
Is Autophagy Inhibition in Combination with Temozolomide a Therapeutically Viable Strategy?



Abstract
:1. Introduction
2. Autophagy Overview
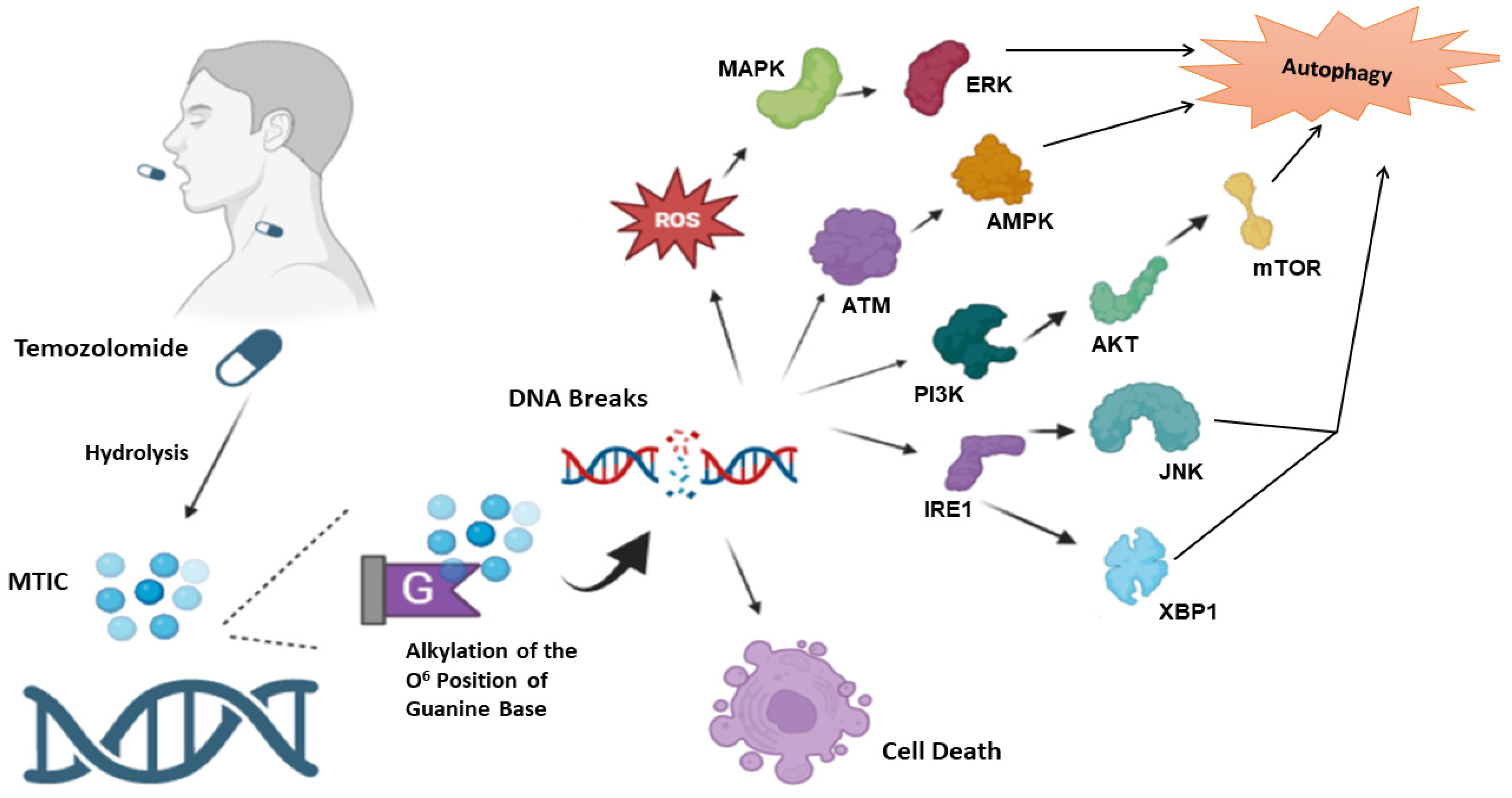
3. Temozolomide
4. Temozolomide and Autophagy
4.1. Glioblastoma
Clinical Trials
4.2. Melanoma
Clinical Trials
5. Conclusions
Funding
Conflicts of Interest
References
- Patel, N.H.; Sohal, S.S.; Manjili, M.H.; Harrell, J.C.; Gewirtz, D.A. The Roles of Autophagy and Senescence in the Tumor Cell Response to Radiation. Radiat. Res. 2020, 194, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Gewirtz, D.A. Is Autophagy Always a Barrier to Cisplatin Therapy? Biomolecules 2022, 12, 463. [Google Scholar] [CrossRef]
- Xu, J.; Elshazly, A.M.; Gewirtz, D.A. The Cytoprotective, Cytotoxic and Nonprotective Functional Forms of Autophagy Induced by Microtubule Poisons in Tumor Cells—Implications for Autophagy Modulation as a Therapeutic Strategy. Biomedicines 2022, 10, 1632. [Google Scholar] [CrossRef]
- Finnegan, R.M.; Elshazly, A.M.; Schoenlein, P.V.; Gewirtz, D.A. Therapeutic Potential for Targeting Autophagy in ER+ Breast Cancer. Cancers 2022, 14, 4289. [Google Scholar] [CrossRef] [PubMed]
- ELSHAZLY, A.-M.; NGUYEN, T.-V.-V.; GEWIRTZ, D.-A. Is autophagy induction by PARP inhibitors a target for therapeutic benefit? Oncol. Res. 2022, 30, 1–12. [Google Scholar] [CrossRef]
- Elshazly, A.M.; Wright, P.A.; Xu, J.; Gewirtz, D.A. Topoisomerase I poisons-induced autophagy: Cytoprotective, Cytotoxic or Non-protective. Autophagy Rep. 2023, 2, 1–16. [Google Scholar] [CrossRef]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4, a008813. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Rangel, M.; Kong, J.; Bhatt, V.; Khayati, K.; Guo, J.Y. Autophagy and tumorigenesis. FEBS J. 2022, 289, 7177–7198. [Google Scholar] [CrossRef]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of Mammalian Autophagy in Physiology and Pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Towers, C.G.; Thorburn, A. Therapeutic Targeting of Autophagy. EBioMedicine 2016, 14, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Benoit, A.; Hasmim, M.; Duhem, C.; Vogin, G.; Berchem, G.; Noman, M.Z.; Janji, B. Targeting Cytoprotective Autophagy to Enhance Anticancer Therapies. Front. Oncol. 2021, 11, 626309. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Coly, P.M.; Gandolfo, P.; Castel, H.; Morin, F. The Autophagy Machinery: A New Player in Chemotactic Cell Migration. Front. Neurosci. 2017, 11, 78. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A. The four faces of autophagy: Implications for cancer therapy. Cancer Res. 2014, 74, 647–651. [Google Scholar] [CrossRef]
- Ulasov, I.; Fares, J.; Timashev, P.; Lesniak, M.S. Editing Cytoprotective Autophagy in Glioma: An Unfulfilled Potential for Therapy. Trends Mol. Med. 2020, 26, 252–262. [Google Scholar] [CrossRef]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Dando, I.; Strippoli, R.; Kumar, S.; Somoza, A.; Cordani, M.; Tafani, M. Nanomaterials for Autophagy-Related miRNA-34a Delivery in Cancer Treatment. Front. Pharmacol. 2020, 11, 1141. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Park, M.N.; Rahman, M.H.; Rashid, M.M.; Islam, R.; Uddin, M.J.; Hannan, M.A.; Kim, B. p53 Modulation of Autophagy Signaling in Cancer Therapies: Perspectives Mechanism and Therapeutic Targets. Front. Cell Dev. Biol. 2022, 10, 761080. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef]
- Grochans, S.; Cybulska, A.M.; Simińska, D.; Korbecki, J.; Kojder, K.; Chlubek, D.; Baranowska-Bosiacka, I. Epidemiology of Glioblastoma Multiforme-Literature Review. Cancers 2022, 14, 2412. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro-Oncology 2014, 16, 896–913. [Google Scholar] [CrossRef]
- Birzu, C.; French, P.; Caccese, M.; Cerretti, G.; Idbaih, A.; Zagonel, V.; Lombardi, G. Recurrent Glioblastoma: From Molecular Landscape to New Treatment Perspectives. Cancers 2020, 13, 47. [Google Scholar] [CrossRef]
- Wesolowski, J.R.; Rajdev, P.; Mukherji, S.K. Temozolomide (Temodar). Am. J. Neuroradiol. 2010, 31, 1383–1384. [Google Scholar] [CrossRef]
- Agarwala, S.S.; Kirkwood, J.M. Temozolomide, a novel alkylating agent with activity in the central nervous system, may improve the treatment of advanced metastatic melanoma. Oncologist 2000, 5, 144–151. [Google Scholar] [CrossRef]
- Villano, J.L.; Seery, T.E.; Bressler, L.R. Temozolomide in malignant gliomas: Current use and future targets. Cancer Chemother. Pharmacol. 2009, 64, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Kyrtopoulos, S.A.; Anderson, L.M.; Chhabra, S.K.; Souliotis, V.L.; Pletsa, V.; Valavanis, C.; Georgiadis, P. DNA adducts and the mechanism of carcinogenesis and cytotoxicity of methylating agents of environmental and clinical significance. Cancer Detect. Prev. 1997, 21, 391–405. [Google Scholar]
- Margison, G.P.; Santibáñez-Koref, M.F. O6-alkylguanine-DNA alkyltransferase: Role in carcinogenesis and chemotherapy. BioEssays News Rev. Mol. Cell. Dev. Biol. 2002, 24, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Mojas, N.; Lopes, M.; Jiricny, J. Mismatch repair-dependent processing of methylation damage gives rise to persistent single-stranded gaps in newly replicated DNA. Genes Dev. 2007, 21, 3342–3355. [Google Scholar] [CrossRef]
- Elshazly, A.M.; Gewirtz, D.A. An overview of resistance to Human epidermal growth factor receptor 2 (Her2) targeted therapies in breast cancer. Cancer Drug Resist. 2022, 5, 472–486. [Google Scholar] [CrossRef]
- Singh, N.; Miner, A.; Hennis, L.; Mittal, S. Mechanisms of temozolomide resistance in glioblastoma—A comprehensive review. Cancer Drug Resist. 2021, 4, 17–43. [Google Scholar] [CrossRef]
- Lin, C.J.; Lee, C.C.; Shih, Y.L.; Lin, T.Y.; Wang, S.H.; Lin, Y.F.; Shih, C.M. Resveratrol enhances the therapeutic effect of temozolomide against malignant glioma in vitro and in vivo by inhibiting autophagy. Free. Radic. Biol. Med. 2012, 52, 377–391. [Google Scholar] [CrossRef]
- Filippi-Chiela, E.C.; Bueno e Silva, M.M.; Thomé, M.P.; Lenz, G. Single-cell analysis challenges the connection between autophagy and senescence induced by DNA damage. Autophagy 2015, 11, 1099–1113. [Google Scholar] [CrossRef]
- Chen, C.M.; Syu, J.P.; Way, T.D.; Huang, L.J.; Kuo, S.C.; Lin, C.T.; Lin, C.L. BC3EE2,9B, a synthetic carbazole derivative, upregulates autophagy and synergistically sensitizes human GBM8901 glioblastoma cells to temozolomide. Int. J. Mol. Med. 2015, 36, 1244–1252. [Google Scholar] [CrossRef]
- Zou, Y.; Wang, Q.; Wang, W. MutL homolog 1 contributes to temozolomide-induced autophagy via ataxia-telangiectasia mutated in glioma. Mol. Med. Rep. 2015, 11, 4591–4596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, M.; Kawaguchi, T.; Berger, M.S.; Pieper, R.O. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ. 2007, 14, 548–558. [Google Scholar] [CrossRef]
- Knizhnik, A.V.; Roos, W.P.; Nikolova, T.; Quiros, S.; Tomaszowski, K.H.; Christmann, M.; Kaina, B. Survival and death strategies in glioma cells: Autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS ONE 2013, 8, e55665. [Google Scholar] [CrossRef]
- Branch, P.; Aquilina, G.; Bignami, M.; Karran, P. Defective mismatch binding and a mutator phenotype in cells tolerant to DNA damage. Nature 1993, 362, 652–654. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Ziouta, A.; Ochs, K.; Coquerelle, T. Chromosomal instability, reproductive cell death and apoptosis induced by O6-methylguanine in Mex-, Mex+ and methylation-tolerant mismatch repair compromised cells: Facts and models. Mutat. Res. 1997, 381, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Hickman, M.J.; Samson, L.D. Role of DNA mismatch repair and p53 in signaling induction of apoptosis by alkylating agents. Proc. Natl. Acad. Sci. USA 1999, 96, 10764–10769. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Nikolova, T.; Quiros, S.; Naumann, S.C.; Kiedron, O.; Zdzienicka, M.Z.; Kaina, B. Brca2/Xrcc2 dependent HR, but not NHEJ, is required for protection against O(6)-methylguanine triggered apoptosis, DSBs and chromosomal aberrations by a process leading to SCEs. DNA Repair 2009, 8, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Caporali, S.; Falcinelli, S.; Starace, G.; Russo, M.T.; Bonmassar, E.; Jiricny, J.; D’Atri, S. DNA damage induced by temozolomide signals to both ATM and ATR: Role of the mismatch repair system. Mol. Pharmacol. 2004, 66, 478–491. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef]
- Patel, N.H.; Bloukh, S.; Alwohosh, E.; Alhesa, A.; Saleh, T.; Gewirtz, D.A. Autophagy and senescence in cancer therapy. Adv. Cancer Res. 2021, 150, 1–74. [Google Scholar] [CrossRef]
- Gewirtz, D.A. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy 2009, 5, 1232–1234. [Google Scholar] [CrossRef] [Green Version]
- Gewirtz, D.A. Autophagy and senescence. Autophagy 2013, 9, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Goehe, R.W.; Di, X.; Sharma, K.; Bristol, M.L.; Henderson, S.C.; Valerie, K.; Rodier, F.; Davalos, A.R.; Gewirtz, D.A. The autophagy-senescence connection in chemotherapy: Must tumor cells (self) eat before they sleep? J. Pharmacol. Exp. Ther. 2012, 343, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Dong, X.; Li, H.; Wang, H.; Jiang, Q.; Liu, L.; Wang, L.; Dong, J. Nicardipine sensitizes temozolomide by inhibiting autophagy and promoting cell apoptosis in glioma stem cells. Aging 2021, 13, 6820–6831. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Chen, J.H.; Tsai, C.F.; Wu, C.T.; Wu, M.H.; Chang, P.C.; Yeh, W.L. Nicardipine Inhibits Breast Cancer Migration via Nrf2/HO-1 Axis and Matrix Metalloproteinase-9 Regulation. Front. Pharmacol. 2021, 12, 710978. [Google Scholar] [CrossRef]
- Kim, S.; Choi, S.; Kang, D. Quantitative and qualitative analysis of autophagy flux using imaging. BMB Rep. 2020, 53, 241–247. [Google Scholar] [CrossRef]
- Ando, S.; Moyama, C.; Kojima, N.; Fujita, M.; Ohta, K.; Kohno, Y.; Ii, H.; Nakata, S. JCI-20679 suppresses autophagy and enhances temozolomide-mediated growth inhibition of glioblastoma cells. Biochem. Biophys. Res. Commun. 2022, 591, 62–67. [Google Scholar] [CrossRef]
- Stevens, F.J.; Argon, Y. Protein folding in the ER. Semin. Cell Dev. Biol. 1999, 10, 443–454. [Google Scholar] [CrossRef]
- Ryabaya, O.; Prokofieva, A.; Khochenkov, D.; Abramov, I.; Zasedatelev, A.; Stepanova, E. Inhibition of endoplasmic reticulum stress-induced autophagy sensitizes melanoma cells to temozolomide treatment. Oncol. Rep. 2018, 40, 385–394. [Google Scholar] [CrossRef]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Weissman, A.M. The Unfolded Protein Response, Degradation from Endoplasmic Reticulum and Cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006, 4, e423. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ni, M.; Lee, B.; Barron, E.; Hinton, D.R.; Lee, A.S. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 2008, 15, 1460–1471. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Cho, H.Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg. Focus 2014, 37, E12. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Kanzawa, T.; Germano, I.M.; Komata, T.; Ito, H.; Kondo, Y.; Kondo, S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004, 11, 448–457. [Google Scholar] [CrossRef]
- Lee, S.W.; Kim, H.K.; Lee, N.H.; Yi, H.Y.; Kim, H.S.; Hong, S.H.; Hong, Y.K.; Joe, Y.A. The synergistic effect of combination temozolomide and chloroquine treatment is dependent on autophagy formation and p53 status in glioma cells. Cancer Lett. 2015, 360, 195–204. [Google Scholar] [CrossRef]
- Torres, S.; Lorente, M.; Rodríguez-Fornés, F.; Hernández-Tiedra, S.; Salazar, M.; García-Taboada, E.; Barcia, J.; Guzmán, M.; Velasco, G. A combined preclinical therapy of cannabinoids and temozolomide against glioma. Mol. Cancer Ther. 2011, 10, 90–103. [Google Scholar] [CrossRef]
- Bristol, M.L.; Emery, S.M.; Maycotte, P.; Thorburn, A.; Chakradeo, S.; Gewirtz, D.A.J.J.o.P.; Therapeutics, E. Autophagy inhibition for chemosensitization and radiosensitization in cancer: Do the preclinical data support this therapeutic strategy? J. Pharmacol. Exp. Ther. 2013, 344, 544–552. [Google Scholar]
- Young, M.M.; Takahashi, Y.; Khan, O.; Park, S.; Hori, T.; Yun, J.; Sharma, A.K.; Amin, S.; Hu, C.D.; Zhang, J.; et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J. Biol. Chem. 2012, 287, 12455–12468. [Google Scholar] [CrossRef] [PubMed]
- Caserta, T.M.; Smith, A.N.; Gultice, A.D.; Reedy, M.A.; Brown, T.L. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis Int. J. Program. Cell Death 2003, 8, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Liu, L.; Herman, J.G.; Stupp, R.; Wick, W.; Weller, M.; Mehta, M.P.; Gilbert, M.R. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 4189–4199. [Google Scholar] [CrossRef]
- Massi, P.; Vaccani, A.; Bianchessi, S.; Costa, B.; Macchi, P.; Parolaro, D. The non-psychoactive cannabidiol triggers caspase activation and oxidative stress in human glioma cells. Cell. Mol. Life Sci. 2006, 63, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- Vaccani, A.; Massi, P.; Colombo, A.; Rubino, T.; Parolaro, D. Cannabidiol inhibits human glioma cell migration through a cannabinoid receptor-independent mechanism. Br. J. Pharmacol. 2005, 144, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef]
- Compter, I.; Eekers, D.B.P.; Hoeben, A.; Rouschop, K.M.A.; Reymen, B.; Ackermans, L.; Beckervordersantforth, J.; Bauer, N.J.C.; Anten, M.M.; Wesseling, P.; et al. Chloroquine combined with concurrent radiotherapy and temozolomide for newly diagnosed glioblastoma: A phase IB trial. Autophagy 2021, 17, 2604–2612. [Google Scholar] [CrossRef]
- Rangwala, R.; Leone, R.; Chang, Y.C.; Fecher, L.A.; Schuchter, L.M.; Kramer, A.; Tan, K.S.; Heitjan, D.F.; Rodgers, G.; Gallagher, M.; et al. Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1369–1379. [Google Scholar] [CrossRef]
- Makita, K.; Hara, H.; Sano, E.; Okamoto, Y.; Ochiai, Y.; Harada, T.; Ueda, T.; Nakayama, T.; Aizawa, S.; Yoshino, A. Interferon-β sensitizes human malignant melanoma cells to temozolomide-induced apoptosis and autophagy. Int. J. Oncol. 2019, 54, 1864–1874. [Google Scholar] [CrossRef]
- Ryabaya, O.O.; Inshakov, A.N.; Egorova, A.V.; Emelyanova, M.A.; Nasedkina, T.V.; Zasedatelev, A.S.; Khochenkov, D.A.; Stepanova, E.V. Autophagy inhibitors chloroquine and LY294002 enhance temozolomide cytotoxicity on cutaneous melanoma cell lines in vitro. Anti-Cancer Drugs 2017, 28, 307–315. [Google Scholar] [CrossRef]
- Allavena, G.; Del Bello, B.; Tini, P.; Volpi, N.; Valacchi, G.; Miracco, C.; Pirtoli, L.; Maellaro, E. Trehalose inhibits cell proliferation and amplifies long-term temozolomide- and radiation-induced cytotoxicity in melanoma cells: A role for autophagy and premature senescence. J. Cell. Physiol. 2019, 234, 11708–11721. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.; Kohli, L.; Klocke, B.J.; Roth, K.A. Chloroquine-induced autophagic vacuole accumulation and cell death in glioma cells is p53 independent. Neuro-Oncology 2010, 12, 473–481. [Google Scholar] [CrossRef]
- Richardson, P.J.; Ottaviani, S.; Prelle, A.; Stebbing, J.; Casalini, G.; Corbellino, M.J.J.o.N. CNS penetration of potential anti-COVID-19 drugs. J. Neurol. 2020, 267, 1880–1882. [Google Scholar]
- McChesney, E.W. Animal toxicity and pharmacokinetics of hydroxychloroquine sulfate. Am. J. Med. 1983, 75, 11–18. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, X.Y.; Wang, W.W.; Wang, J. Hydroxychloroquine Increased Anxiety-Like Behaviors and Disrupted the Expression of Some Related Genes in the Mouse Brain. Front. Pharmacol. 2021, 12. [Google Scholar] [CrossRef]
- Manzo, C.; Gareri, P.; Castagna, A. Psychomotor Agitation Following Treatment with Hydroxychloroquine. Drug Saf. Case Rep. 2017, 4, 6. [Google Scholar] [CrossRef]
- Naddaf, E.; Paul, P.; AbouEzzeddine, O.F. Chloroquine and Hydroxychloroquine Myopathy: Clinical Spectrum and Treatment Outcomes. Front. Neurol. 2021, 11, 616075. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Winkler, J.D. Lys05: A new lysosomal autophagy inhibitor. Autophagy 2012, 8, 1383–1384. [Google Scholar] [CrossRef]
- Molero-Valenzuela, A.; Fontova, P.; Alonso-Carrillo, D.; Carreira-Barral, I.; Torres, A.A.; García-Valverde, M.; Benítez-García, C.; Pérez-Tomás, R.; Quesada, R.; Soto-Cerrato, V. A Novel Late-Stage Autophagy Inhibitor That Efficiently Targets Lysosomes Inducing Potent Cytotoxic and Sensitizing Effects in Lung Cancer. Cancers 2022, 14, 3387. [Google Scholar] [CrossRef]
- Ye, H.; Chen, M.; Cao, F.; Huang, H.; Zhan, R.; Zheng, X. Chloroquine, an autophagy inhibitor, potentiates the radiosensitivity of glioma initiating cells by inhibiting autophagy and activating apoptosis. BMC Neurol. 2016, 16, 178. [Google Scholar] [CrossRef]
- Zhuang, W.; Li, B.; Long, L.; Chen, L.; Huang, Q.; Liang, Z. Induction of autophagy promotes differentiation of glioma-initiating cells and their radiosensitivity. Int. J. Cancer 2011, 129, 2720–2731. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Title | Reference |
|---|---|
| Hydroxychloroquine, Radiation, and Temozolomide Treating Patients with Newly Diagnosed Glioblastoma Multiforme. | [76] [NCT00486603] |
| Chloroquine combined with concurrent radiotherapy and temozolomide for newly diagnosed glioblastoma: a phase IB trial. | [77] |
| A Phase II Randomized Controlled Trial for the Addition of Chloroquine, an Autophagy Inhibitor, to Concurrent Chemoradiation for Newly Diagnosed Glioblastoma. | [NCT02432417] |
| TN-TC11G (THC+CBD) Combination with Temozolomide and Radiotherapy in Patients With Newly-diagnosed Glioblastoma (GEINOCANN). | [NCT03529448] |
| The Addition of Chloroquine to Chemoradiation for Glioblastoma (CHLOROBRAIN). | [NCT02378532] |
| The Addition of Chloroquine to Chemoradiation for Glioblastoma, | [NCT02432417] |
| Phase I trial of hydroxychloroquine with dose-intense temozolomide in patients with advanced solid tumors and melanoma. | [78] |
| Drug | Cell Line | Autophagy Modulation | Autophagy Function | References |
|---|---|---|---|---|
| Temozolomide | U251 glioma cell line and p53-mutant glioma cell lines, U373 and SF188. | 3-MA, and Beclin-1 knockdown mediated via shRNA | Cytoprotective | [42] |
| LN-229 glioblastoma and U87-MG astrocytoma cell lines. | 3-MA | Cytoprotective | [43] | |
| Temozolomide and nicardipine | Glioma stem cells (GSCs) including surgical specimen derived SU4 and SU5 cell lines. | Rapamycin | Cytoprotective | [54] |
| Temozolomide and JCI-20679 | Murine primary glioblastoma cells, U251, T98, A172 and U87MG human glioblastoma cell lines. | Bafilomycin A1 | Cytoprotective | [57] |
| Temozolomide | Resistant cell lines (U251-TMZR, LN229-TMZR, U87-TMZR, and TuBECs) and their sensitive counterparts (U251-TMZS, LN229-TMZS, U87-TMZS, and BECs). In vivo. | CQ, 3-MA, and knockdown of Beclin 1 mediated by siRNA | Cytoprotective | [65] |
| Temozolomide | U373-MG cell line | 3-Methyladenine (3-MA), and bafilomycin A1 | Cytotoxic | [67] |
| Temozolomide | U87-MG (wild type p53), U373 (mutant p53) glioma cell lines, and P53-overexpressing U87 mutant cell line. | CQ, bafilomycin A1, 3-MA, and knockdown of Beclin 1 mediated by siRNA | Cytotoxic in U87-MG cells Dependent upon p53 status, non-protective in U373 (mutant p53) glioma cell lines and p53-overexpressing U87 mutant cell line | [68] |
| Temozolomide, Δ9-tetrahydrocannabinol (THC) and Cannabidiol (CBD) | U87MG, LN405, HG14, HG19, T98G, HG2, HG21, U373, A172, SW1783 cells, T98G and U87-MG based tumor xenografts. | 3-MA, or genetically using siRNA directed to Atg1 | Cytotoxic form of autophagy TMZ did not induce autophagy | [69] |
| Temozolomide and interferon (IFN-β) | Melanoma cell lines including A375 and CRL-1579 cells | NA | Cytotoxic | [79] |
| Temozolomide | Mel MTP, Mel Z, Mel IL, Mel Ksen, and Mel Rac melanoma cell lines. | Chloroquine and LY294002 (LY) | Cytoprotective | [80] |
| Temozolomide and trehalose with and without radiation | A375 and SK-Mel-28 melanoma cells | NA | TMZ did not induce autophagy | [81] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elshazly, A.M.; Gewirtz, D.A. Is Autophagy Inhibition in Combination with Temozolomide a Therapeutically Viable Strategy? Cells 2023, 12, 535. https://doi.org/10.3390/cells12040535
Elshazly AM, Gewirtz DA. Is Autophagy Inhibition in Combination with Temozolomide a Therapeutically Viable Strategy? Cells. 2023; 12(4):535. https://doi.org/10.3390/cells12040535
Chicago/Turabian StyleElshazly, Ahmed M., and David A. Gewirtz. 2023. "Is Autophagy Inhibition in Combination with Temozolomide a Therapeutically Viable Strategy?" Cells 12, no. 4: 535. https://doi.org/10.3390/cells12040535
APA StyleElshazly, A. M., & Gewirtz, D. A. (2023). Is Autophagy Inhibition in Combination with Temozolomide a Therapeutically Viable Strategy? Cells, 12(4), 535. https://doi.org/10.3390/cells12040535