
Cholesteryl Hemiazelate Present in Cardiovascular Disease Patients Causes Lysosome Dysfunction in Murine Fibroblasts

 , , , ,
, , , ,  ,
,  ,
,  and
and 
Abstract
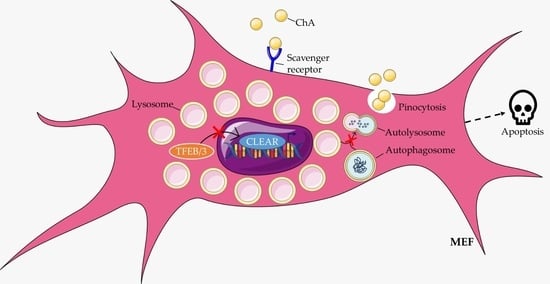
:
1. Introduction
2. Materials and Methods
2.1. Liposome Preparation
2.2. Cell Culture and Lipid Treatment
2.3. Fluorescence Microscopy, Staining, Image Acquisition and Analysis
2.4. Western Blotting
2.5. LDH Assay
2.6. Total and Free Cholesterol and Cholesteryl Ester Measurement
2.7. qRT-PCR
2.8. LAL Activity Assay
2.9. Endocytosis Evaluation by Flow Cytometry
2.10. Micropinocytosis Evaluation by Flow Cytometry
2.11. Statistical Analysis
3. Results
3.1. Cholesteryl Hemiazelate Is Sufficient to Cause Lysosome Dysfunction in Mouse Embryonic Fibroblasts
3.2. Cholesteryl Hemiazelate Inhibits Autophagy in Mouse Embryonic Fibroblasts
3.3. Cholesteryl Hemiazelate Causes Apoptosis in Mouse Embryonic Fibroblasts
3.4. The Presence of TFEB/3 Is Required for the Internalization of Cholesteryl Hemiazelate
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, A.S.; Vellarikkal, S.K.; Edelman, E.R.; Nguyen, L.; Subramanian, A.; Ellinor, P.T.; Regev, A.; Kathiresan, S.; Gupta, R.M. Single-Cell Analysis of the Normal Mouse Aorta Reveals Functionally Distinct Endothelial Cell Populations. Circulation 2019, 140, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Tillie, R.J.H.A.; van Kuijk, K.; Sluimer, J.C. Fibroblasts in atherosclerosis: Heterogeneous and plastic participants. Curr. Opin. Lipidol. 2020, 31, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Kuret, T.; Sodin-Šemrl, S. The Role of Fibroblasts in Atherosclerosis Progression. In Fibroblasts-Advances in Inflammation, Autoimmunity and Cancer; Bertoncelj, M.F., Lakota, K., Blumenberg, M., Eds.; Intech Open: London, UK, 2021; p. 136. [Google Scholar]
- Wang, J.; Wang, Y.; Wang, J.; Guo, X.; Chan, E.C.; Jiang, F. Adventitial Activation in the Pathogenesis of Injury-Induced Arterial Remodeling: Potential Implications in Transplant Vasculopathy. Am. J. Pathol. 2018, 188, 838–845. [Google Scholar] [CrossRef]
- Goel, S.A.; Guo, L.W.; Liu, B.; Kent, K.C. Mechanisms of post-intervention arterial remodelling. Cardiovasc. Res. 2012, 96, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Coen, M.; Gabbiani, G.; Bochaton-Piallat, M.L. Myofibroblast-mediated adventitial remodeling: An underestimated player in arterial pathology. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2391–2396. [Google Scholar] [CrossRef] [PubMed]
- Christen, T.; Verin, V.; Bochaton-Piallat, M.L.; Popowski, Y.; Ramaekers, F.; Debruyne, P.; Camenzind, E.; Van Eys, G.; Gabbiani, G. Mechanisms of neointima formation and remodeling in the porcine coronary artery. Circulation 2001, 103, 882–888. [Google Scholar] [CrossRef]
- Singh, S.; Torzewski, M. Fibroblasts and Their Pathological Functions in the Fibrosis of Aortic Valve Sclerosis and Atherosclerosis. Biomolecules 2019, 9, 472. [Google Scholar] [CrossRef]
- Brokopp, C.E.; Schoenauer, R.; Richards, P.; Bauer, S.; Lohmann, C.; Emmert, M.Y.; Weber, B.; Winnik, S.; Aikawa, E.; Graves, K.; et al. Fibroblast activation protein is induced by inflammation and degrades type I collagen in thin-cap fibroatheromata. Eur. Heart J. 2011, 32, 2713–2722. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Yeager, M.E.; El Kasmi, K.C.; Nozik-Grayck, E.; Gerasimovskaya, E.V.; Li, M.; Riddle, S.R.; Frid, M.G. The adventitia: Essential regulator of vascular wall structure and function. Annu. Rev. Physiol. 2013, 75, 23–47. [Google Scholar] [CrossRef] [PubMed]
- Maiellaro, K.; Taylor, W.R. The role of the adventitia in vascular inflammation. Cardiovasc. Res. 2007, 75, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, J.T.; Tallquist, M.D. Tracking Adventitial Fibroblast Contribution to Disease. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; O’Brien, J.E.; Fard, A.; Mannion, J.D.; Wang, D.; Zalewski, A. Adventitial myofibroblasts contribute to neointimal formation in injured porcine coronary arteries. Circulation 1996, 94, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Siow, R.C.M.; Mallawaarachchi, C.M.; Weissberg, P.L. Migration of adventitial myofibroblasts following vascular balloon injury: Insights from in vivo gene transfer to rat carotid arteries. Cardiovasc. Res. 2003, 59, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.; Weber, J.; Nusser-Stein, S.; Pahla, J.; Zhang, H.E.; Mohammed, S.A.; Oppi, S.; Gaul, D.S.; Paneni, F.; Tailleux, A.; et al. Deletion of fibroblast activation protein provides atheroprotection. Cardiovasc. Res. 2021, 117, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.A.; Ramos, C.; Machado-Oliveira, G.; Vieira, O.V. Lysosome (Dys)function in Atherosclerosis—A Big Weight on the Shoulders of a Small Organelle. Front. Cell Dev. Biol. 2021, 9, 658995. [Google Scholar] [CrossRef]
- Steinberg, D.; Witztum, J.L. Lipoproteins and atherogenesis. Current concepts. JAMA 1990, 264, 3047–3052. [Google Scholar] [CrossRef]
- Matthiesen, R.; Lauber, C.; Sampaio, J.L.; Domingues, N.; Alves, L.; Gerl, M.J.; Almeida, M.S.; Rodrigues, G.; Araújo Gonçalves, P.; Ferreira, J.; et al. Shotgun mass spectrometry-based lipid profiling identifies and distinguishes between chronic inflammatory diseases. EBioMedicine 2021, 70, 103504. [Google Scholar] [CrossRef]
- Domingues, N.; Gaifem, J.; Matthiesen, R.; Saraiva, D.P.; Bento, L.; Marques, A.R.A.; Soares, M.I.L.; Sampaio, J.; Klose, C.; Surma, M.A.; et al. Cholesteryl hemiazelate identified in CVD patients causes in vitro and in vivo inflammation. J. Lipid Res. 2023, 64, 100419. [Google Scholar] [CrossRef]
- Alves, L.S.; Marques, A.R.A.; Padrão, N.; Carvalho, F.A.; Ramalho, J.; Lopes, C.S.; Soares, M.I.L.; Futter, C.E.; Pinho, E.; Melo, T.M.V.D.; et al. Cholesteryl hemiazelate causes lysosome dysfunction impacting vascular smooth muscle cell homeostasis. J. Cell Sci. 2022, 135, jcs254631. [Google Scholar] [CrossRef]
- Domingues, N.; Marques, A.R.A.; Calado, R.D.A.; Ferreira, I.S.; Ramos, C.; Ramalho, J.; Soares, M.I.L.; Pereira, T.; Oliveira, L.; Vicente, J.R.; et al. Oxidized cholesteryl ester induces exocytosis of dysfunctional lysosomes in lipidotic macrophages. Traffic 2023, 24, 284–307. [Google Scholar] [CrossRef]
- Boullier, A.; Bird, D.A.; Chang, M.K.; Dennis, E.A.; Friedman, P.; Gillotte-Taylor, K.; Hörkkö, S.; Palinski, W.; Quehenberger, O.; Shaw, P.; et al. Scavenger Receptors, Oxidized LDL, and Atherosclerosis. Ann. N. Y. Acad. Sci. 2001, 947, 214–223. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of OxLDL and Its Impact on Cardiovascular Health: Focus on Atherosclerosis. Front. Pharmacol. 2021, 11, 613780. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; da Costa Martins, P.A.; Bitsch, N.; Pintelon, I.; de Meyer, G.R.Y.; Martinet, W.; Schrijvers, D.M. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy 2015, 11, 2014–2032. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; Moulis, M.; Roth, L.; Martinet, W.; Vindis, C.; Bennett, M.R.; De Meyer, G.R.Y. Vascular smooth muscle cell death, autophagy and senescence in atherosclerosis. Cardiovasc. Res. 2018, 114, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.C.; Chen, C.P.; Wefler, V.; Raftery, A. A Stable Reagent for the Liebermann-Burchard Reaction: Application to Rapid Serum Cholesterol Determination. Anal. Chem. 1961, 33, 1405–1407. [Google Scholar] [CrossRef]
- Martina, J.A.; Diab, H.I.; Brady, O.A.; Puertollano, R. TFEB and TFE3 are novel components of the integrated stress response. EMBO J. 2016, 35, 479–495. [Google Scholar] [CrossRef] [PubMed]
- Tol, M.J.; van der Lienden, M.J.C.; Gabriel, T.L.; Hagen, J.J.; Scheij, S.; Veenendaal, T.; Klumperman, J.; Donker-Koopman, W.E.; Verhoeven, A.J.; Overkleeft, H.; et al. HEPES activates a MiT/TFE-dependent lysosomal-autophagic gene network in cultured cells: A call for caution. Autophagy 2018, 14, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef]
- Bright, N.A.; Davis, L.J.; Luzio, J.P. Endolysosomes Are the Principal Intracellular Sites of Acid Hydrolase Activity. Curr. Biol. 2016, 26, 2233–2245. [Google Scholar] [CrossRef] [PubMed]
- Ameis, D.; Merkel, M.; Eckerskorn, C.; Greten, H. Purification, characterization and molecular cloning of human hepatic lysosomal acid lipase. Eur. J. Biochem. 1994, 219, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Zschenker, O.; Oezden, D.; Ameis, D. Lysosomal acid lipase as a preproprotein. J. Biochem. 2004, 136, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Sung, T.; Lin, N.; Abraham, R.T.; Jessen, B.A. Lysosomal adaptation: How cells respond to lysosomotropic compounds. PLoS ONE 2017, 12, e0173771. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.Y.; Alamri, A.H.; Valapala, M. Nutrient deprivation and lysosomal stress induce activation of TFEB in retinal pigment epithelial cells. Cell. Mol. Biol. Lett. 2019, 24, 33. [Google Scholar] [CrossRef] [PubMed]
- Ni, D.; Mo, Z.; Yi, G. Recent insights into atherosclerotic plaque cell autophagy. Exp. Biol. Med. (Maywood) 2021, 246, 2553–2558. [Google Scholar] [CrossRef]
- Razani, B.; Feng, C.; Coleman, T.; Emanuel, R.; Wen, H.; Hwang, S.; Ting, J.P.; Virgin, H.W.; Kastan, M.B.; Semenkovich, C.F. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012, 15, 534–544. [Google Scholar] [CrossRef]
- Tang, Y.; Wu, H.; Shao, B.; Wang, Y.; Liu, C.; Guo, M. Celosins inhibit atherosclerosis in ApoE−/− mice and promote autophagy flow. J. Ethnopharmacol. 2018, 215, 74–82. [Google Scholar] [CrossRef]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef]
- Nakatogawa, H. Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol. 2020, 21, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [PubMed]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Estronca, L.M.B.B.; Silva, J.C.P.; Sampaio, J.L.; Shevchenko, A.; Verkade, P.; Vaz, A.D.N.; Vaz, W.L.C.; Vieira, O.V. Molecular etiology of atherogenesis–in vitro induction of lipidosis in macrophages with a new LDL model. PLoS ONE 2012, 7, e34822. [Google Scholar] [CrossRef] [PubMed]
- Bern, M.; Sand, K.M.K.; Nilsen, J.; Sandlie, I.; Andersen, J.T. The role of albumin receptors in regulation of albumin homeostasis: Implications for drug delivery. J. Control Release 2015, 211, 144–162. [Google Scholar] [CrossRef] [PubMed]
- Miller, Y.I. Toll-like receptors and atherosclerosis: Oxidized LDL as an endogenous Toll-like receptor ligand. Future Cardiol. 2005, 1, 785–792. [Google Scholar] [CrossRef]
- Choi, S.-H.; Harkewicz, R.; Lee, J.H.; Boullier, A.; Almazan, F.; Li, A.C.; Witztum, J.L.; Bae, Y.S.; Miller, Y.I. Lipoprotein Accumulation in Macrophages via Toll-Like Receptor-4–Dependent Fluid Phase Uptake. Circ. Res. 2009, 104, 1355–1363. [Google Scholar] [CrossRef]
- Enzerink, A.; Vaheri, A. Fibroblast activation in vascular inflammation. J. Thromb. Haemost. 2011, 9, 619–626. [Google Scholar] [CrossRef]
- Hu, D.; Yin, C.; Luo, S.; Habenicht, A.J.R.; Mohanta, S.K. Vascular Smooth Muscle Cells Contribute to Atherosclerosis Immunity. Front. Immunol. 2019, 10, 1101. [Google Scholar] [CrossRef]
- Buckley, C.D.; Pilling, D.; Lord, J.M.; Akbar, A.N.; Scheel-Toellner, D.; Salmon, M. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol. 2001, 22, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science (1979) 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Maxson, M.E.; Grinstein, S. The vacuolar-type H+-ATPase at a glance–more than a proton pump. J. Cell Sci. 2014, 127, 4987–4993. [Google Scholar] [CrossRef] [PubMed]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Martinet, W.; De Meyer, G.R.Y. Autophagy in atherosclerosis: A cell survival and death phenomenon with therapeutic potential. Circ. Res. 2009, 104, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011, 13, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Weissman, I.L.; Leeper, N.J. The Role of Efferocytosis in Atherosclerosis. Circulation 2017, 135, 476–489. [Google Scholar] [CrossRef]
- Doran, A.C.; Yurdagul, A.; Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 2020, 20, 254–267. [Google Scholar] [CrossRef]
- Wang, L.; Li, H.; Tang, Y.; Yao, P. Potential Mechanisms and Effects of Efferocytosis in Atherosclerosis. Front. Endocrinol. 2021, 11, 585285. [Google Scholar] [CrossRef]
- Li, M.; Wang, Z.; Wang, P.; Li, H.; Yang, L. TFEB: A Emerging Regulator in Lipid Homeostasis for Atherosclerosis. Front. Physiol. 2021, 12, 639920. [Google Scholar] [CrossRef]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Jürgen Klisch, T.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Pastore, N.; Vainshtein, A.; Klisch, T.J.; Armani, A.; Huynh, T.; Herz, N.J.; Polishchuk, E.V.; Sandri, M.; Ballabio, A. TFE3 regulates whole-body energy metabolism in cooperation with TFEB. EMBO Mol. Med. 2017, 9, 605–621. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, X.; Zheng, L.; Kou, J.; Zhong, Z.; Jiang, Y.; Wang, W.; Dong, Z.; Liu, Z.; Han, X.; et al. Hypericin-mediated sonodynamic therapy induces autophagy and decreases lipids in THP-1 macrophage by promoting ROS-dependent nuclear translocation of TFEB. Cell Death Dis. 2016, 7, e2527. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence | |
|---|---|---|
| Forward | Reverse | |
| Tfeb | AGGAGCGGCAGAAGAAAGAC | CAGGTCCTTCTGCATCCTCC |
| Tfe3 | CCTGAAGGCATCTGTGGATT | TGTAGGTCCAGAAGGGCATC |
| Lamp-1 | ACATCAGCCCAAATGACACA | GGCTAGAGCTGGCATTCATC |
| Ctsd | GCTTCCGGTCTTTGACAACCT | CACCAAGCATTAGTTCTCCTCC |
| Ctsb | TCCTTGATCCTTCTTTCTTGCC | ACAGTGCCACACAGCTTCTTC |
| Mcln1 | GCGCCTATGACACCATCAA | TATCCTGGCACTGCTCGAT |
| Lipa | CTAGAATCTGCCAGCAAGCC | AGTATTCACCGAATCCCTCG |
| Atp6v0d2 | CAGAGCTGTACTTCAATGTGGAC | AGGTCTCACACTGCACTAGGT |
| Map1/lc3 | GACGGCTTCCTGTACATGGTTT | TGGAGTCTTACACAGCCATTGC |
| Sqstm1 | 5GTCTTCTGTGCCTGTGCTGGAA | TCTGCTCCACCAGAAGATCCCA |
| Pgk1 | ATGGATGAGGTGGTGAAAGC | CAGTGCTCACATGGCTGACT |
| Gapdh | GGGAAGCCCATCACCATCTTC | AGAGGGGCCATCCACAGTCT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopes, E.; Machado-Oliveira, G.; Simões, C.G.; Ferreira, I.S.; Ramos, C.; Ramalho, J.; Soares, M.I.L.; Melo, T.M.V.D.P.e.; Puertollano, R.; Marques, A.R.A.; et al. Cholesteryl Hemiazelate Present in Cardiovascular Disease Patients Causes Lysosome Dysfunction in Murine Fibroblasts. Cells 2023, 12, 2826. https://doi.org/10.3390/cells12242826
Lopes E, Machado-Oliveira G, Simões CG, Ferreira IS, Ramos C, Ramalho J, Soares MIL, Melo TMVDPe, Puertollano R, Marques ARA, et al. Cholesteryl Hemiazelate Present in Cardiovascular Disease Patients Causes Lysosome Dysfunction in Murine Fibroblasts. Cells. 2023; 12(24):2826. https://doi.org/10.3390/cells12242826
Chicago/Turabian StyleLopes, Elizeth, Gisela Machado-Oliveira, Catarina Guerreiro Simões, Inês S. Ferreira, Cristiano Ramos, José Ramalho, Maria I. L. Soares, Teresa M. V. D. Pinho e Melo, Rosa Puertollano, André R. A. Marques, and et al. 2023. "Cholesteryl Hemiazelate Present in Cardiovascular Disease Patients Causes Lysosome Dysfunction in Murine Fibroblasts" Cells 12, no. 24: 2826. https://doi.org/10.3390/cells12242826
APA StyleLopes, E., Machado-Oliveira, G., Simões, C. G., Ferreira, I. S., Ramos, C., Ramalho, J., Soares, M. I. L., Melo, T. M. V. D. P. e., Puertollano, R., Marques, A. R. A., & Vieira, O. V. (2023). Cholesteryl Hemiazelate Present in Cardiovascular Disease Patients Causes Lysosome Dysfunction in Murine Fibroblasts. Cells, 12(24), 2826. https://doi.org/10.3390/cells12242826