
Simultaneous Increases in Intracellular Sodium and Tonicity Boost Antimicrobial Activity of Macrophages

 ,
,
Abstract
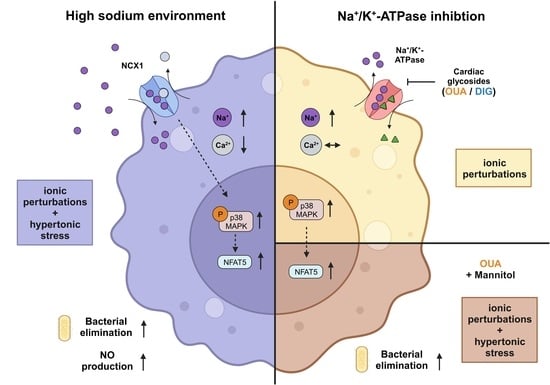
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Generation and Cultivation of Macrophages and Cell Lines
2.3. Macrophage Stimulation and Infection Assay
2.4. E. coli Growth Curves
2.5. Quantification of Nitrite Concentration and Lactate Dehydrogenase Activity
2.6. Immunoblotting
2.7. Measurement of Intracellular Na+ and K+ Levels with Atomic Absorption Spectrometry (AAS)
2.8. Rubidium Incorporation Measurements with Atomic Absorption Spectrometry
2.9. Determination of Intracellular Ca2+-Levels with Epifluorescence Microscopy
2.10. IL-1ß Quantification via Enzyme-Linked Immunosorbent Assay
2.11. RNA Isolation, Reverse Transcription, Real-Time PCR, and Relative Quantification
2.12. Statistical Analyses
3. Results
3.1. CGs Induce Intracellular Na+ Accumulation Similar to HS Conditions
3.2. Loss of Potassium and Increase in IL-1β Production upon CG but Not HS Treatment
3.3. Cardiac Glycosides Do Not Influence Intracellular Calcium Levels
3.4. Cardiac Glycosides Induce p-p38/MAPK Signaling, Whereas NFAT5 Expression Remains Unchanged
3.5. Heterogeneous Effects of CGs and HS towards NO Production and the Elimination of Intracellular E. coli
3.6. Intracellular Na+ Accumulation and Hypertonic Stress Increase NFAT5 Expression and Antibacterial Activity
4. Discussion
5. Conclusions
6. Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jantsch, J.; Schatz, V.; Friedrich, D.; Schröder, A.; Kopp, C.; Siegert, I.; Maronna, A.; Wendelborn, D.; Linz, P.; Binger, K.J.; et al. Cutaneous Na+ storage strengthens the antimicrobial barrier function of the skin and boosts macrophage-driven host defense. Cell Metab. 2015, 21, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, L.; Guais, A.; Pooya, M.; Abolhassani, M. Is inflammation a consequence of extracellular hyperosmolarity? J. Inflamm. 2009, 6, 21. [Google Scholar] [CrossRef]
- Kopp, C.; Beyer, C.; Linz, P.; Dahlmann, A.; Hammon, M.; Jantsch, J.; Neubert, P.; Rosenhauer, D.; Müller, D.N.; Cavallaro, A.; et al. Na+ deposition in the fibrotic skin of systemic sclerosis patients detected by 23Na-magnetic resonance imaging. Rheumatology 2017, 56, 556–560. [Google Scholar] [CrossRef]
- Matthias, J.; Maul, J.; Noster, R.; Meinl, H.; Chao, Y.-Y.; Gerstenberg, H.; Jeschke, F.; Gasparoni, G.; Welle, A.; Walter, J.; et al. Sodium chloride is an ionic checkpoint for human TH2 cells and shapes the atopic skin microenvironment. Sci. Transl. Med. 2019, 11, eaau0683. [Google Scholar] [CrossRef] [PubMed]
- Maifeld, A.; Wild, J.; Karlsen, T.V.; Rakova, N.; Wistorf, E.; Linz, P.; Jung, R.; Birukov, A.; Gimenez-Rivera, V.-A.; Wilck, N.; et al. Skin Sodium Accumulates in Psoriasis and Reflects Disease Severity. J. Investig. Dermatol. 2022, 142, 166–178.e8. [Google Scholar] [CrossRef] [PubMed]
- Titze, J.; Lang, R.; Ilies, C.; Schwind, K.H.; Kirsch, K.A. Osmotically inactive skin Na+ storage in rats. Am. J. Physiol. Renal Physiol. 2003, 285, F1108–F1117. [Google Scholar] [CrossRef] [PubMed]
- Machnik, A.; Neuhofer, W.; Jantsch, J.; Dahlmann, A.; Tammela, T.; Machura, K.; Park, J.-K.; Beck, F.-X.; Müller, D.N.; Derer, W.; et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat. Med. 2009, 15, 545–552. [Google Scholar] [CrossRef]
- Jobin, K.; Müller, D.N.; Jantsch, J.; Kurts, C. Sodium and its manifold impact on our immune system. Trends Immunol. 2021, 42, 469–479. [Google Scholar] [CrossRef]
- Neubert, P.; Schröder, A.; Müller, D.N.; Jantsch, J. Interplay of Na+ Balance and Immunobiology of Dendritic Cells. Front. Immunol. 2019, 10, 599. [Google Scholar] [CrossRef]
- Schatz, V.; Neubert, P.; Schröder, A.; Binger, K.; Gebhard, M.; Müller, D.N.; Luft, F.C.; Titze, J.; Jantsch, J. Elementary immunology: Na+ as a regulator of immunity. Pediatr. Nephrol. 2017, 32, 201–210. [Google Scholar] [CrossRef]
- Wilck, N.; Balogh, A.; Markó, L.; Bartolomaeus, H.; Müller, D.N. The role of sodium in modulating immune cell function. Nat. Rev. Nephrol. 2019, 15, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.N.; Wilck, N.; Haase, S.; Kleinewietfeld, M.; Linker, R.A. Sodium in the microenvironment regulates immune responses and tissue homeostasis. Nat. Rev. Immunol. 2019, 19, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Gurusamy, D.; Clever, D.; Eil, R.; Restifo, N.P. Novel “Elements” of Immune Suppression within the Tumor Microenvironment. Cancer Immunol. Res. 2017, 5, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.N.; Geisberger, S.; Kleinewietfeld, M.; Jantsch, J. Salt sensitivity includes effects on immune cell signalling and metabolism. Nat. Rev. Immunol. 2023, 23, 341–342. [Google Scholar] [CrossRef] [PubMed]
- Kleinewietfeld, M.; Manzel, A.; Titze, J.; Kvakan, H.; Yosef, N.; Linker, R.A.; Muller, D.N.; Hafler, D.A. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 2013, 496, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Yosef, N.; Thalhamer, T.; Zhu, C.; Xiao, S.; Kishi, Y.; Regev, A.; Kuchroo, V.K. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 2013, 496, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Matthias, J.; Heink, S.; Picard, F.; Zeiträg, J.; Kolz, A. Salt generates antiinflammatory Th17 cells but amplifies pathogenicity in proinflammatory cytokine microenvironments. J. Clin. Investig. 2020, 130, 4587–4600. [Google Scholar] [CrossRef]
- Côrte-Real, B.F.; Hamad, I.; Arroyo Hornero, R.; Geisberger, S.; Roels, J.; Van Zeebroeck, L.; Dyczko, A.; van Gisbergen, M.W.; Kurniawan, H.; Wagner, A.; et al. Sodium perturbs mitochondrial respiration and induces dysfunctional Tregs. Cell Metab. 2023, 35, 299–315.e8. [Google Scholar] [CrossRef]
- Krampert, L.; Bauer, K.; Ebner, S.; Neubert, P.; Ossner, T.; Weigert, A.; Schatz, V.; Toelge, M.; Schröder, A.; Herrmann, M.; et al. High Na+ Environments Impair Phagocyte Oxidase-Dependent Antibacterial Activity of Neutrophils. Front. Immunol. 2021, 12, 712948. [Google Scholar] [CrossRef]
- Mazzitelli, I.; Bleichmar, L.; Melucci, C.; Gerber, P.P.; Toscanini, A.; Cuestas, M.L.; Diaz, F.E.; Geffner, J. High Salt Induces a Delayed Activation of Human Neutrophils. Front. Immunol. 2022, 13, 831844. [Google Scholar] [CrossRef]
- Neubert, P.; Homann, A.; Wendelborn, D.; Bär, A.-L.; Krampert, L.; Trum, M.; Schröder, A.; Ebner, S.; Weichselbaum, A.; Schatz, V.; et al. NCX1 represents an ionic Na+ sensing mechanism in macrophages. PLoS Biol. 2020, 18, e3000722. [Google Scholar] [CrossRef] [PubMed]
- Baartscheer, A.; Schumacher, C.A.; Coronel, R.; Fiolet, J.W.T. The Driving Force of the Na/Ca-Exchanger during Metabolic Inhibition. Front. Physiol. 2011, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Philipson, K.D.; Nicoll, D.A. Sodium-calcium exchange. Curr. Opin. Cell Biol. 1992, 4, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Philipson, K.D.; Nicoll, D.A. Sodium-Calcium Exchange: A Molecular Perspective. Annu. Rev. Physiol. 2000, 62, 111–133. [Google Scholar] [CrossRef]
- Skou, J.C. The Influence of some Cations on an Adenosine Triphosphatase from Pheripheral Nerves. Biochim. Biophys. Acta 1957, 23, 394–401. [Google Scholar] [CrossRef]
- Skou, J.C. Further Investigations on a Mg++ + Na+-Activated Adenosintriphosphatase, Possibly Related to the Active, Linked Transport of Na+ and K+ Across the Nerve Membrane. Biochim. Biophys. Acta 1960, 42, 6–23. [Google Scholar] [CrossRef]
- Therien, A.G.; Blostein, R. Mechanisms of sodium pump regulation. Am. J. Physiol. Cell Physiol. 2000, 279, C541–C566. [Google Scholar] [CrossRef]
- Schatzmann, H.J. Herzglykoside als Hemmstoffe fur den aktiven Kalium und Natrium-Transport durch die Erythrocytemembran. Cardiac glycosides as inhibitors of active potassium and sodium transport through the erythrocyte membrane. Helv. Physiol. Pharmacol. Acta 1953, 11, 346–354. [Google Scholar]
- McMurray, J.J.V.; Adamopoulos, S.; Anker, S.D.; Auricchio, A.; Böhm, M.; Dickstein, K.; Falk, V.; Filippatos, G.; Fonseca, C.; Gomez-Sanchez, M.A.; et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2012, 33, 1787–1847. [Google Scholar]
- Cheminay, C.; Schoen, M.; Hensel, M.; Wandersee-Steinhäuser, A.; Ritter, U. Migration of Salmonella typhimurium-harboring bone marrow-derived dendritic cells towards the chemokines CCL19 and CCL21. Microb. Pathog. 2002, 32, 207–218. [Google Scholar] [CrossRef]
- Schleicher, U.; Bogdan, C. Generation, culture and flow-cytometric characterization of primary mouse macrophages. Methods Mol. Biol. 2009, 531, 203–224. [Google Scholar] [PubMed]
- Ralph, P.; Nakoinz, I. Antibody-dependent killing of erythrocyte and tumor targets by macrophage-related cell lines: Enhancement by PPD and LPS. J. Immunol. 1977, 119, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Raschke, W.C.; Baird, S.; Ralph, P.; Nakoinz, I. Functional macrophage cell lines transformed by Abelson leukemia virus. Cell 1978, 15, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Neubert, P.; Weichselbaum, A.; Reitinger, C.; Schatz, V.; Schröder, A. HIF1A and NFAT5 coordinate Na+-boosted antibacterial defense via enhanced autophagy and autolysosomal targeting. Autophagy 2019, 15, 1899–1916. [Google Scholar] [CrossRef] [PubMed]
- Geisberger, S.; Bartolomaeus, H.; Neubert, P.; Willebrand, R.; Zasada, C. Salt Transiently Inhibits Mitochondrial Energetics in Mononuclear Phagocytes. Circulation 2021, 144, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Gill, R.; Wicks, D.; Despotovski, S.; Liang, D. Development of an HTS assay for Na+, K+-ATPase using nonradioactive rubidium ion uptake. Assay Drug Dev. Technol. 2004, 2, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Motulsky, H.J.; Brown, R.E. Detecting outliers when fitting data with nonlinear regression—A new method based on robust nonlinear regression and the false discovery rate. BMC Bioinform. 2006, 7, 123. [Google Scholar] [CrossRef]
- Zhang, W.-C.; Zheng, X.-J.; Du, L.-J.; Sun, J.-Y.; Shen, Z.-X.; Shi, C.; Sun, S.; Zhang, Z.; Chen, X.-Q.; Qin, M.; et al. High salt primes a specific activation state of macrophages, M(Na). Cell Res. 2015, 25, 893–910. [Google Scholar] [CrossRef]
- Castillo, J.P.; Rui, H.; Basilio, D.; Das, A.; Roux, B. Mechanism of potassium ion uptake by the Na(+)/K(+)-ATPase. Nat. Commun. 2015, 6, 7622. [Google Scholar] [CrossRef]
- Perregaux, D.; Gabel, C.A. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J. Biol. Chem. 1994, 269, 15195–15203. [Google Scholar] [CrossRef]
- Ferrari, D.; Pizzirani, C.; Adinolfi, E.; Lemoli, R.M.; Curti, A.; Idzko, M.; Panther, E.; Di Virgilio, F. The P2X7 receptor: A key player in IL-1 processing and release. J. Immunol. 2006, 176, 3877–3883. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Kanneganti, T.-D.; Dubyak, G.R.; Núñez, G. Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J. Biol. Chem. 2007, 282, 18810–18818. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Staiano, R.I.; Granata, F.; Secondo, A.; Petraroli, A.; Loffredo, S. Expression and function of Na+/Ca2+ exchangers 1 and 3 in human macrophages and monocytes. Eur. J. Immunol. 2009, 39, 1405–1418. [Google Scholar] [CrossRef]
- Desai, B.N.; Leitinger, N. Purinergic and calcium signaling in macrophage function and plasticity. Front. Immunol. 2014, 5, 580. [Google Scholar] [CrossRef]
- Shapiro, L.; Dinarello, C.A. Osmotic regulation of cytokine synthesis in vitro. Proc. Natl. Acad. Sci. USA 1995, 92, 12230–12234. [Google Scholar] [CrossRef]
- Binger, K.J.; Gebhardt, M.; Heinig, M.; Rintisch, C.; Schroeder, A.; Neuhofer, W.; Hilgers, K.; Manzel, A.; Schwartz, C.; Kleinewietfeld, M.; et al. High salt reduces the activation of IL-4- and IL-13-stimulated macrophages. J. Clin. Investig. 2015, 125, 4223–4238. [Google Scholar] [CrossRef]
- Hucke, S.; Eschborn, M.; Liebmann, M.; Herold, M.; Freise, N.; Engbers, A.; Ehling, P.; Meuth, S.G.; Roth, J.; Kuhlmann, T.; et al. Sodium chloride promotes pro-inflammatory macrophage polarization thereby aggravating CNS autoimmunity. J. Autoimmun. 2016, 67, 90–101. [Google Scholar] [CrossRef]
- Choi, S.Y.; Lee-Kwon, W.; Kwon, H.M. The evolving role of TonEBP as an immunometabolic stress protein. Nat. Rev. Nephrol. 2020, 16, 352–364. [Google Scholar] [CrossRef]
- Bogdan, C. Nitric oxide and the immune response. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, C. Nitric oxide synthase in innate and adaptive immunity: An update. Trends Immunol. 2015, 36, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Flannagan, R.S.; Cosío, G.; Grinstein, S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 2009, 7, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.-H.; Hong, B.-K.; Choi, S.Y.; Moo Kwon, H.; Cho, C.-S.; Yi, E.C.; Kim, W.-U. Reactive oxygen species regulate context-dependent inhibition of NFAT5 target genes. Exp. Mol. Med. 2013, 45, e32. [Google Scholar] [CrossRef] [PubMed]
- Naguro, I.; Umeda, T.; Kobayashi, Y.; Maruyama, J.; Hattori, K.; Shimizu, Y.; Kataoka, K.; Kim-Mitsuyama, S.; Uchida, S.; Vandewalle, A.; et al. ASK3 responds to osmotic stress and regulates blood pressure by suppressing WNK1-SPAK/OSR1 signaling in the kidney. Nat. Commun. 2012, 3, 1285. [Google Scholar] [CrossRef]
- Xie, Z.; Askari, A. Na(+)/K(+)-ATPase as a signal transducer. Eur. J. Biochem. 2002, 269, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.; Madan, N.; Ye, Q.; Duan, Q.; Li, Z.; Wang, S.; Si, S.; Xie, Z. Identification of a mutant α1 Na/K-ATPase that pumps but is defective in signal transduction. J. Biol. Chem. 2013, 288, 13295–13304. [Google Scholar] [CrossRef]
- Škubník, J.; Pavlíčková, V.; Rimpelová, S. Cardiac Glycosides as Immune System Modulators. Biomolecules 2021, 11, 659. [Google Scholar] [CrossRef]
- Huh, J.R.; Leung, M.W.L.; Huang, P.; Ryan, D.A.; Krout, M.R. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORγt activity. Nature 2011, 472, 486–490. [Google Scholar] [CrossRef]
- Karaś, K.; Sałkowska, A.; Sobalska-Kwapis, M.; Walczak-Drzewiecka, A.; Strapagiel, D. Digoxin, an Overlooked Agonist of RORγ/RORγT. Front. Pharmacol. 2018, 9, 1460. [Google Scholar] [CrossRef]
- Kobayashi, M.; Usui-Kawanishi, F.; Karasawa, T.; Kimura, H.; Watanabe, S.; Mise, N.; Kayama, F.; Kasahara, T.; Hasebe, N.; Takahashi, M. The cardiac glycoside ouabain activates NLRP3 inflammasomes and promotes cardiac inflammation and dysfunction. PLoS ONE 2017, 12, e0176676. [Google Scholar] [CrossRef] [PubMed]
- Ip, W.K.E.; Medzhitov, R. Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat. Commun. 2015, 6, 6931. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Mechanisms contributing to the cardiac inotropic effect of Na pump inhibition and reduction of extracellular Na. J. Gen. Physiol. 1987, 90, 479–504. [Google Scholar] [CrossRef] [PubMed]
- Barry, W.H.; Bridge, J.H. Intracellular calcium homeostasis in cardiac myocytes. Circulation 1993, 87, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Altamirano, J.; Li, Y.; DeSantiago, J.; Piacentino, V.; Houser, S.R. The inotropic effect of cardioactive glycosides in ventricular myocytes requires Na+-Ca2+ exchanger function. J. Physiol. 2006, 575, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Gallin, E.K.; Gallin, J.I. Interaction of chemotactic factors with human macrophages. Induction of transmembrane potential changes. J. Cell Biol. 1977, 75, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Gallin, E.K.; Livengood, D.R. Demonstration of an electrogenic Na+-K+ pump in mouse spleen macrophages. Am. J. Physiol. 1983, 245, C184–C188. [Google Scholar] [CrossRef]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wülfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522.e20. [Google Scholar] [CrossRef]
- Pinnell, J.; Turner, S.; Howell, S. Cardiac muscle physiology. Contin. Educ. Anaesth. Crit. Care Pain 2007, 7, 85–88. [Google Scholar] [CrossRef]
- Fontana, J.M.; Burlaka, I.; Khodus, G.; Brismar, H.; Aperia, A. Calcium oscillations triggered by cardiotonic steroids. FEBS J. 2013, 280, 5450–5455. [Google Scholar] [CrossRef]
- Grant, A.O. Cardiac ion channels. Circ. Arrhythm. Electrophysiol. 2009, 2, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Lee, J.D.; Bibbs, L.; Ulevitch, R.J. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 1994, 265, 808–811. [Google Scholar] [CrossRef]
- Lopez-Rodríguez, C.; Aramburu, J.; Rakeman, A.S.; Rao, A. NFAT5, a constitutively nuclear NFAT protein that does not cooperate with Fos and Jun. Proc. Natl. Acad. Sci. USA 1999, 96, 7214–7219. [Google Scholar] [CrossRef]
- Miyakawa, H.; Woo, S.K.; Dahl, S.C.; Handler, J.S.; Kwon, H.M. Tonicity-responsive enhancer binding protein, a rel-like protein that stimulates transcription in response to hypertonicity. Proc. Natl. Acad. Sci. USA 1999, 96, 2538–2542. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues-Mascarenhas, S.; Da Silva de Oliveira, A.; Amoedo, N.D.; Affonso-Mitidieri, O.R.; Rumjanek, F.D.; Rumjanek, V.M. Modulation of the immune system by ouabain. Ann. N. Y. Acad. Sci. 2009, 1153, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante-Silva, L.H.A.; Carvalho, D.C.M.; Almeida Lima É de Rodrigues-Mascarenhas, S. Ouabain inhibits p38 activation in mice neutrophils. Inflammopharmacology 2021, 29, 1829–1833. [Google Scholar] [CrossRef]
- Valente, R.C.; Nascimento, C.R.; Araujo, E.G.; Rumjanek, V.M. mCD14 expression in human monocytes is downregulated by ouabain via transactivation of epithelial growth factor receptor and activation of p38 mitogen-activated protein kinase. Neuroimmunomodulation 2009, 16, 228–236. [Google Scholar] [CrossRef]
- Watanabe, K.; Morishita, K.; Zhou, X.; Shiizaki, S.; Uchiyama, Y.; Koike, M.; Naguro, I.; Ichijo, H. Cells recognize osmotic stress through liquid-liquid phase separation lubricated with poly(ADP-ribose). Nat. Commun. 2021, 12, 1353. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krampert, L.; Ossner, T.; Schröder, A.; Schatz, V.; Jantsch, J. Simultaneous Increases in Intracellular Sodium and Tonicity Boost Antimicrobial Activity of Macrophages. Cells 2023, 12, 2816. https://doi.org/10.3390/cells12242816
Krampert L, Ossner T, Schröder A, Schatz V, Jantsch J. Simultaneous Increases in Intracellular Sodium and Tonicity Boost Antimicrobial Activity of Macrophages. Cells. 2023; 12(24):2816. https://doi.org/10.3390/cells12242816
Chicago/Turabian StyleKrampert, Luka, Thomas Ossner, Agnes Schröder, Valentin Schatz, and Jonathan Jantsch. 2023. "Simultaneous Increases in Intracellular Sodium and Tonicity Boost Antimicrobial Activity of Macrophages" Cells 12, no. 24: 2816. https://doi.org/10.3390/cells12242816
APA StyleKrampert, L., Ossner, T., Schröder, A., Schatz, V., & Jantsch, J. (2023). Simultaneous Increases in Intracellular Sodium and Tonicity Boost Antimicrobial Activity of Macrophages. Cells, 12(24), 2816. https://doi.org/10.3390/cells12242816