell Carcinoma. Sci. Rep. 2020, 10, 9578. [Google Scholar] [CrossRef] [PubMed]
- Biswas, N.K.; Das, C.; Das, S.; Maitra, A.; Nair, S.; Gupta, T.; D’Cruz, A.K.; Sarin, R.; Majumder, P.P. Lymph Node Metastasis in Oral Cancer Is Strongly Associated with Chromosomal Instability and DNA Repair Defects. Int. J. Cancer 2019, 145, 2568–2579. [Google Scholar] [CrossRef]
- Lu, C.; Mahajan, A.; Hong, S.-H.; Galli, S.; Zhu, S.; Tilan, J.U.; Abualsaud, N.; Adnani, M.; Chung, S.; Elmansy, N.; et al. Hypoxia-Activated Neuropeptide Y/Y5 Receptor/RhoA Pathway Triggers Chromosomal Instability and Bone Metastasis in Ewing Sarcoma. Nat. Commun. 2022, 13, 2323. [Google Scholar] [CrossRef]
- Tan, Z.; Chan, Y.J.A.; Chua, Y.J.K.; Rutledge, S.D.; Pavelka, N.; Cimini, D.; Rancati, G. Environmental Stresses Induce Karyotypic Instability in Colorectal Cancer Cells. Mol. Biol. Cell 2019, 30, 42–55. [Google Scholar] [CrossRef]
- Vargas-Rondón, N.; Pérez-Mora, E.; Villegas, V.E.; Rondón-Lagos, M. Role of Chromosomal Instability and Clonal Heterogeneity in the Therapy Response of Breast Cancer Cell Lines. Cancer Biol. Med. 2020, 17, 970–985. [Google Scholar] [CrossRef]
- Sugaya, K. Chromosome Instability Caused by Mutations in the Genes Involved in Transcription and Splicing. RNA Biol. 2019, 16, 1521–1525. [Google Scholar] [CrossRef]
- Panatta, E.; Butera, A.; Mammarella, E.; Pitolli, C.; Mauriello, A.; Leist, M.; Knight, R.A.; Melino, G.; Amelio, I. Metabolic Regulation by P53 Prevents R-Loop-Associated Genomic Instability. Cell Rep. 2022, 41, 111568. [Google Scholar] [CrossRef]
- Kuzyk, A.; Mai, S. C-MYC-Induced Genomic Instability. Cold Spring Harb. Perspect. Med. 2014, 4, a014373. [Google Scholar] [CrossRef]
- Henriques, R.; Magyar, Z.; Monardes, A.; Khan, S.; Zalejski, C.; Orellana, J.; Szabados, L.; de la Torre, C.; Koncz, C.; Bögre, L. Arabidopsis S6 Kinase Mutants Display Chromosome Instability and Altered RBR1-E2F Pathway Activity. EMBO J. 2010, 29, 2979–2993. [Google Scholar] [CrossRef] [PubMed]
- Samper, E.; Nicholls, D.G.; Melov, S. Mitochondrial Oxidative Stress Causes Chromosomal Instability of Mouse Embryonic Fibroblasts. Aging Cell 2003, 2, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.K.; Raghuram, G.V.; Panwar, H.; Jain, D.; Pandey, H.; Maudar, K.K. Mitochondrial Oxidative Stress Elicits Chromosomal Instability after Exposure to Isocyanates in Human Kidney Epithelial Cells. Free Radic. Res. 2009, 43, 718–728. [Google Scholar] [CrossRef]
- Goetz, E.M.; Shankar, B.; Zou, Y.; Morales, J.C.; Luo, X.; Araki, S.; Bachoo, R.; Mayo, L.D.; Boothman, D.A. ATM-Dependent IGF-1 Induction Regulates Secretory Clusterin Expression after DNA Damage and in Genetic Instability. Oncogene 2011, 30, 3745–3754. [Google Scholar] [CrossRef] [PubMed]
- Agustinus, A.S.; Al-Rawi, D.; Dameracharla, B.; Raviram, R.; Jones, B.S.C.L.; Stransky, S.; Scipioni, L.; Luebeck, J.; Di Bona, M.; Norkunaite, D.; et al. Epigenetic Dysregulation from Chromosomal Transit in Micronuclei. Nature 2023, 619, 176–183. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor Microenvironment Complexity and Therapeutic Implications at a Glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Bizzarri, M.; Cucina, A. Tumor and the Microenvironment: A Chance to Reframe the Paradigm of Carcinogenesis? Biomed. Res. Int. 2014, 2014, 934038. [Google Scholar] [CrossRef]
- Mironchik, Y.; Winnard, P.T.J.; Vesuna, F.; Kato, Y.; Wildes, F.; Pathak, A.P.; Kominsky, S.; Artemov, D.; Bhujwalla, Z.; Van Diest, P.; et al. Twist Overexpression Induces in Vivo Angiogenesis and Correlates with Chromosomal Instability in Breast Cancer. Cancer Res. 2005, 65, 10801–10809. [Google Scholar] [CrossRef]
- Adams, S.D.; Csere, J.; D’angelo, G.; Carter, E.P.; Romao, M.; Arnandis, T.; Dodel, M.; Kocher, H.M.; Grose, R.; Raposo, G.; et al. Centrosome Amplification Mediates Small Extracellular Vesicle Secretion via Lysosome Disruption. Curr. Biol. 2021, 31, 1403–1416.e7. [Google Scholar] [CrossRef] [PubMed]
- Xian, S.; Dosset, M.; Almanza, G.; Searles, S.; Sahani, P.; Waller, T.C.; Jepsen, K.; Carter, H.; Zanetti, M. The Unfolded Protein Response Links Tumor Aneuploidy to Local Immune Dysregulation. EMBO Rep. 2021, 22, e52509. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor Aneuploidy Correlates with Markers of Immune Evasion and with Reduced Response to Immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.H.; Wang, M.; Zhu, H.; Phan, S.H.; Andrechak, J.C.; Chang, A.H.; Dooling, L.J.; Tobin, M.P.; Marchena, T.; Discher, D.E. Chromosomal Instability Can Favor Macrophage-Mediated Immune Response and Induce a Broad, Vaccination-like Anti-Tumor IgG Response. bioRxiv 2023. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef]
- de Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and Temporal Diversity in Genomic Instability Processes Defines Lung Cancer Evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef]
- Zhang, J.T.; Dong, S.; Ji, L.Y.; Zhou, J.Y.; Chen, Z.H.; Su, J.; Zhu, Q.G.; Wang, M.M.; Ke, E.E.; Sun, H.; et al. Intratumoral Genetic and Immune Microenvironmental Heterogeneity in T4N0M0 (Diameter ≥ 7 Cm) Non-Small Cell Lung Cancers. Thorac. Cancer 2022, 13, 1333–1341. [Google Scholar] [CrossRef]
- Zhao, X.; Cohen, E.E.W.; William, W.N.J.; Bianchi, J.J.; Abraham, J.P.; Magee, D.; Spetzler, D.B.; Gutkind, J.S.; Alexandrov, L.B.; Cavenee, W.K.; et al. Somatic 9p24.1 Alterations in HPV(-) Head and Neck Squamous Cancer Dictate Immune Microenvironment and Anti-PD-1 Checkpoint Inhibitor Activity. Proc. Natl. Acad. Sci. USA 2022, 119, e2213835119. [Google Scholar] [CrossRef]
- Schubert, M.; Hong, C.; Jilderda, L.J.; Rueda, M.R.; Tijhuis, A.E.; Simon, J.E.; Bakker, P.L.; Cooper, J.L.; Damaskou, A.; Wardenaar, R.; et al. Cancer Tolerance to Chromosomal Instability Is Driven by Stat1 Inactivation in Vivo. bioRxiv 2021. [Google Scholar] [CrossRef]
- Tripathi, R.; Modur, V.; Senovilla, L.; Kroemer, G.; Komurov, K. Suppression of Tumor Antigen Presentation during Aneuploid Tumor Evolution Contributes to Immune Evasion. Oncoimmunology 2019, 8, 1657374. [Google Scholar] [CrossRef]
- Pal, S.; Bhattacharjee, A.; Ali, A.; Mandal, N.C.; Mandal, S.C.; Pal, M. Chronic Inflammation and Cancer: Potential Chemoprevention through Nuclear Factor Kappa B and P53 Mutual Antagonism. J. Inflamm. 2014, 11, 23. [Google Scholar] [CrossRef]
- Sokač, M.; Ahrenfeldt, J.; Litchfield, K.; Watkins, T.B.K.; Knudsen, M.; Dyrskjøt, L.; Jakobsen, M.R.; Birkbak, N.J. Classifying CGAS-STING Activity Links Chromosomal Instability with Immunotherapy Response in Metastatic Bladder Cancer. Cancer Res. Commun. 2022, 2, 762–771. [Google Scholar] [CrossRef]
- Andriani, G.A.; Almeida, V.P.; Faggioli, F.; Mauro, M.; Tsai, W.L.; Santambrogio, L.; Maslov, A.; Gadina, M.; Campisi, J.; Vijg, J.; et al. Whole Chromosome Instability Induces Senescence and Promotes SASP. Sci. Rep. 2016, 6, 35218. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between Cancer-Associated Fibroblasts and Immune Cells in the Tumor Microenvironment: New Findings and Future Perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef]
- Liu, X.; Yao, L.; Qu, J.; Liu, L.; Lu, N.; Wang, J.; Zhang, J. Cancer-Associated Fibroblast Infiltration in Gastric Cancer: The Discrepancy in Subtypes Pathways and Immunosuppression. J. Transl. Med. 2021, 19, 325. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Wu, M.; Arcand, S.L.; Lavallée, S.; Hébert, J.; Tonin, P.N.; Basik, M. Breast Carcinoma–Associated Fibroblasts Rarely Contain P53 Mutations or Chromosomal Aberrations. Cancer Res. 2010, 70, 5770–5777. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Fischbach, S.R.; Bronk, S.F.; Hirsova, P.; Krishnan, A.; Dhanasekaran, R.; Smadbeck, J.B.; Smoot, R.L.; Vasmatzis, G.; Gores, G.J. YAP-Associated Chromosomal Instability and Cholangiocarcinoma in Mice. Oncotarget 2018, 9, 5892–5905. [Google Scholar] [CrossRef]
- Zhou, Y.-H.; Afrasiabi, K.; Linskey, M.E. Extracellular Control of Chromosomal Instability and Maintenance of Intra-Tumoral Heterogeneity. J. Cancer Metastasis Treat. 2018, 4, 41. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The Extracellular Matrix Modulates the Hallmarks of Cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef]
- Sullivan, W.J.; Mullen, P.J.; Schmid, E.W.; Flores, A.; Momcilovic, M.; Sharpley, M.S.; Jelinek, D.; Whiteley, A.E.; Maxwell, M.B.; Wilde, B.R.; et al. Extracellular Matrix Remodeling Regulates Glucose Metabolism through TXNIP Destabilization. Cell 2018, 175, 117–132.e21. [Google Scholar] [CrossRef]
- Barresi, V.; Cinnirella, G.; Valenti, G.; Spampinato, G.; Musso, N.; Castorina, S.; Condorelli, D.F. Gene Expression Profiles in Genome Instability-Based Classes of Colorectal Cancer. BMC Cancer 2018, 18, 1265. [Google Scholar] [CrossRef]
- López-Carrasco, A.; Martín-Vañó, S.; Burgos-Panadero, R.; Monferrer, E.; Berbegall, A.P.; Fernández-Blanco, B.; Navarro, S.; Noguera, R. Impact of Extracellular Matrix Stiffness on Genomic Heterogeneity in MYCN-Amplified Neuroblastoma Cell Line. J. Exp. Clin. Cancer Res. 2020, 39, 226. [Google Scholar] [CrossRef]
- Bazzan, E.; Tinè, M.; Casara, A.; Biondini, D.; Semenzato, U.; Cocconcelli, E.; Balestro, E.; Damin, M.; Radu, C.M.; Turato, G.; et al. Critical Review of the Evolution of Extracellular Vesicles’ Knowledge: From 1946 to Today. Int. J. Mol. Sci. 2021, 22, 6417. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding Light on the Cell Biology of Extracellular Vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Lima, L.G.; Ham, S.; Shin, H.; Chai, E.P.Z.; Lek, E.S.H.; Lobb, R.J.; Müller, A.F.; Mathivanan, S.; Yeo, B.; Choi, Y.; et al. Tumor Microenvironmental Cytokines Bound to Cancer Exosomes Determine Uptake by Cytokine Receptor-Expressing Cells and Biodistribution. Nat. Commun. 2021, 12, 3543. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour Exosome Integrins Determine Organotropic Metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Hurley, J.; Roberts, D.; Chakrabortty, S.K.; Enderle, D.; Noerholm, M.; Breakefield, X.O.; Skog, J.K. Exosome-Based Liquid Biopsies in Cancer: Opportunities and Challenges. Ann. Oncol. 2021, 32, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Fordjour, F.K.; Daaboul, G.G.; Gould, S.J. A Shared Pathway of Exosome Biogenesis Operates at Plasma and Endosome Membranes. J. Biol. Chem. 2019, 298, 102394. [Google Scholar] [CrossRef]
- Martins, Á.M.; Lopes, T.M.; Diniz, F.; Pires, J.; Osório, H.; Pinto, F.; Freitas, D.; Reis, C.A. Differential Protein and Glycan Packaging into Extracellular Vesicles in Response to 3D Gastric Cancer Cellular Organization. Adv. Sci. 2023, 10, e2300588. [Google Scholar] [CrossRef]
- Bao, S.; Hu, T.; Liu, J.; Su, J.; Sun, J.; Ming, Y.; Li, J.; Wu, N.; Chen, H.; Zhou, M. Genomic Instability-Derived Plasma Extracellular Vesicle-MicroRNA Signature as a Minimally Invasive Predictor of Risk and Unfavorable Prognosis in Breast Cancer. J. Nanobiotechnol. 2021, 19, 22. [Google Scholar] [CrossRef]
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.-R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef]
- Xie, B.; Liang, X.; Yue, W.; Ma, J.; Li, X.; Zhang, N.; Wang, P.; Liu, C.; Shi, X.; Qiao, J.; et al. Targeting Cytokinesis Bridge Proteins to Kill High-CIN Type Tumors. Fundam. Res. 2021, 1, 752–766. [Google Scholar] [CrossRef]
- Dai, C.; Sun, F.; Zhu, C.; Hu, X. Tumor Environmental Factors Glucose Deprivation and Lactic Acidosis Induce Mitotic Chromosomal Instability--an Implication in Aneuploid Human Tumors. PLoS ONE 2013, 8, e63054. [Google Scholar] [CrossRef]
- Shaukat, Z.; Liu, D.; Choo, A.; Hussain, R.; O’Keefe, L.; Richards, R.; Saint, R.; Gregory, S.L. Chromosomal Instability Causes Sensitivity to Metabolic Stress. Oncogene 2015, 34, 4044–4055. [Google Scholar] [CrossRef]
- Liu, B.; Carlson, R.J.; Pires, I.S.; Gentili, M.; Feng, E.; Hellier, Q.; Schwartz, M.A.; Blainey, P.C.; Irvine, D.J.; Hacohen, N. Human STING Is a Proton Channel. Science 2023, 381, 508–514. [Google Scholar] [CrossRef]
- Hu, X.; Chao, M.; Wu, H. Central Role of Lactate and Proton in Cancer Cell Resistance to Glucose Deprivation and Its Clinical Translation. Signal Transduct. Target. Ther. 2017, 2, 16047. [Google Scholar] [CrossRef]
- Thompson, L.L.; Jeusset, L.M.-P.; Lepage, C.C.; McManus, K.J. Evolving Therapeutic Strategies to Exploit Chromosome Instability in Cancer. Cancers 2017, 9, 151. [Google Scholar] [CrossRef]
- Dhital, B.; Santasusagna, S.; Kirthika, P.; Xu, M.; Li, P.; Carceles-Cordon, M.; Soni, R.K.; Li, Z.; Hendrickson, R.C.; Schiewer, M.J.; et al. Harnessing Transcriptionally Driven Chromosomal Instability Adaptation to Target Therapy-Refractory Lethal Prostate Cancer. Cell Rep. Med. 2023, 4, 100937. [Google Scholar] [CrossRef]
- Cosenza, M.R.; Krämer, A. Centrosome Amplification, Chromosomal Instability and Cancer: Mechanistic, Clinical and Therapeutic Issues. Chromosome Res. 2016, 24, 105–126. [Google Scholar] [CrossRef] [PubMed]
- Shoshani, O.; Bakker, B.; de Haan, L.; Tijhuis, A.E.; Wang, Y.; Kim, D.H.; Maldonado, M.; Demarest, M.A.; Artates, J.; Zhengyu, O.; et al. Transient Genomic Instability Drives Tumorigenesis through Accelerated Clonal Evolution. Genes. Dev. 2021, 35, 1093–1108. [Google Scholar] [CrossRef] [PubMed]
- Schukken, K.M.K.M.; Lin, Y.-C.; Bakker, P.P.L.; Schubert, M.; Preuss, S.F.S.F.; Simon, J.E.J.E.; van den Bos, H.; Storchova, Z.; Colome-Tatche, M.; Bastians, H.; et al. Altering Microtubule Dynamics Is Synergistically Toxic with Spindle Assembly Checkpoint Inhibition. Life Sci. Alliance 2020, 3, e201900499. [Google Scholar] [CrossRef]
- Marquis, C.; Fonseca, C.L.; Queen, K.A.; Wood, L.; Vandal, S.E.; Malaby, H.L.H.; Clayton, J.E.; Stumpff, J. Chromosomally Unstable Tumor Cells Specifically Require KIF18A for Proliferation. Nat. Commun. 2021, 12, 1213. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, M.; Mustachio, L.M.; Chen, Y.; Chen, Z.; Liu, X.; Wei, C.-H.; Roszik, J.; Kittai, A.S.; Danilov, A.V.; Zhang, X.; et al. A Novel CDK2/9 Inhibitor CYC065 Causes Anaphase Catastrophe and Represses Proliferation, Tumorigenesis, and Metastasis in Aneuploid Cancers. Mol. Cancer Ther. 2021, 20, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Vaja, R. Single-Cell RNA Sequencing of Glioblastoma Cancer Stem Cell Lines Reveals That Chromosomal Instability Leads to Disturbed Extra-Cellular Matrix Organization. Adv. Tech. Biol. Med. 2022, 10, 359. [Google Scholar] [CrossRef]
- Deng, M.; Lin, J.; Nowsheen, S.; Liu, T.; Zhao, Y.; Villalta, P.W.; Sicard, D.; Tschumperlin, D.J.; Lee, S.; Kim, J.; et al. Extracellular Matrix Stiffness Determines DNA Repair Efficiency and Cellular Sensitivity to Genotoxic Agents. Sci. Adv. 2023, 6, eabb2630. [Google Scholar] [CrossRef]
- Beernaert, B.; Parkes, E.E. CGAS-STING Signalling in Cancer: Striking a Balance with Chromosomal Instability. Biochem. Soc. Trans. 2023, 51, 539–555. [Google Scholar] [CrossRef]
- Zhu, Y.; An, X.; Zhang, X.; Qiao, Y.; Zheng, T.; Li, X. STING: A Master Regulator in the Cancer-Immunity Cycle. Mol. Cancer 2019, 18, 152. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, S.; Guerrero-Haughton, E.; Foijer, F. Chromosomal Instability-Driven Cancer Progression: Interplay with the Tumour Microenvironment and Therapeutic Strategies. Cells 2023, 12, 2712. https://doi.org/10.3390/cells12232712
Zheng S, Guerrero-Haughton E, Foijer F. Chromosomal Instability-Driven Cancer Progression: Interplay with the Tumour Microenvironment and Therapeutic Strategies. Cells. 2023; 12(23):2712. https://doi.org/10.3390/cells12232712
Chicago/Turabian StyleZheng, Siqi, Erika Guerrero-Haughton, and Floris Foijer. 2023. "Chromosomal Instability-Driven Cancer Progression: Interplay with the Tumour Microenvironment and Therapeutic Strategies" Cells 12, no. 23: 2712. https://doi.org/10.3390/cells12232712
APA StyleZheng, S., Guerrero-Haughton, E., & Foijer, F. (2023). Chromosomal Instability-Driven Cancer Progression: Interplay with the Tumour Microenvironment and Therapeutic Strategies. Cells, 12(23), 2712. https://doi.org/10.3390/cells12232712