

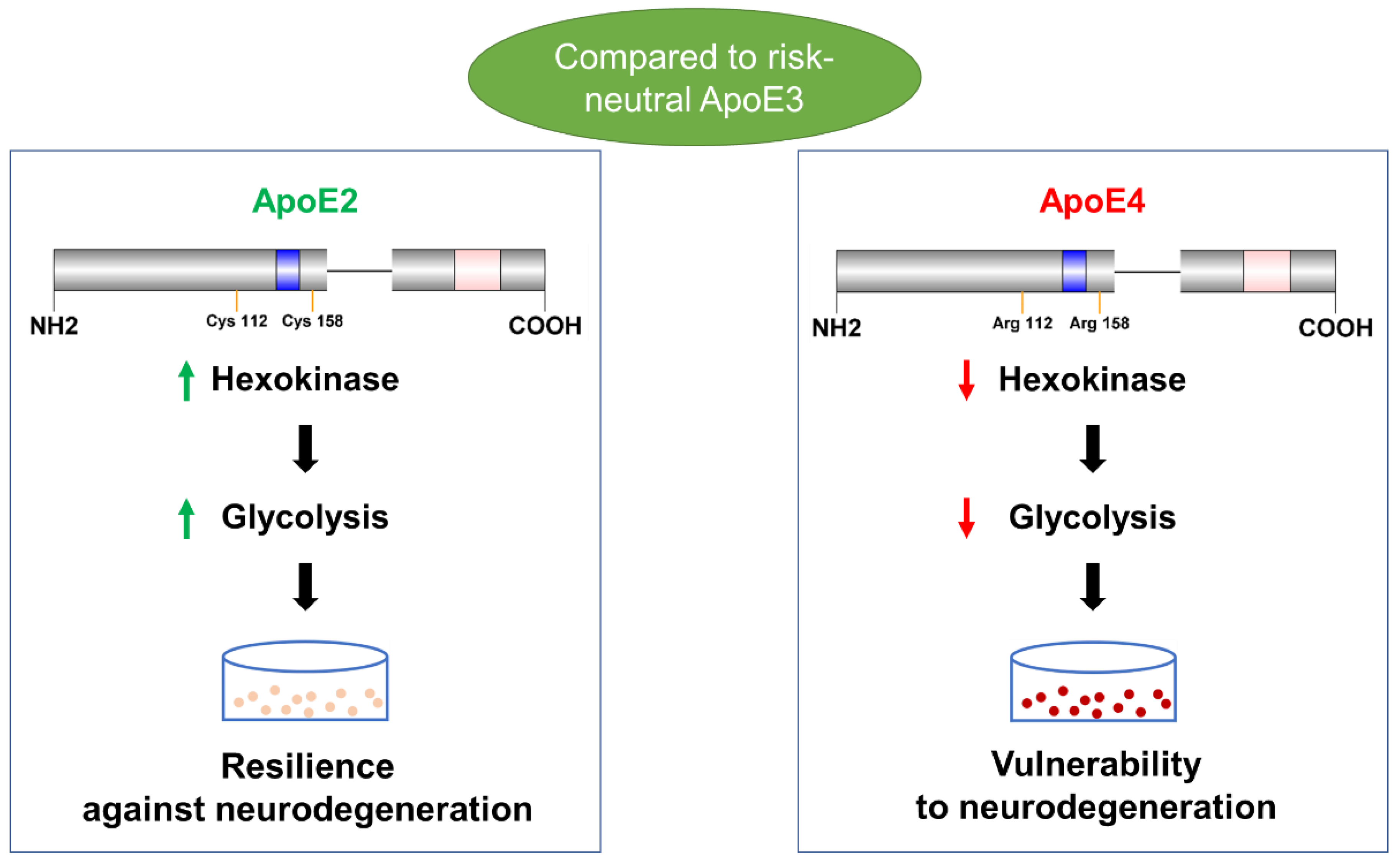
Opposing Effects of ApoE2 and ApoE4 on Glycolytic Metabolism in Neuronal Aging Supports a Warburg Neuroprotective Cascade against Alzheimer’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Models
2.1.1. Generation and Maintenance of N2a-hApoE Stable Cell Lines
2.1.2. Neuronal Differentiation of N2a-hApoE Stable Cell Lines with Retinoic Acid
2.2. Animals
2.3. Western Immunoblotting
2.4. Hexokinase Activity Assay
2.5. Glycolytic Stress Test
2.6. LIVE/DEAD Cell Assay
2.7. Phase Contrast Imaging
2.8. Metabolic Activity Assay
2.9. Transfection of ApoE2 in N2a-ApoE4 Cells
2.10. Protein Carbonylation Assay
2.11. Statistical Analyses
3. Results
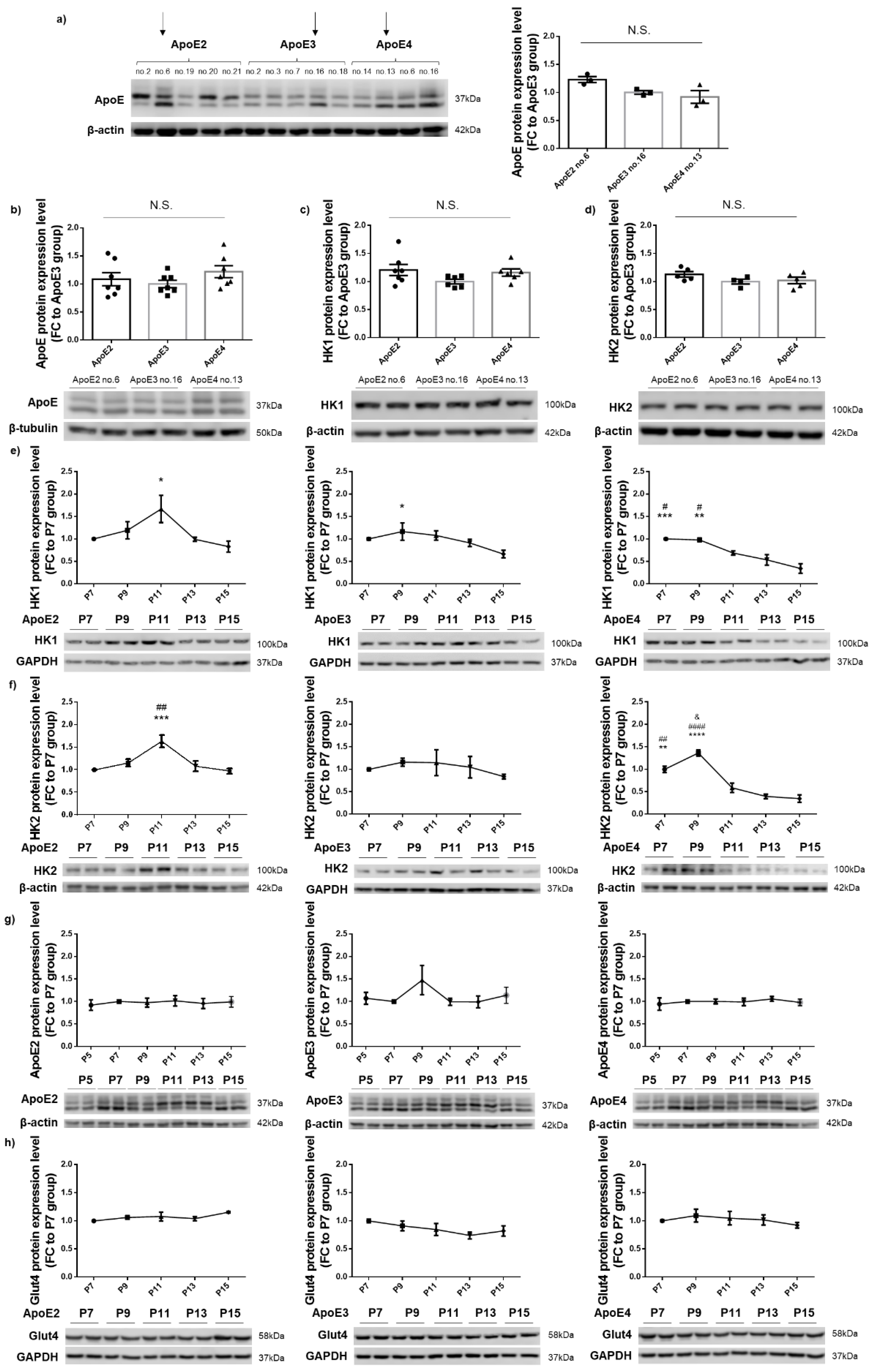
3.1. HK Expression Remained Relatively Stable in ApoE2- and ApoE3-Expressing Cells but Exhibited a Gradual Decrease with Increasing Passages in ApoE4-Expressing Cells
3.2. ApoE2 Upregulated Whereas ApoE4 Downregulated HK, Which Occurred Concurrently with Downregulation of PFKP and PKM1 by ApoE2 and Upregulation by ApoE4
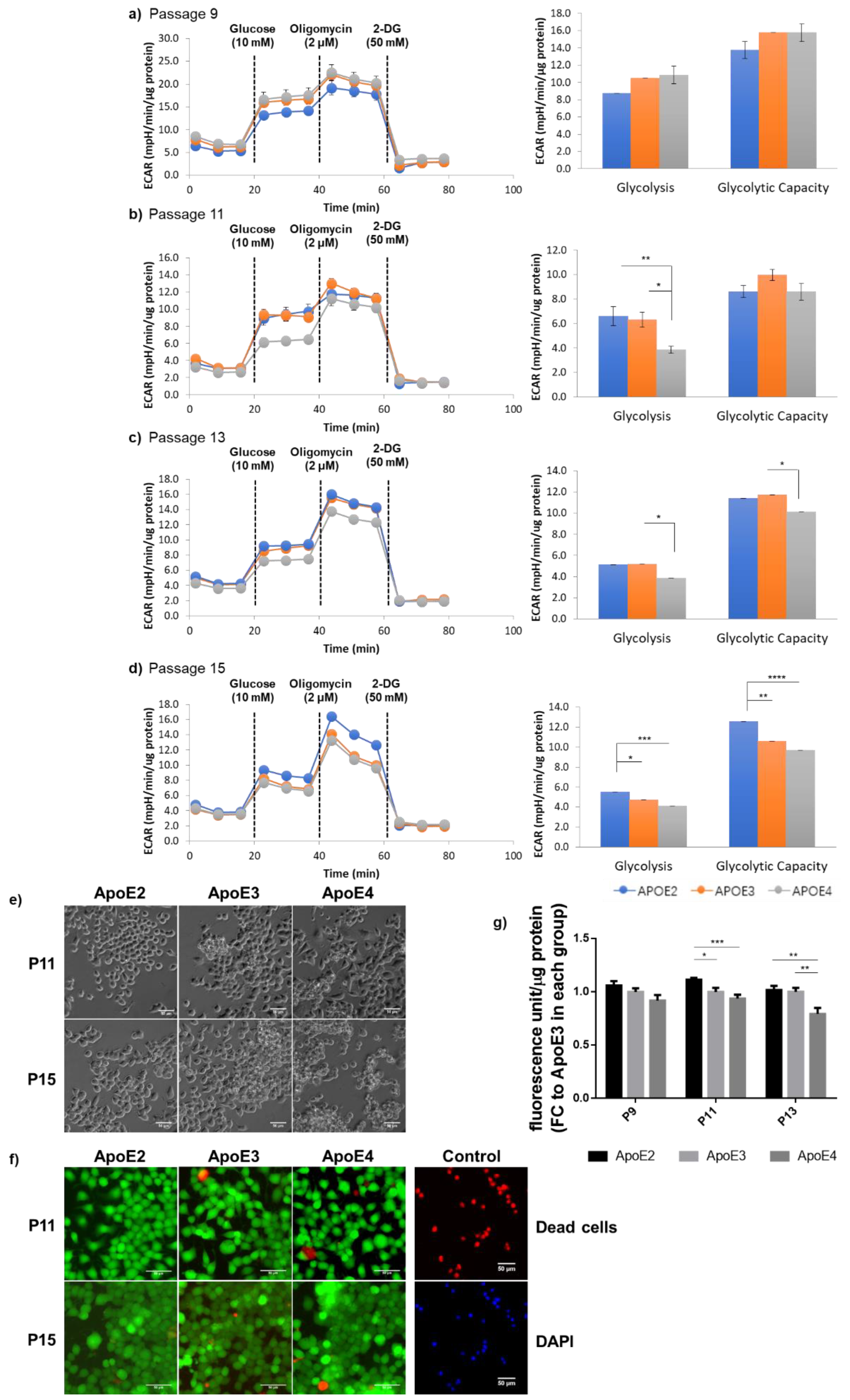
3.3. ApoE2-Expressing Cells Exhibited Enhanced Glycolytic Activity When Compared to ApoE4-Expressing Cells, Which Correlated with the General State of Cellular Health
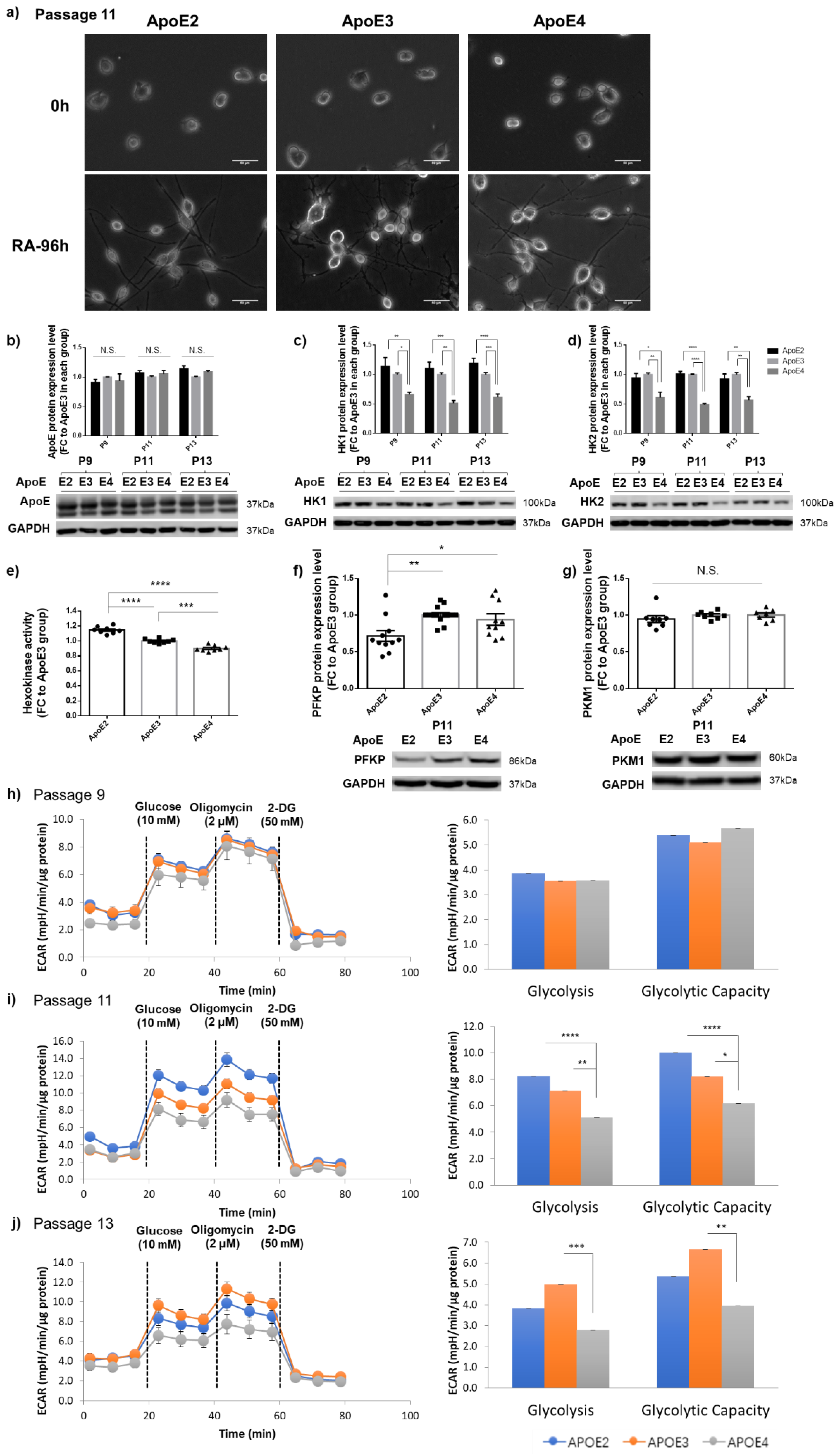
3.4. Differential Regulation of HK and Glycolytic Activity by ApoE Isoforms Was Also Observed in Differentiated Neuron-like Cells
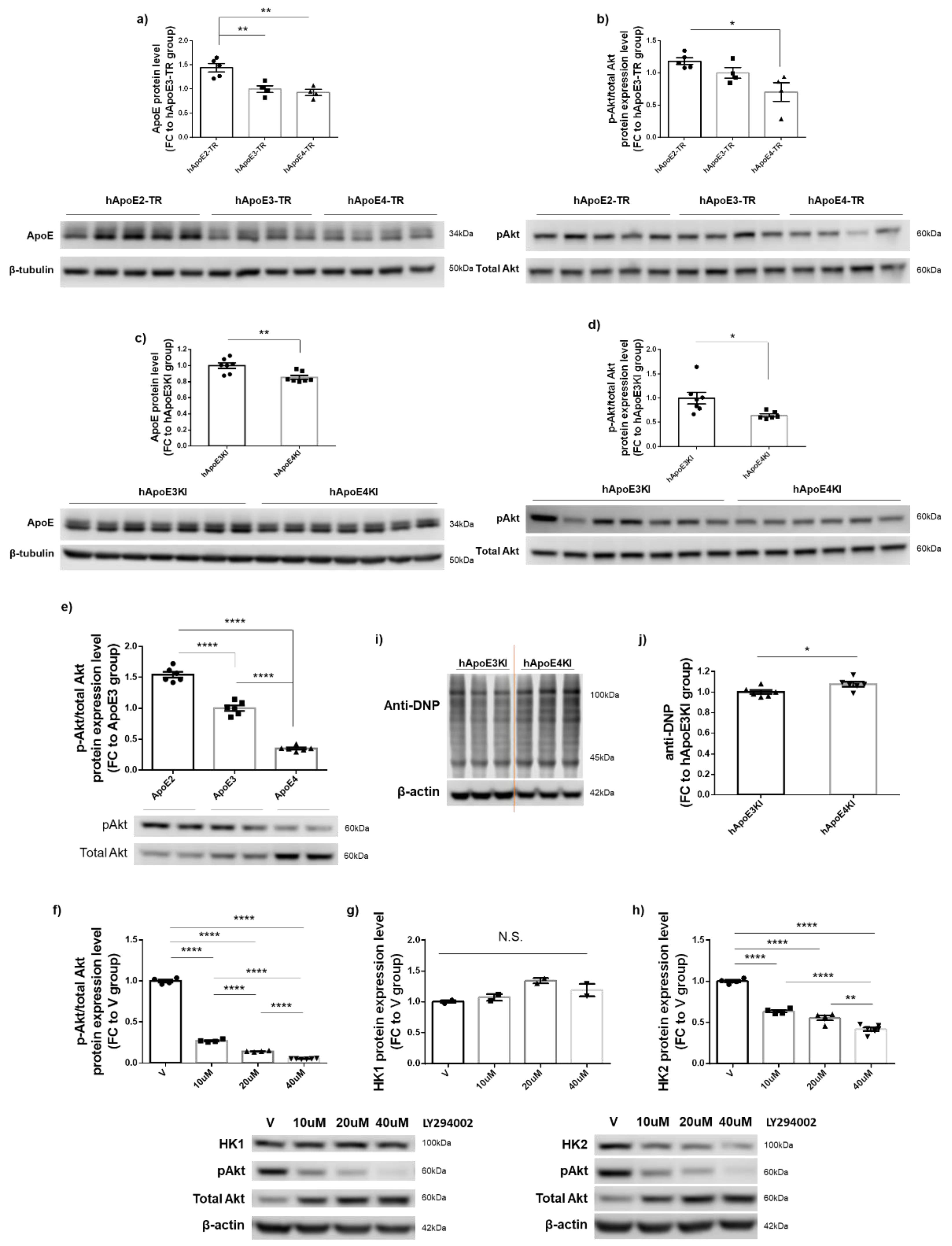
3.5. Introduction of ApoE2 Ameliorated HK Deficits and Further Improved Glycolytic Metabolism in ApoE4-Expressing Cells
3.6. PI3K/Akt Signaling Activity Appeared to Be Responsible for ApoE2-Mediated Upregulation of HK2 but Not of HK1 Expression
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mahley, R.W.; Huang, Y. Apolipoprotein e sets the stage: Response to injury triggers neuropathology. Neuron 2012, 76, 871–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahley, R.W.; Rall, S.C., Jr. Apolipoprotein E: Far more than a lipid transport protein. Annu. Rev. Genom. Hum. Genet. 2000, 1, 507–537. [Google Scholar] [CrossRef]
- Liu, C.-C.; Liu, C.-C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Association. 2020 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. 2021, 17, 391. [Google Scholar]
- Morris, C.M.; Benjamin, R.; Leake, A.; McArthur, F.K.; Candy, J.M.; Ince, P.G.; Torvik, A.; Bjertness, E.; Edwardson, J.A. Effect of apolipoprotein E genotype on Alzheimer’s disease neuropathology in a cohort of elderly Norwegians. Neurosci. Lett. 1995, 201, 45–47. [Google Scholar] [CrossRef]
- Nagy, Z.; Esiri, M.M.; Jobst, K.A.; Johnston, C.; Litchfield, S.; Sim, E.; Smith, A.D. Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer’s disease. Neuroscience 1995, 69, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.S.; Bienias, J.L.; Berry-Kravis, E.; Evans, D.A.; Bennett, D.A. The apolipoprotein E epsilon 2 allele and decline in episodic memory. J. Neurol. Neurosurg. Psychiatry 2002, 73, 672–677. [Google Scholar] [CrossRef]
- Mosconi, L.; De Santi, S.; Li, J.; Tsui, W.H.; Li, Y.; Boppana, M.; Laska, E.; Rusinek, H.; de Leon, M.J. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol. Aging 2008, 29, 676–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.; Marcus, D.L.; Landman, J.; Kooby, D.; Frey, W., 2nd; Freedman, M.L. Brain glucose metabolism in Alzheimer’s disease. Am. J. Med. Sci. 1994, 308, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A. Chapter Five—APOE and metabolic dysfunction in Alzheimer’s disease. In International Review of Neurobiology; Söderbom, G., Esterline, R., Oscarsson, J., Mattson, M.P., Eds.; Academic Press: Cambridge, MA, USA, 2020; Volume 154, pp. 131–151. [Google Scholar]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc. 2018, 14, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Vlassenko, A.G.; Gordon, B.A.; Goyal, M.S.; Su, Y.; Blazey, T.M.; Durbin, T.J.; Couture, L.E.; Christensen, J.J.; Jafri, H.; Morris, J.C.; et al. Aerobic glycolysis and tau deposition in preclinical Alzheimer’s disease. Neurobiol. Aging 2018, 67, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Theurey, P.; Connolly, N.M.C.; Fortunati, I.; Basso, E.; Lauwen, S.; Ferrante, C.; Moreira Pinho, C.; Joselin, A.; Gioran, A.; Bano, D.; et al. Systems biology identifies preserved integrity but impaired metabolism of mitochondria due to a glycolytic defect in Alzheimer’s disease neurons. Aging Cell 2019, 18, e12924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Alshakhshir, N.; Zhao, L. Glycolytic Metabolism, Brain Resilience, and Alzheimer’s Disease. Front. Neurosci. 2021, 15, 662242. [Google Scholar] [CrossRef]
- Newington, J.T.; Pitts, A.; Chien, A.; Arseneault, R.; Schubert, D.; Cumming, R.C. Amyloid beta resistance in nerve cell lines is mediated by the Warburg effect. PLoS ONE 2011, 6, e19191. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, L.; De Santi, S.; Brys, M.; Tsui, W.H.; Pirraglia, E.; Glodzik-Sobanska, L.; Rich, K.E.; Switalski, R.; Mehta, P.D.; Pratico, D.; et al. Hypometabolism and altered cerebrospinal fluid markers in normal apolipoprotein E E4 carriers with subjective memory complaints. Biol. Psychiatry 2008, 63, 609–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiman, E.M.; Caselli, R.J.; Yun, L.S.; Chen, K.; Bandy, D.; Minoshima, S.; Thibodeau, S.N.; Osborne, D. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N. Engl. J. Med. 1996, 334, 752–758. [Google Scholar] [CrossRef]
- Harris, R.A.; Lone, A.; Lim, H.; Martinez, F.; Frame, A.K.; Scholl, T.J.; Cumming, R.C. Aerobic Glycolysis Is Required for Spatial Memory Acquisition But Not Memory Retrieval in Mice. Eneuro 2019, 6, 0389-18.2019. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Zhang, X.; Zhao, L. Human ApoE Isoforms Differentially Modulate Brain Glucose and Ketone Body Metabolism: Implications for Alzheimer’s Disease Risk Reduction and Early Intervention. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 6665–6681. [Google Scholar] [CrossRef]
- Wu, L. Perturbed Brain Energy Metabolism in Alzheimer’s Disease and Diabetes. Ph.D. Thesis, University of Kansas, Lawrence, KS, USA, 2017. Available online: https://kuscholarworks.ku.edu/handle/1808/25391 (accessed on 16 November 2017).
- Tremblay, R.G.; Sikorska, M.; Sandhu, J.K.; Lanthier, P.; Ribecco-Lutkiewicz, M.; Bani-Yaghoub, M. Differentiation of mouse Neuro 2A cells into dopamine neurons. J. Neurosci. Methods 2010, 186, 60–67. [Google Scholar] [CrossRef]
- Wu, P.Y.; Lin, Y.C.; Chang, C.L.; Lu, H.T.; Chin, C.H.; Hsu, T.T.; Chu, D.; Sun, S.H. Functional decreases in P2X7 receptors are associated with retinoic acid-induced neuronal differentiation of Neuro-2a neuroblastoma cells. Cell. Signal. 2009, 21, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.M.; Mace, B.E.; Maeda, N.; Schmechel, D.E. Marked regional differences of brain human apolipoprotein E expression in targeted replacement mice. Neuroscience 2004, 124, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Zheng, H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 89. [Google Scholar] [CrossRef] [Green Version]
- Ding, F.; Yao, J.; Rettberg, J.R.; Chen, S.; Brinton, R.D. Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: Implication for bioenergetic intervention. PLoS ONE 2013, 8, e79977. [Google Scholar] [CrossRef] [Green Version]
- Poorman, R.A.; Randolph, A.; Kemp, R.G.; Heinrikson, R.L. Evolution of phosphofructokinase—Gene duplication and creation of new effector sites. Nature 1984, 309, 467–469. [Google Scholar] [CrossRef]
- Fernandes, P.M.; Kinkead, J.; McNae, I.; Michels, P.A.M.; Walkinshaw, M.D. Biochemical and transcript level differences between the three human phosphofructokinases show optimisation of each isoform for specific metabolic niches. Biochem. J. 2020, 477, 4425–4441. [Google Scholar] [CrossRef]
- Agostini, M.; Romeo, F.; Inoue, S.; Niklison-Chirou, M.V.; Elia, A.J.; Dinsdale, D.; Morone, N.; Knight, R.A.; Mak, T.W.; Melino, G. Metabolic reprogramming during neuronal differentiation. Cell Death Differ. 2016, 23, 1502–1514. [Google Scholar] [CrossRef] [Green Version]
- Carbonell, J.; Marco, R.; Felíu, J.E.; Sols, A. Pyruvate Kinase. Eur. J. Biochem. 1973, 37, 148–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [Green Version]
- Szablewski, L. Glucose Transporters in Brain: In Health and in Alzheimer’s Disease. J. Alzheimers Dis. JAD 2017, 55, 1307–1320. [Google Scholar] [CrossRef]
- Huang, Y.A.; Zhou, B.; Nabet, A.M.; Wernig, M.; Südhof, T.C. Differential Signaling Mediated by ApoE2, ApoE3, and ApoE4 in Human Neurons Parallels Alzheimer’s Disease Risk. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 7408–7427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laffont, I.; Takahashi, M.; Shibukawa, Y.; Honke, K.; Shuvaev, V.V.; Siest, G.; Visvikis, S.; Taniguchi, N. Apolipoprotein E activates Akt pathway in neuro-2a in an isoform-specific manner. Biochem. Biophys. Res. Commun. 2002, 292, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Miyamoto, S. Hexokinase II integrates energy metabolism and cellular protection: Akting on mitochondria and TORCing to autophagy. Cell Death Differ. 2015, 22, 248–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riddell, D.R.; Zhou, H.; Atchison, K.; Warwick, H.K.; Atkinson, P.J.; Jefferson, J.; Xu, L.; Aschmies, S.; Kirksey, Y.; Hu, Y.; et al. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 11445–11453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.J.; Carini, M.; Butterfield, D.A. Protein carbonylation. Antioxid. Redox. Signal. 2010, 12, 323–325. [Google Scholar] [CrossRef]
- Li, Z.; Shue, F.; Zhao, N.; Shinohara, M.; Bu, G. APOE2: Protective mechanism and therapeutic implications for Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 63. [Google Scholar] [CrossRef]
- Drzezga, A.; Riemenschneider, M.; Strassner, B.; Grimmer, T.; Peller, M.; Knoll, A.; Wagenpfeil, S.; Minoshima, S.; Schwaiger, M.; Kurz, A. Cerebral glucose metabolism in patients with AD and different APOE Genotypes. Neurology 2005, 64, 102. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 486–510. [Google Scholar] [CrossRef]
- Goyal, M.S.; Hawrylycz, M.; Miller, J.A.; Snyder, A.Z.; Raichle, M.E. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab. 2014, 19, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Moriyama, Y.; Maeda, M.; Futai, M. The role of V-ATPase in neuronal and endocrine systems. J. Exp. Biol. 1992, 172, 171. [Google Scholar] [CrossRef]
- Kohio, H.P.; Adamson, A.L. Glycolytic control of vacuolar-type ATPase activity: A mechanism to regulate influenza viral infection. Virology 2013, 444, 301–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, S. Glucose activates H+-ATPase in kidney epithelial cells. Am. J. Physiol.-Cell Physiol. 2004, 287, C97–C105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woody, S.K.; Zhou, H.; Ibrahimi, S.; Dong, Y.; Zhao, L. Human ApoE ɛ2 Promotes Regulatory Mechanisms of Bioenergetic and Synaptic Function in Female Brain: A Focus on V-type H +-ATPase. J. Alzheimers Dis. 2016, 53, 1015–1031. [Google Scholar] [CrossRef] [Green Version]
- Saraiva, L.M.; Seixas da Silva, G.S.; Galina, A.; da-Silva, W.S.; Klein, W.L.; Ferreira, S.T.; De Felice, F.G. Amyloid-β triggers the release of neuronal hexokinase 1 from mitochondria. PLoS ONE 2010, 5, e15230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorbi, S.; Mortilla, M.; Piacentini, S.; Tonini, S.; Amaducci, L. Altered hexokinase activity in skin cultured fibroblasts and leukocytes from Alzheimer’s disease patients. Neurosci. Lett. 1990, 117, 165–168. [Google Scholar] [CrossRef]
- Bigl, M.; Bleyl, A.-D.; Zedlick, D.; Arendt, T.; Bigl, V.; Eschrich, K. Changes of Activity and Isozyme Pattern of Phosphofructokinase in the Brains of Patients with Alzheimer’s Disease. J. Neurochem. 1996, 67, 1164–1171. [Google Scholar] [CrossRef]
- Bigl, M.; Brückner, M.K.; Arendt, T.; Bigl, V.; Eschrich, K. Activities of key glycolytic enzymes in the brains of patients with Alzheimer’s disease. J. Neural Transm. 1999, 106, 499–511. [Google Scholar] [CrossRef]
- Jayasena, T.; Poljak, A.; Braidy, N.; Smythe, G.; Raftery, M.; Hill, M.; Brodaty, H.; Trollor, J.; Kochan, N.; Sachdev, P. Upregulation of glycolytic enzymes, mitochondrial dysfunction and increased cytotoxicity in glial cells treated with Alzheimer’s disease plasma. PLoS ONE 2015, 10, e0116092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, N.; Liu, C.C.; Van Ingelgom, A.J.; Martens, Y.A.; Linares, C.; Knight, J.A.; Painter, M.M.; Sullivan, P.M.; Bu, G. Apolipoprotein E4 Impairs Neuronal Insulin Signaling by Trapping Insulin Receptor in the Endosomes. Neuron 2017, 96, 115–129.e115. [Google Scholar] [CrossRef] [Green Version]
- Orr, A.L.; Kim, C.; Jimenez-Morales, D.; Newton, B.W.; Johnson, J.R.; Krogan, N.J.; Swaney, D.L.; Mahley, R.W. Neuronal Apolipoprotein E4 Expression Results in Proteome-Wide Alterations and Compromises Bioenergetic Capacity by Disrupting Mitochondrial Function. J. Alzheimers Dis. JAD 2019, 68, 991–1011. [Google Scholar] [CrossRef] [Green Version]
- Williams, H.C.; Farmer, B.C.; Piron, M.A.; Walsh, A.E.; Bruntz, R.C.; Gentry, M.S.; Sun, R.C.; Johnson, L.A. APOE alters glucose flux through central carbon pathways in astrocytes. Neurobiol. Dis. 2020, 136, 104742. [Google Scholar] [CrossRef]
- Farmer, B.C.; Williams, H.C.; Devanney, N.A.; Piron, M.A.; Nation, G.K.; Carter, D.J.; Walsh, A.E.; Khanal, R.; Young, L.E.A.; Kluemper, J.C.; et al. APOΕ4 lowers energy expenditure in females and impairs glucose oxidation by increasing flux through aerobic glycolysis. Mol. Neurodegener. 2021, 16, 62. [Google Scholar] [CrossRef] [PubMed]
- Qi, G.; Mi, Y.; Shi, X.; Gu, H.; Brinton, R.D.; Yin, F. ApoE4 Impairs Neuron-Astrocyte Coupling of Fatty Acid Metabolism. Cell Rep. 2021, 34, 108572. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konttinen, H.; Cabral-da-Silva, M.E.C.; Ohtonen, S.; Wojciechowski, S.; Shakirzyanova, A.; Caligola, S.; Giugno, R.; Ishchenko, Y.; Hernández, D.; Fazaludeen, M.F.; et al. PSEN1ΔE9, APPswe, and APOE4 Confer Disparate Phenotypes in Human iPSC-Derived Microglia. Stem Cell Rep. 2019, 13, 669–683. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.; Xiao, N.; Zeng, G.; Bi, D.; Dai, X.; Mi, X.; Ye, Q.; Chen, X.; Zhang, J. APOE4 genotype exacerbates the depression-like behavior of mice during aging through ATP decline. Transl. Psychiatry 2021, 11, 507. [Google Scholar] [CrossRef]
- Li, J.-M.; Liu, C.; Hu, X.; Cai, Y.; Ma, C.; Luo, X.-G.; Yan, X.-X. Inverse correlation between Alzheimer’s disease and cancer: Implication for a strong impact of regenerative propensity on neurodegeneration? BMC Neurol. 2014, 14, 211. [Google Scholar] [CrossRef] [Green Version]
- Lanni, C.; Masi, M.; Racchi, M.; Govoni, S. Cancer and Alzheimer’s disease inverse relationship: An age-associated diverging derailment of shared pathways. Mol. Psychiatry 2021, 26, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Skin cancer—Protective effect against Alzheimer disease? Nat. Rev. Neurol. 2013, 9, 358. [CrossRef]
- Alzheimer’s role of breast-cancer gene. Nature 2015, 528, 10. [CrossRef] [Green Version]
- Beal, E. Cancer link to Alzheimer disease, but not vascular dementia. Nat. Rev. Neurol. 2010, 6, 124. [Google Scholar] [CrossRef]
- Majd, S.; Power, J.; Majd, Z. Alzheimer’s Disease and Cancer: When Two Monsters Cannot Be Together. Front. Neurosci. 2019, 13, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Demetrius, L.A.; Magistretti, P.J.; Pellerin, L. Alzheimer’s disease: The amyloid hypothesis and the Inverse Warburg effect. Front. Physiol. 2014, 5, 522. [Google Scholar] [CrossRef] [Green Version]
- Demetrius, L.A.; Simon, D.K. An inverse-Warburg effect and the origin of Alzheimer’s disease. Biogerontology 2012, 13, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Liu, M.; Li, L.; Chen, L. Involvement of the Warburg effect in non-tumor diseases processes. J. Cell Physiol. 2018, 233, 2839–2849. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Atlante, A.; de Bari, L.; Bobba, A.; Amadoro, G. A disease with a sweet tooth: Exploring the Warburg effect in Alzheimer’s disease. Biogerontology 2017, 18, 301–319. [Google Scholar] [CrossRef]
- Zebhauser, P.T.; Berthele, A.; Goldhardt, O.; Diehl-Schmid, J.; Priller, J.; Ortner, M.; Grimmer, T. Cerebrospinal fluid lactate levels along the Alzheimer’s disease continuum and associations with blood-brain barrier integrity, age, cognition, and biomarkers. Alzheimers Res. Ther. 2022, 14, 61. [Google Scholar] [CrossRef] [PubMed]
- Palmer, A.M. The activity of the pentose phosphate pathway is increased in response to oxidative stress in Alzheimer’s disease. J. Neural. Transm. 1999, 106, 317–328. [Google Scholar] [CrossRef]
- Bermejo, P.; Martín-Aragón, S.; Benedí, J.; Susín, C.; Felici, E.; Gil, P.; Ribera, J.M.; Villar, A.M. Peripheral levels of glutathione and protein oxidation as markers in the development of Alzheimer’s disease from Mild Cognitive Impairment. Free Radic. Res. 2008, 42, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.I.; Malkov, A.E.; Waseem, T.; Mukhtarov, M.; Buldakova, S.; Gubkina, O.; Zilberter, M.; Zilberter, Y. Glycolysis and oxidative phosphorylation in neurons and astrocytes during network activity in hippocampal slices. J. Cereb. Blood Flow. Metab. 2014, 34, 397–407. [Google Scholar] [CrossRef]
- Liu, F.; Shi, J.; Tanimukai, H.; Gu, J.; Gu, J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain 2009, 132, 1820–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawal, P.; Zhao, L. Sialometabolism in Brain Health and Alzheimer’s Disease. Front. Neurosci. 2021, 15, 648617. [Google Scholar] [CrossRef]
- Moon, H.J.; Haroutunian, V.; Zhao, L. Human apolipoprotein E isoforms are differentially sialylated and the sialic acid moiety in ApoE2 attenuates ApoE2-Aβ interaction and Aβ fibrillation. Neurobiol. Dis. 2022, 164, 105631. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.B.; Hay, N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene 2006, 25, 4683–4696. [Google Scholar] [CrossRef] [Green Version]
- Pastorino, J.G.; Shulga, N.; Hoek, J.B. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J. Biol. Chem. 2002, 277, 7610–7618. [Google Scholar] [CrossRef] [Green Version]
- Chiara, F.; Castellaro, D.; Marin, O.; Petronilli, V.; Brusilow, W.S.; Juhaszova, M.; Sollott, S.J.; Forte, M.; Bernardi, P.; Rasola, A. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS ONE 2008, 3, e1852. [Google Scholar] [CrossRef] [Green Version]
- Abu-Hamad, S.; Zaid, H.; Israelson, A.; Nahon, E.; Shoshan-Barmatz, V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: Mapping the site of binding. J. Biol. Chem. 2008, 283, 13482–13490. [Google Scholar] [CrossRef] [Green Version]
- Miura, T.; Tanno, M. The mPTP and its regulatory proteins: Final common targets of signalling pathways for protection against necrosis. Cardiovasc. Res. 2012, 94, 181–189. [Google Scholar] [CrossRef] [Green Version]
- da-Silva, W.S.; Gómez-Puyou, A.; de Gómez-Puyou, M.T.; Moreno-Sanchez, R.; De Felice, F.G.; de Meis, L.; Oliveira, M.F.; Galina, A. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: Steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J. Biol. Chem. 2004, 279, 39846–39855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.Y.; Chen, Y.F.; Chan, H.C.; Chung, C.H.; Peng, H.Y.; Ho, Y.C.; Chen, C.H.; Chang, K.C.; Tang, C.H.; Lee, A.S. Role of apolipoprotein E in electronegative low-density lipoprotein-induced mitochondrial dysfunction in cardiomyocytes. Metabolism 2020, 107, 154227. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Gottesdiener, A.J.; Parmar, M.; Li, M.; Kaminsky, S.M.; Chiuchiolo, M.J.; Sondhi, D.; Sullivan, P.M.; Holtzman, D.M.; Crystal, R.G.; et al. Intracerebral adeno-associated virus gene delivery of apolipoprotein E2 markedly reduces brain amyloid pathology in Alzheimer’s disease mouse models. Neurobiol. Aging 2016, 44, 159–172. [Google Scholar] [CrossRef]
- Hudry, E.; Dashkoff, J.; Roe, A.D.; Takeda, S.; Koffie, R.M.; Hashimoto, T.; Scheel, M.; Spires-Jones, T.; Arbel-Ornath, M.; Betensky, R.; et al. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci. Transl. Med. 2013, 5, 212ra161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theendakara, V.; Peters-Libeu, C.A.; Bredesen, D.E.; Rao, R.V. Transcriptional Effects of ApoE4: Relevance to Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 5243–5254. [Google Scholar] [CrossRef] [PubMed]
- Parcon, P.A.; Balasubramaniam, M.; Ayyadevara, S.; Jones, R.A.; Liu, L.; Shmookler Reis, R.J.; Barger, S.W.; Mrak, R.E.; Griffin, W.S.T. Apolipoprotein E4 inhibits autophagy gene products through direct, specific binding to CLEAR motifs. Alzheimers Dement. J. Alzheimers Assoc. 2018, 14, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Li, P.; Zhou, Y.G.; Ye, J. Altered Energy Metabolism During Early Optic Nerve Crush Injury: Implications of Warburg-Like Aerobic Glycolysis in Facilitating Retinal Ganglion Cell Survival. Neurosci. Bull. 2020, 36, 761–777. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Wu, L.; Swerdlow, R.H.; Zhao, L. Opposing Effects of ApoE2 and ApoE4 on Glycolytic Metabolism in Neuronal Aging Supports a Warburg Neuroprotective Cascade against Alzheimer’s Disease. Cells 2023, 12, 410. https://doi.org/10.3390/cells12030410
Zhang X, Wu L, Swerdlow RH, Zhao L. Opposing Effects of ApoE2 and ApoE4 on Glycolytic Metabolism in Neuronal Aging Supports a Warburg Neuroprotective Cascade against Alzheimer’s Disease. Cells. 2023; 12(3):410. https://doi.org/10.3390/cells12030410
Chicago/Turabian StyleZhang, Xin, Long Wu, Russell H. Swerdlow, and Liqin Zhao. 2023. "Opposing Effects of ApoE2 and ApoE4 on Glycolytic Metabolism in Neuronal Aging Supports a Warburg Neuroprotective Cascade against Alzheimer’s Disease" Cells 12, no. 3: 410. https://doi.org/10.3390/cells12030410
APA StyleZhang, X., Wu, L., Swerdlow, R. H., & Zhao, L. (2023). Opposing Effects of ApoE2 and ApoE4 on Glycolytic Metabolism in Neuronal Aging Supports a Warburg Neuroprotective Cascade against Alzheimer’s Disease. Cells, 12(3), 410. https://doi.org/10.3390/cells12030410