
C1q and Tumor Necrosis Factor Related Protein 9 Protects from Diabetic Cardiomyopathy by Alleviating Cardiac Insulin Resistance and Inflammation

 , , , , , , and
, , , , , , and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Echocardiography
2.3. Serial 18F-FDG PET-CT Imaging
2.4. Image Processing and Analysis
2.5. Serum Analysis
2.6. Isolation of Adult Cardiomyocytes
2.7. Seahorse Extracellular Flux Analysis
2.8. Western Blot Analysis
2.9. Histology
2.10. RNA Sequencing and Bioinformatics
2.11. Statistics
3. Results
3.1. High-Fat Diet Leads to Peripheral Insulin Resistance in CTRP9 KO Mice
3.2. Reduced CTRP9 Levels Trigger Impaired Diastolic Heart Function during HFD
3.3. CTRP9 Knockout Leads to Impaired Cardiac Glucose Utilization In Vivo
3.4. Cardiac Overexpression of CTRP9 Leads to Impaired Peripheral Glucose Sensitivity
3.5. Overexpression of CTRP9 Improved Diastolic Cardiac Function and Increased Cardiac Glucose Metabolism
3.6. Histological Examination Revealed Increased Cardiomyocyte Hypertrophy upon CTRP9 Knock-Out and CTRP9 Overexpression
3.7. RNA Sequencing Reveals Anti-Inflammatory Effects of CTRP9 Overexpression during HFD Administration
4. Discussion
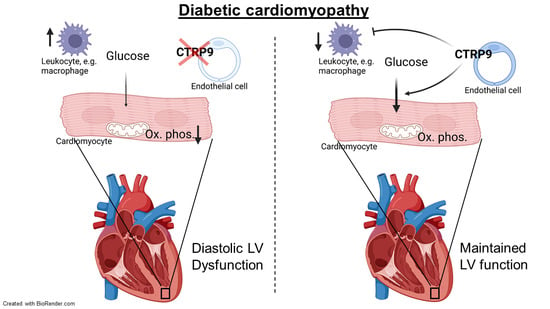
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef]
- Matheus, A.S.; Tannus, L.R.; Cobas, R.A.; Palma, C.C.; Negrato, C.A.; Gomes, M.B. Impact of diabetes on cardiovascular disease: An update. Int. J. Hypertens. 2013, 2013, 653789. [Google Scholar] [CrossRef]
- Kannel, W.B.; Hjortland, M.; Castelli, W.P. Role of diabetes in congestive heart failure: The Framingham study. Am. J. Cardiol. 1974, 34, 29–34. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Seldin, M.M.; Tan, S.Y.; Wong, G.W. Metabolic function of the CTRP family of hormones. Rev. Endocr. Metab. Disord. 2014, 15, 111–123. [Google Scholar] [CrossRef]
- Appari, M.; Breitbart, A.; Brandes, F.; Szaroszyk, M.; Froese, N.; Korf-Klingebiel, M.; Mohammadi, M.M.; Grund, A.; Scharf, G.M.; Wang, H.; et al. C1q-TNF-Related Protein-9 Promotes Cardiac Hypertrophy and Failure. Circ. Res. 2017, 120, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.W.; Krawczyk, S.A.; Kitidis-Mitrokostas, C.; Ge, G.; Spooner, E.; Hug, C.; Gimeno, R.; Lodish, H.F. Identification and characterization of CTRP9, a novel secreted glycoprotein, from adipose tissue that reduces serum glucose in mice and forms heterotrimers with adiponectin. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 241–258. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Lei, X.; Petersen, P.S.; Aja, S.; Wong, G.W. Targeted deletion of C1q/TNF-related protein 9 increases food intake, decreases insulin sensitivity, and promotes hepatic steatosis in mice. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E779–E790. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.M.; Wei, Z.; Seldin, M.M.; Byerly, M.S.; Aja, S.; Wong, G.W. CTRP9 transgenic mice are protected from diet-induced obesity and metabolic dysfunction. Am. J. Physiology. Regul. Integr. Comp. Physiol. 2013, 305, R522–R533. [Google Scholar] [CrossRef] [PubMed]
- Kambara, T.; Ohashi, K.; Shibata, R.; Ogura, Y.; Maruyama, S.; Enomoto, T.; Uemura, Y.; Shimizu, Y.; Yuasa, D.; Matsuo, K.; et al. CTRP9 protein protects against myocardial injury following ischemia-reperfusion through AMP-activated protein kinase (AMPK)-dependent mechanism. J. Biol. Chem. 2012, 287, 18965–18973. [Google Scholar] [CrossRef]
- Kambara, T.; Shibata, R.; Ohashi, K.; Matsuo, K.; Hiramatsu-Ito, M.; Enomoto, T.; Yuasa, D.; Ito, M.; Hayakawa, S.; Ogawa, H.; et al. C1q/Tumor Necrosis Factor-Related Protein 9 Protects against Acute Myocardial Injury through an Adiponectin Receptor I-AMPK-Dependent Mechanism. Mol. Cell. Biol. 2015, 35, 2173–2185. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yi, W.; Yuan, Y.; Lau, W.B.; Yi, D.; Wang, X.; Wang, Y.; Su, H.; Wang, X.; Gao, E.; et al. C1q/tumor necrosis factor-related protein-9, a novel adipocyte-derived cytokine, attenuates adverse remodeling in the ischemic mouse heart via protein kinase A activation. Circulation 2013, 128, S113–S120. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yuan, Y.; Wang, X.M.; Lau, W.B.; Wang, Y.; Wang, X.; Gao, E.; Koch, W.J.; Ma, X.L. Inhibition of CTRP9, a novel and cardiac-abundantly expressed cell survival molecule, by TNFalpha-initiated oxidative signaling contributes to exacerbated cardiac injury in diabetic mice. Basic Res. Cardiol. 2013, 108, 315. [Google Scholar] [CrossRef]
- Li, L.; Aslam, M.; Siegler, B.H.; Niemann, B.; Rohrbach, S. Comparative Analysis of CTRP-Mediated Effects on Cardiomyocyte Glucose Metabolism: Cross Talk between AMPK and Akt Signaling Pathway. Cells 2021, 10, 905. [Google Scholar] [CrossRef]
- Grund, A.; Szaroszyk, M.; Korf-Klingebiel, M.; Malek Mohammadi, M.; Trogisch, F.A.; Schrameck, U.; Gigina, A.; Tiedje, C.; Gaestel, M.; Kraft, T.; et al. TIP30 counteracts cardiac hypertrophy and failure by inhibiting translational elongation. EMBO Mol. Med. 2019, 11, e10018. [Google Scholar] [CrossRef] [PubMed]
- Szaroszyk, M.; Kattih, B.; Martin-Garrido, A.; Trogisch, F.A.; Dittrich, G.M.; Grund, A.; Abouissa, A.; Derlin, K.; Meier, M.; Holler, T.; et al. Skeletal muscle derived Musclin protects the heart during pathological overload. Nat. Commun. 2022, 13, 149. [Google Scholar] [CrossRef]
- Ram, R.; Mickelsen, D.M.; Theodoropoulos, C.; Blaxall, B.C. New approaches in small animal echocardiography: Imaging the sounds of silence. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1765–H1780. [Google Scholar] [CrossRef]
- Thackeray, J.T.; Pietzsch, S.; Stapel, B.; Ricke-Hoch, M.; Lee, C.W.; Bankstahl, J.P.; Scherr, M.; Heineke, J.; Scharf, G.; Haghikia, A.; et al. Insulin supplementation attenuates cancer-induced cardiomyopathy and slows tumor disease progression. JCI Insight 2017, 2, e93098. [Google Scholar] [CrossRef]
- Thackeray, J.T.; Bankstahl, J.P.; Bengel, F.M. Impact of Image-Derived Input Function and Fit Time Intervals on Patlak Quantification of Myocardial Glucose Uptake in Mice. J. Nucl. Med. 2015, 56, 1615–1621. [Google Scholar] [CrossRef]
- Louch, W.E.; Sheehan, K.A.; Wolska, B.M. Methods in cardiomyocyte isolation, culture, and gene transfer. J. Mol. Cell. Cardiol. 2011, 51, 288–298. [Google Scholar] [CrossRef]
- Battiprolu, P.K.; Hojayev, B.; Jiang, N.; Wang, Z.V.; Luo, X.; Iglewski, M.; Shelton, J.M.; Gerard, R.D.; Rothermel, B.A.; Gillette, T.G.; et al. Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J. Clin. Investig. 2012, 122, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Werfel, S.; Jungmann, A.; Lehmann, L.; Ksienzyk, J.; Bekeredjian, R.; Kaya, Z.; Leuchs, B.; Nordheim, A.; Backs, J.; Engelhardt, S.; et al. Rapid and highly efficient inducible cardiac gene knockout in adult mice using AAV-mediated expression of Cre recombinase. Cardiovasc. Res. 2014, 104, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Riehle, C.; Bauersachs, J. Of mice and men: Models and mechanisms of diabetic cardiomyopathy. Basic Res. Cardiol. 2018, 114, 2. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, R.H.; Abel, E.D. Basic Mechanisms of Diabetic Heart Disease. Circ. Res. 2020, 126, 1501–1525. [Google Scholar] [CrossRef]
- Zuo, A.; Zhao, X.; Li, T.; Li, J.; Lei, S.; Chen, J.; Xu, D.; Song, C.; Liu, T.; Li, C.; et al. CTRP9 knockout exaggerates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy through inhibiting the LKB1/AMPK pathway. J. Cell. Mol. Med. 2020, 24, 2635–2647. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, A.P.; Leite-Moreira, A.F.; Balligand, J.L.; Bauersachs, J.; Dawson, D.; de Boer, R.A.; de Windt, L.J.; Falcao-Pires, I.; Fontes-Carvalho, R.; Franz, S.; et al. An integrative translational approach to study heart failure with preserved ejection fraction: A position paper from the Working Group on Myocardial Function of the European Society of Cardiology. Eur. J. Heart Fail. 2018, 20, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Grueter, C.E.; van Rooij, E.; Johnson, B.A.; Deleon, S.M.; Sutherland, L.B.; Qi, X.; Gautron, L.; Elmquist, J.K.; Bassel-Duby, R.; Olson, E.N. A Cardiac MicroRNA Governs Systemic Energy Homeostasis by Regulation of MED13. Cell 2012, 149, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Sengenes, C.; Berlan, M.; De Glisezinski, I.; Lafontan, M.; Galitzky, J. Natriuretic peptides: A new lipolytic pathway in human adipocytes. FASEB J. 2000, 14, 1345–1351. [Google Scholar] [CrossRef]
- Wu, H.K.; Zhang, Y.; Cao, C.M.; Hu, X.; Fang, M.; Yao, Y.; Jin, L.; Chen, G.; Jiang, P.; Zhang, S.; et al. Glucose-Sensitive Myokine/Cardiokine MG53 Regulates Systemic Insulin Response and Metabolic Homeostasis. Circulation 2019, 139, 901–914. [Google Scholar] [CrossRef]
- Lopaschuk, G.D. Fatty Acid Oxidation and Its Relation with Insulin Resistance and Associated Disorders. Ann. Nutr. Metab. 2016, 68 (Suppl. S3), 15–20. [Google Scholar] [CrossRef]
- How, O.J.; Aasum, E.; Kunnathu, S.; Severson, D.L.; Myhre, E.S.; Larsen, T.S. Influence of substrate supply on cardiac efficiency, as measured by pressure-volume analysis in ex vivo mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2979–H2985. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, S.; Li, L.; Novoyatleva, T.; Niemann, B.; Knapp, F.; Molenda, N.; Schulz, R. Impact of PCSK9 on CTRP9-Induced Metabolic Effects in Adult Rat Cardiomyocytes. Front. Physiol. 2021, 12, 593862. [Google Scholar] [CrossRef] [PubMed]
- Boudina, S.; Sena, S.; Theobald, H.; Sheng, X.; Wright, J.J.; Hu, X.X.; Aziz, S.; Johnson, J.I.; Bugger, H.; Zaha, V.G.; et al. Mitochondrial energetics in the heart in obesity-related diabetes: Direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 2007, 56, 2457–2466. [Google Scholar] [CrossRef] [PubMed]
- Echtay, K.S.; Roussel, D.; St-Pierre, J.; Jekabsons, M.B.; Cadenas, S.; Stuart, J.A.; Harper, J.A.; Roebuck, S.J.; Morrison, A.; Pickering, S.; et al. Superoxide activates mitochondrial uncoupling proteins. Nature 2002, 415, 96–99. [Google Scholar] [CrossRef]
- Wende, A.R.; Symons, J.D.; Abel, E.D. Mechanisms of lipotoxicity in the cardiovascular system. Curr. Hypertens. Rep. 2012, 14, 517–531. [Google Scholar] [CrossRef]
- Jabs, M.; Rose, A.J.; Lehmann, L.H.; Taylor, J.; Moll, I.; Sijmonsma, T.P.; Herberich, S.E.; Sauer, S.W.; Poschet, G.; Federico, G.; et al. Inhibition of Endothelial Notch Signaling Impairs Fatty Acid Transport and Leads to Metabolic and Vascular Remodeling of the Adult Heart. Circulation 2018, 137, 2592–2608. [Google Scholar] [CrossRef]
- Verma, S.K.; Deshmukh, V.; Liu, P.; Nutter, C.A.; Espejo, R.; Hung, M.L.; Wang, G.S.; Yeo, G.W.; Kuyumcu-Martinez, M.N. Reactivation of fetal splicing programs in diabetic hearts is mediated by protein kinase C signaling. J. Biol. Chem. 2013, 288, 35372–35386. [Google Scholar] [CrossRef]
- Nutter, C.A.; Jaworski, E.A.; Verma, S.K.; Deshmukh, V.; Wang, Q.; Botvinnik, O.B.; Lozano, M.J.; Abass, I.J.; Ijaz, T.; Brasier, A.R.; et al. Dysregulation of RBFOX2 Is an Early Event in Cardiac Pathogenesis of Diabetes. Cell Rep. 2016, 15, 2200–2213. [Google Scholar] [CrossRef]
- Bajpai, A.; Tilley, D.G. The Role of Leukocytes in Diabetic Cardiomyopathy. Front. Physiol. 2018, 9, 1547. [Google Scholar] [CrossRef]
- Rao, X.; Zhong, J.; Sun, Q. The heterogenic properties of monocytes/macrophages and neutrophils in inflammatory response in diabetes. Life Sci. 2014, 116, 59–66. [Google Scholar] [CrossRef]
- Govindappa, P.K.; Patil, M.; Garikipati, V.N.S.; Verma, S.K.; Saheera, S.; Narasimhan, G.; Zhu, W.; Kishore, R.; Zhang, J.; Krishnamurthy, P. Targeting exosome-associated human antigen R attenuates fibrosis and inflammation in diabetic heart. FASEB J. 2020, 34, 2238–2251. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.J.; Zhang, Z.; Jung, D.Y.; Jun, J.Y.; Ma, Z.; Jones, K.E.; Chan, S.Y.; Kim, J.K. Nutrient stress activates inflammation and reduces glucose metabolism by suppressing AMP-activated protein kinase in the heart. Diabetes 2009, 58, 2536–2546. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Zhang, H.; Xu, T.; Hao, J.; Chen, X.; Sun, S.; Yang, J.; Sun, J.; Lin, H.; Guo, H. Identification and verification of immune-related biomarkers and immune infiltration in diabetic heart failure. Front. Cardiovasc. Med. 2022, 9, 931066. [Google Scholar] [CrossRef] [PubMed]
- Hayward, S.L.; Bautista-Lopez, N.; Suzuki, K.; Atrazhev, A.; Dickie, P.; Elliott, J.F. CD4 T cells play major effector role and CD8 T cells initiating role in spontaneous autoimmune myocarditis of HLA-DQ8 transgenic IAb knockout nonobese diabetic mice. J. Immunol. 2006, 176, 7715–7725. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Prabhu, S.D.; Bansal, S.S. CD4(+) T-lymphocytes exhibit biphasic kinetics post-myocardial infarction. Front. Cardiovasc. Med. 2022, 9, 992653. [Google Scholar] [CrossRef] [PubMed]
- Westermann, D.; Van Linthout, S.; Dhayat, S.; Dhayat, N.; Escher, F.; Bucker-Gartner, C.; Spillmann, F.; Noutsias, M.; Riad, A.; Schultheiss, H.P.; et al. Cardioprotective and anti-inflammatory effects of interleukin converting enzyme inhibition in experimental diabetic cardiomyopathy. Diabetes 2007, 56, 1834–1841. [Google Scholar] [CrossRef]
- Westermann, D.; Van Linthout, S.; Dhayat, S.; Dhayat, N.; Schmidt, A.; Noutsias, M.; Song, X.Y.; Spillmann, F.; Riad, A.; Schultheiss, H.P.; et al. Tumor necrosis factor-alpha antagonism protects from myocardial inflammation and fibrosis in experimental diabetic cardiomyopathy. Basic Res. Cardiol. 2007, 102, 500–507. [Google Scholar] [CrossRef]
- Huang, Z.; Zhao, D.; Wang, Y.; Li, X.; Li, J.; Han, J.; Jiang, L.; Ai, F.; Zhou, Z. C1q/TNF-related protein 9 decreases cardiomyocyte hypoxia/reoxygenation-induced inflammation by inhibiting the TLR4/MyD88/NF-kappaB signaling pathway. Exp. Ther. Med. 2021, 22, 1139. [Google Scholar] [CrossRef]
- Chu, P.Y.; Walder, K.; Horlock, D.; Williams, D.; Nelson, E.; Byrne, M.; Jandeleit-Dahm, K.; Zimmet, P.; Kaye, D.M. CXCR4 Antagonism Attenuates the Development of Diabetic Cardiac Fibrosis. PLoS ONE 2015, 10, e0133616. [Google Scholar] [CrossRef]
- Schilling, J.D.; Machkovech, H.M.; Kim, A.H.; Schwendener, R.; Schaffer, J.E. Macrophages modulate cardiac function in lipotoxic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1366–H1373. [Google Scholar] [CrossRef]
- Lin, Y.; Tang, Y.; Wang, F. The Protective Effect of HIF-1alpha in T Lymphocytes on Cardiac Damage in Diabetic Mice. Ann. Clin. Lab. Sci. 2016, 46, 32–43. [Google Scholar] [PubMed]
- Abdullah, C.S.; Li, Z.; Wang, X.; Jin, Z.Q. Depletion of T lymphocytes ameliorates cardiac fibrosis in streptozotocin-induced diabetic cardiomyopathy. Int. Immunopharmacol. 2016, 39, 251–264. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haustein, R.; Trogisch, F.A.; Keles, M.; Hille, S.; Fuhrmann, M.; Weinzierl, N.; Hemanna, S.; Thackeray, J.; Dou, Y.; Zwadlo, C.; et al. C1q and Tumor Necrosis Factor Related Protein 9 Protects from Diabetic Cardiomyopathy by Alleviating Cardiac Insulin Resistance and Inflammation. Cells 2023, 12, 443. https://doi.org/10.3390/cells12030443
Haustein R, Trogisch FA, Keles M, Hille S, Fuhrmann M, Weinzierl N, Hemanna S, Thackeray J, Dou Y, Zwadlo C, et al. C1q and Tumor Necrosis Factor Related Protein 9 Protects from Diabetic Cardiomyopathy by Alleviating Cardiac Insulin Resistance and Inflammation. Cells. 2023; 12(3):443. https://doi.org/10.3390/cells12030443
Chicago/Turabian StyleHaustein, Ricarda, Felix A. Trogisch, Merve Keles, Susanne Hille, Manuela Fuhrmann, Nina Weinzierl, Shruthi Hemanna, James Thackeray, Yanliang Dou, Carolin Zwadlo, and et al. 2023. "C1q and Tumor Necrosis Factor Related Protein 9 Protects from Diabetic Cardiomyopathy by Alleviating Cardiac Insulin Resistance and Inflammation" Cells 12, no. 3: 443. https://doi.org/10.3390/cells12030443
APA StyleHaustein, R., Trogisch, F. A., Keles, M., Hille, S., Fuhrmann, M., Weinzierl, N., Hemanna, S., Thackeray, J., Dou, Y., Zwadlo, C., Froese, N., Cordero, J., Bengel, F., Müller, O. J., Bauersachs, J., Dobreva, G., & Heineke, J. (2023). C1q and Tumor Necrosis Factor Related Protein 9 Protects from Diabetic Cardiomyopathy by Alleviating Cardiac Insulin Resistance and Inflammation. Cells, 12(3), 443. https://doi.org/10.3390/cells12030443