
Hydroquinone, an Environmental Pollutant, Affects Cartilage Homeostasis through the Activation of the Aryl Hydrocarbon Receptor Pathway


Abstract
:
1. Introduction
2. Material and Methods
2.1. Animals and Collagen-Induced Arthritis (CIA)
2.2. Collagen-Induced Arthritis (CIA) Induction and In Vivo HQ Exposure
2.3. Isolation and Culture of Bovine Articular Chondrocytes and Cartilage Explants
2.4. MTT Assay
2.5. FACS Assay
2.6. Gene Expression
2.7. Micromass Cultures
2.8. Alcian Blue Staining
2.9. Safranin-O Staining
2.10. DMMB Assay
2.11. Quantification of Nitric Oxide by Griess Assay
2.12. Quantification of Reactive Oxygen Species by Dichloro-Dihydro-Fluorescein Diacetate (DCFH-DA) Assay
2.13. Reporter Assay
2.14. Statistical Analysis
3. Results
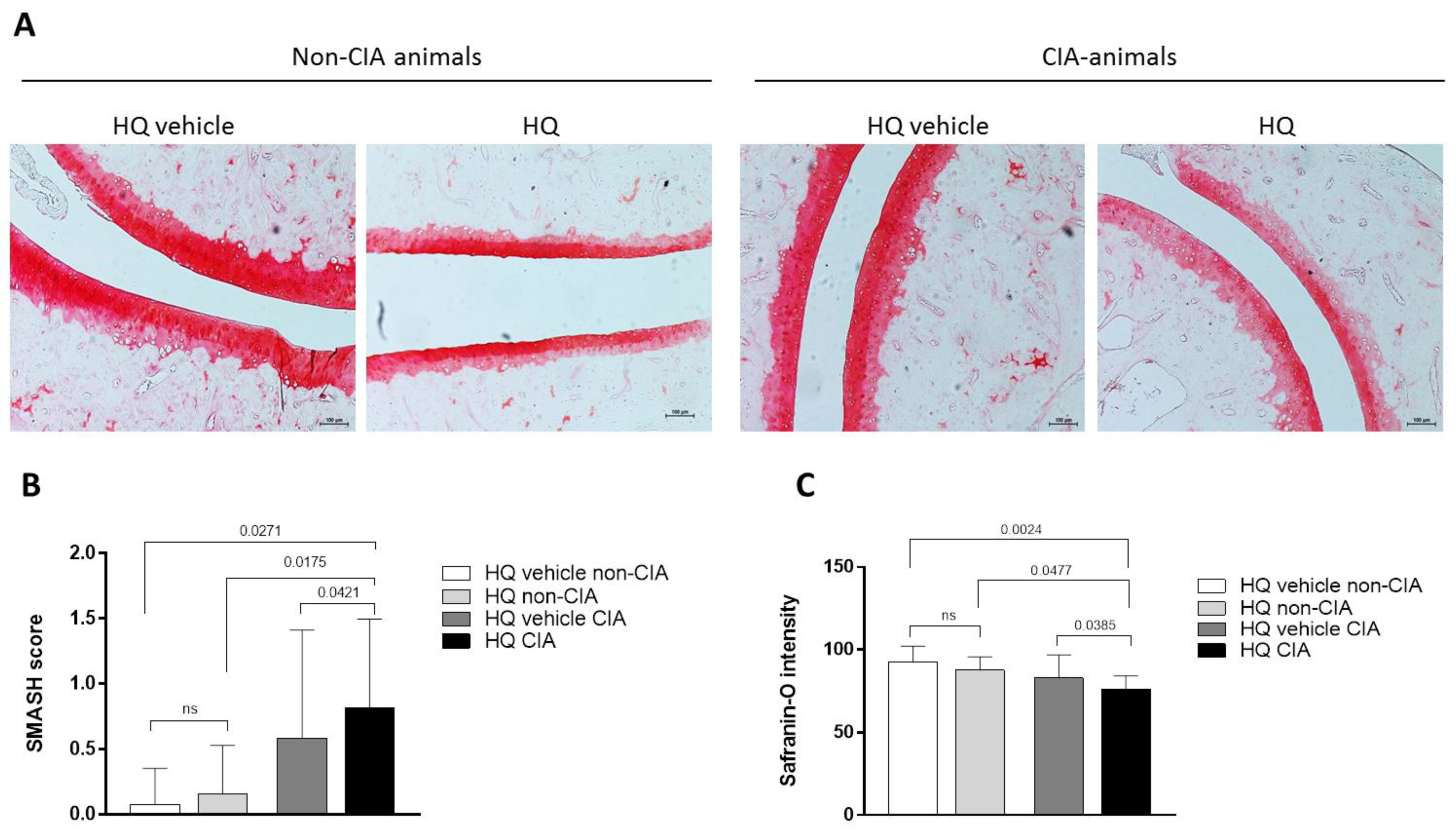
3.1. Exposure to HQ Exacerbates Cartilage Damage in an In Vivo Model of Inflammatory Arthritis
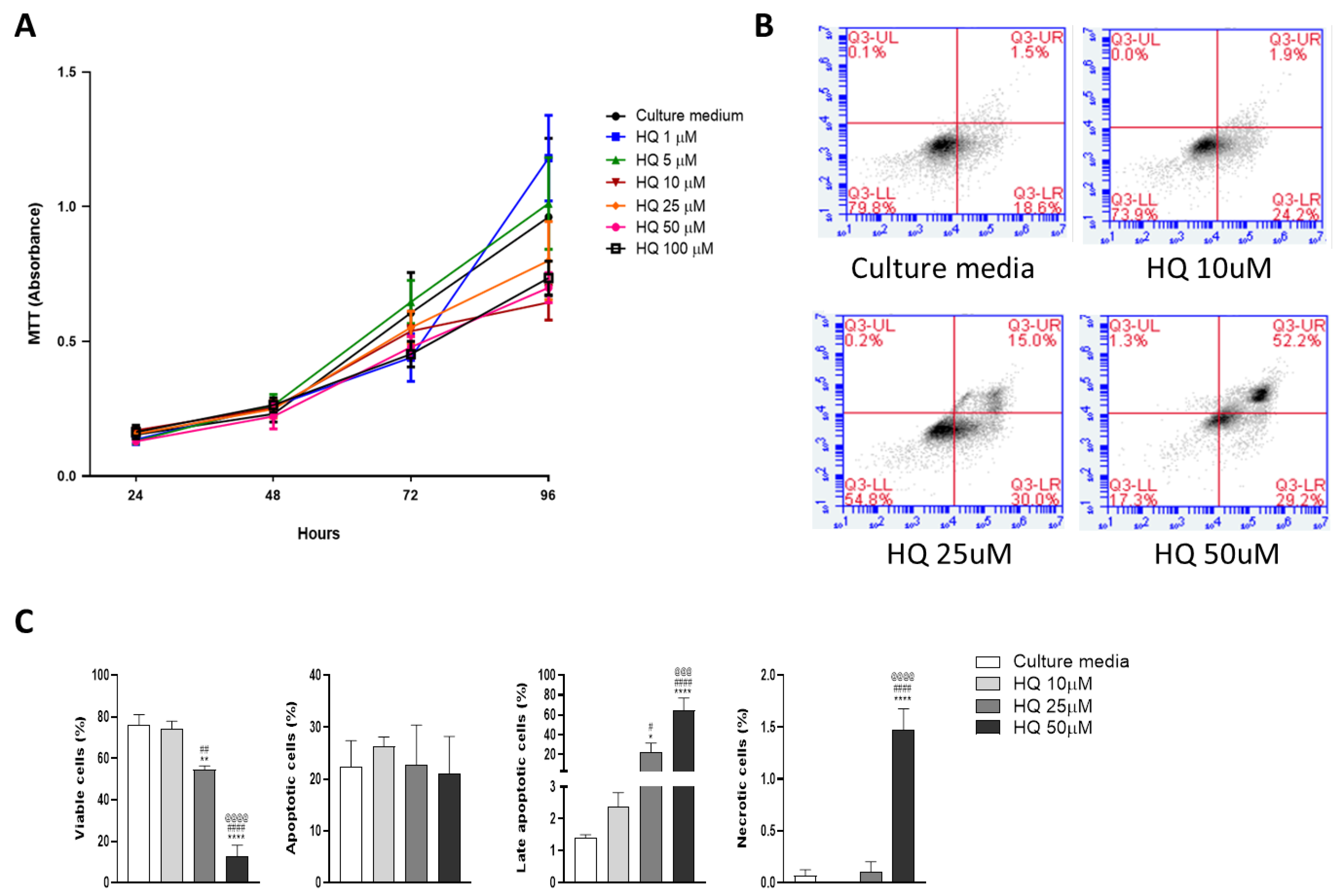
3.2. Low Concentrations of HQ Slow Down Chondrocyte Growth without Compromising Cell Viability
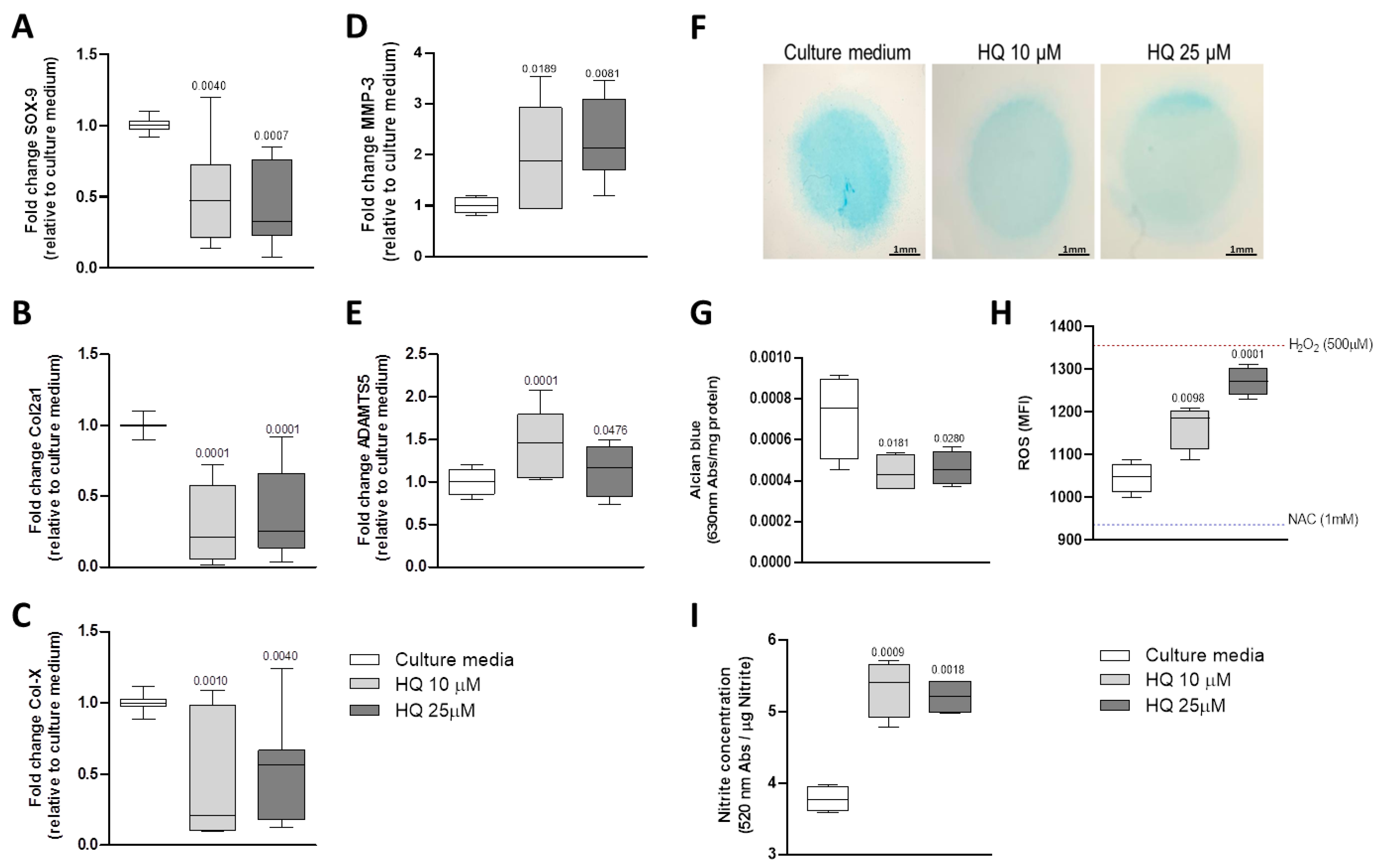
3.3. HQ Modulates the Expression of Phenotypic Markers in Articular Chondrocytes
3.4. HQ Triggers Oxidative Stress in Articular Chondrocytes
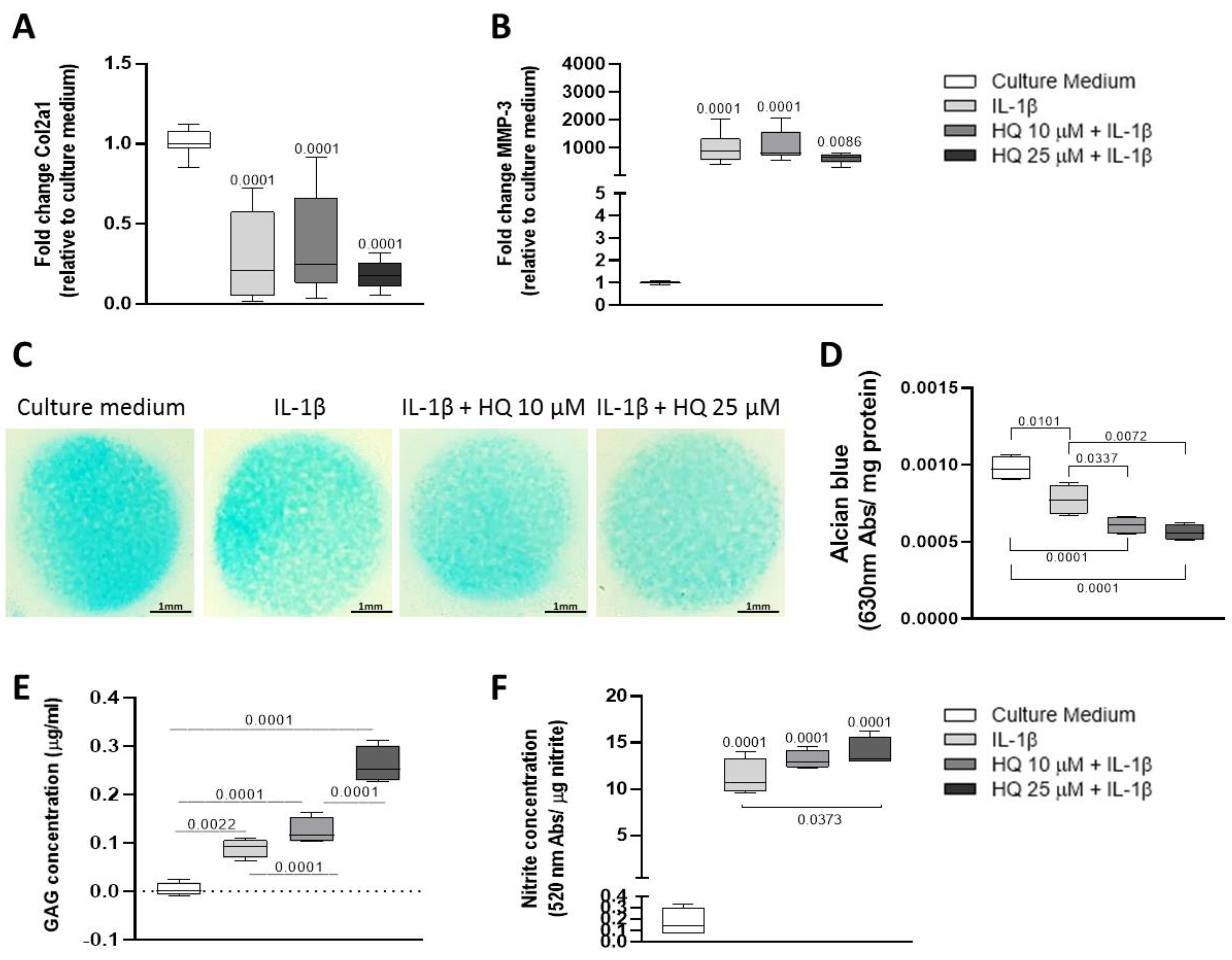
3.5. HQ Enhanced the Pro-Catabolic Activity of IL-1β
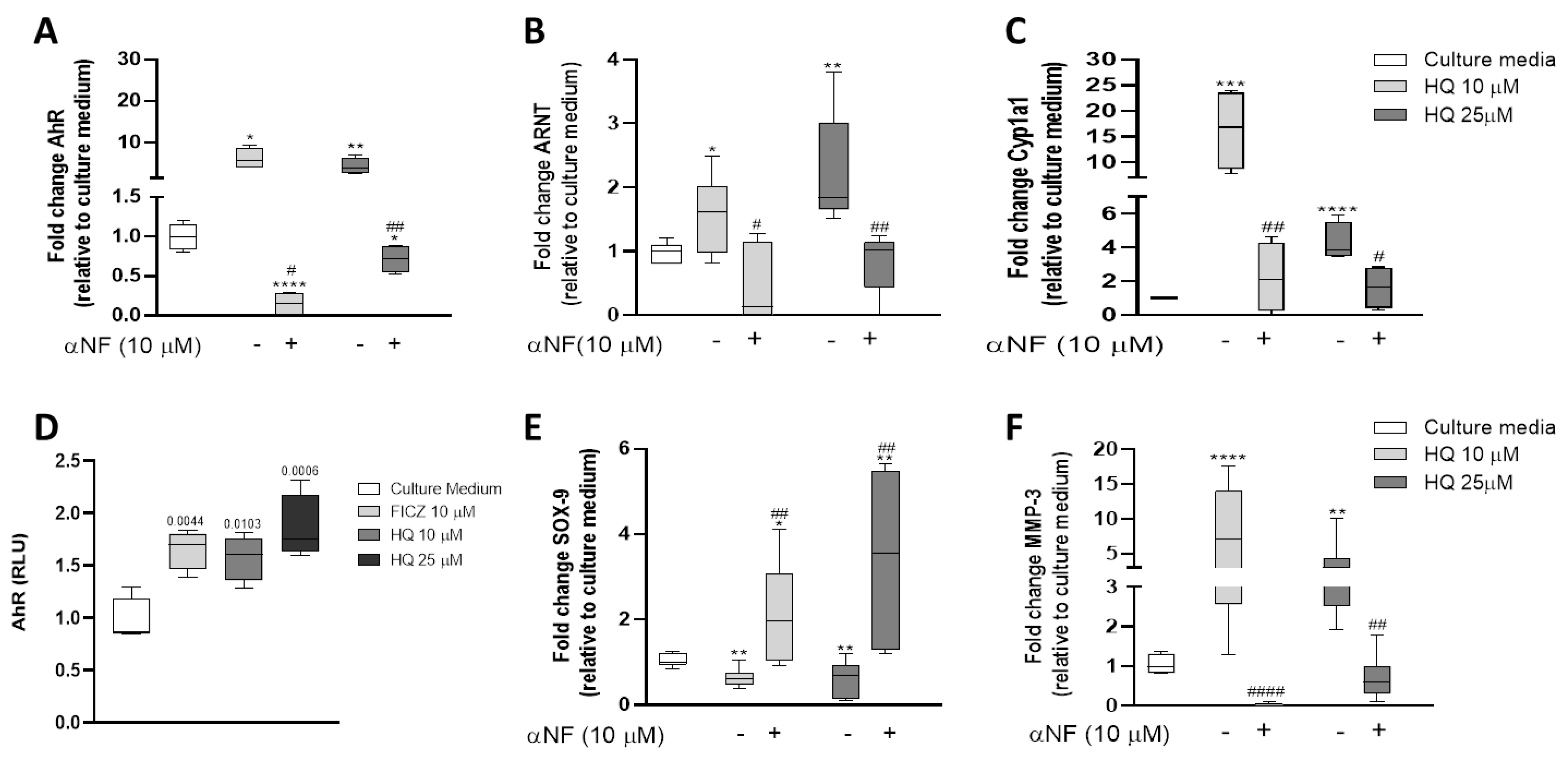
3.6. HQ Activates the AhR Pathway in Articular Chondrocytes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Enguita, F.J.; Leitão, A.L. Hydroquinone: Environmental pollution, toxicity, and microbial answers. Biomed Res. Int. 2013, 2013, 542168. [Google Scholar] [CrossRef] [Green Version]
- Bodnar, J.A.; Morgan, W.T.; Murphy, P.A.; Ogden, M.W. Mainstream smoke chemistry analysis of samples from the 2009 US cigarette market. Regul. Toxicol. Pharmacol. 2012, 64, 35–42. [Google Scholar] [CrossRef]
- McGregor, D. Hydroquinone: An evaluation of the human risks from its carcinogenic and mutagenic properties. Crit. Rev. Toxicol. 2007, 37, 887–914. [Google Scholar] [CrossRef]
- Cho, J.Y. Suppressive effect of hydroquinone, a benzene metabolite, on in vitro inflammatory responses mediated by macrophages, monocytes, and lymphocytes. Mediat. Inflamm. 2008, 2008, 298010. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Yang, J.S.; Yang, E.J.; Kim, I.S. Hydroquinone-induced apoptosis of human lymphocytes through caspase 9/3 pathway. Mol. Biol. Rep. 2012, 39, 6737–6743. [Google Scholar] [CrossRef]
- Li, J.; Jiang, S.; Chen, Y.; Ma, R.; Chen, J.; Qian, S.; Shi, Y.; Han, Y.; Zhang, S.; Yu, K. Benzene metabolite hydroquinone induces apoptosis of bone marrow mononuclear cells through inhibition of β-catenin signaling. Toxicol. Vitr. 2018, 46, 361–369. [Google Scholar] [CrossRef]
- Fogelholm, R.R.; Alho, A.V. Smoking and intervertebral disc degeneration. Med. Hypotheses 2001, 56, 537–539. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Scott, S.C.; Mayo, N. A review of the association between cigarette smoking and the development of nonspecific back pain and related outcomes. Spine 2000, 25, 995–1014. [Google Scholar] [CrossRef]
- Heluany, C.S.; Kupa, L.V.K.; Viana, M.N.; Fernandes, C.M.; Farsky, S.H.P. Hydroquinone exposure worsens the symptomatology of rheumatoid arthritis. Chem. Biol. Interact. 2018, 291, 120–127. [Google Scholar] [CrossRef]
- Heluany, C.S.; Kupa, L.V.K.; Viana, M.N.; Fernandes, C.M.; Silveira, E.L.V.; Farsky, S.H.P. In vivo exposure to hydroquinone during the early phase of collagen-induced arthritis aggravates the disease. Toxicology 2018, 481, 22–30. [Google Scholar] [CrossRef]
- Heluany, C.; Donate, P.; Schneider, A.; Fabris, A.; Gomes, R.; Villas-Boas, I.; Tambourgi, D.; Silva, T.; Trossini, G.; Nalesso, G.; et al. Hydroquinone Exposure Worsens Rheumatoid Arthritis through the Activation of the Aryl Hydrocarbon Receptor and Interleukin-17 Pathways. Antioxidants 2021, 10, 929. [Google Scholar] [CrossRef]
- Lin, Z.; Willers, C.; Xu, J.; Zheng, M.-H. The chondrocyte: Biology and clinical application. Tissue Eng. 2006, 12, 1971–1984. [Google Scholar] [CrossRef]
- Eyre, D. Articular cartilage and changes in Arthritis: Collagen of articular cartilage. Arthritis Res. Ther. 2001, 4, 30. [Google Scholar] [CrossRef] [Green Version]
- Sophia Fox, A.J.; Bedi, A.; Rodeo, S.A. The basic science of articular cartilage: Structure, composition, and function. Sport. Health 2009, 1, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Brand, D.D.; Latham, K.A.; Rosloniec, E.F. Collagen-induced arthritis. Nat. Protoc. 2007, 2, 1269–1275. [Google Scholar] [CrossRef]
- Nalesso, G.; Sherwood, J.; Bertrand, J.; Pap, T.; Ramachandran, M.; De Bari, C.; Pitzalis, C.; Dell’Accio, F. WNT-3A modulates articular chondrocyte phenotype by activating both canonical and noncanonical pathways. J. Cell Biol. 2011, 193, 551–564. [Google Scholar] [CrossRef] [Green Version]
- De Bari, C.; Dell’Accio, F.; Luyten, F.P. Human periosteum-derived cells maintain phenotypic stability and chondrogenic potential throughout expansion regardless of donor age. Arthritis Rheum. 2001, 44, 88–95. [Google Scholar] [CrossRef]
- Hayer, S.; Vervoordeldonk, M.J.; Denis, M.C.; Armaka, M.; Hoffmann, M.; Bäcklund, J.; Nandakumar, K.S.; Niederreiter, B.; Geka, C.; Fischer, A.; et al. ‘SMASH’ recommendations for standardised microscopic arthritis scoring of histological sections from inflammatory arthritis animals models. Ann. Rheum. Dis. 2021, 80, 714–726. [Google Scholar] [CrossRef]
- Farndale, R.W.; Sayers, C.A.; Barrett, A. A direct spectrophotometric microassay for sulphated glycosaminoglycans in cartilage cultures. Connect. Tissue Res. 1982, 9, 247–248. [Google Scholar] [CrossRef]
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef]
- Dell’Accio, F.; De Bari, C.; Luyten, F.P. Molecular markers predictive of the capacity of expanded human articular chondrocytes to form stable cartilage in vivo. Arthritis Rheum. 2001, 44, 1608–1619. [Google Scholar] [CrossRef]
- Sandell, L.J.; Aigner, T. Articular cartilage and changes in arthritis. An introduction: Cell biology of osteoarthritis. Arthritis Res. 2001, 3, 107–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, J.; Dai, W.; Zhang, S.; Sun, L.; Wang, H.; Gao, Y.; Wang, J.; Zhang, F. Quinone-thioether metabolites of hydroquinone play a dual role in promoting a vicious cycle of ROS generation: In vitro and in silico insights. Arch. Toxicol. 2019, 93, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- Henrotin, Y.E.; Bruckner, P.; Pujol, J.P. The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthr. Cartil. 2003, 11, 747–755. [Google Scholar] [CrossRef] [Green Version]
- Amin, A.; Abramson, S.B. The role of nitric oxide in articular cartilage breakdown in osteoarthritis. Curr. Opin. Rheumatol. 1998, 10, 263–268. [Google Scholar] [CrossRef]
- Daheshia, M.; Yao, J.Q. The interleukin 1beta pathway in the pathogenesis of osteoarthritis. J. Rheumatol. 2008, 35, 2306–2312. [Google Scholar] [CrossRef]
- Jenei-Lanzl, Z.; Meurer, A.; Zaucke, F. Interleukin-1β signalling in osteoarthritis—Chondrocytes in focus. Cell. Sig. 2019, 53, 212–223. [Google Scholar] [CrossRef]
- Sellan, J.; Berenbaum, F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat. Rev. Rheumatol. 2010, 6, 625–635. [Google Scholar] [CrossRef]
- Ma, Q.; Baldwin, K.T. 2,3,7,8-tetrachorodibenzo-p-dioxin- induced degradation of aryl hydrocarbon receptor (AhR) by the ubiquitin-proteasome pathway. Role of the transcription activation and DNA binding. J. Biol. Chem. 2000, 275, 8432–8438. [Google Scholar] [CrossRef] [Green Version]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef]
- Finckh, A.; Gilbert, B.; Hodkinson, B.; Bae, S.-C.; Thomas, R.; Deane, K.D.; Alpizar-Rodriguez, D.; Lauper, K. Global epidemiology of rheumatoid arthritis. Nat. Rev. Rheumatol. 2022, 18, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Cui, A.; Li, H.; Wang, D.; Zhong, L.; Chen, Y.; Lu, H. Global, regional prevalence, incidence and risk factors of knee osteoarthritis in population-based studies. EClinicalMedicine 2020, 29, 100587. [Google Scholar] [CrossRef]
- Mort, J.S.; Billinton, C.J. Articular cartilage and changes in arthritis matrix degradation. Arthritis Res. 2001, 3, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Goldring, S.R. Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann. N. Y. Acad. Sci. 2010, 1192, 230–237. [Google Scholar] [CrossRef]
- Arner, E.C. Aggrecanase-mediated cartilage degradation. Curr. Opin. Pharmacol. 2002, 2, 322–329. [Google Scholar] [CrossRef]
- Poole, A.R.; Kobayashi, M.; Yasuda, T.; Laverty, S.; Mwale, F.; Kojima, T.; Sakai, T.; Wahl, C.; El-Maadawy, S.; Webb, G.; et al. Type II collagen degradation and its regulation in articular cartilage in osteoarthritis. Ann. Rheum. Dis. 2001, 61, ii78–ii81. [Google Scholar] [CrossRef] [Green Version]
- Bortoluzzi, A.; Furini, F.; Sciré, C.A. Osteoarthritis and its management—Epidemiology, nutritional aspects and environmental factors. Autoimmun. Rev. 2018, 17, 07–1114. [Google Scholar] [CrossRef]
- Suantawee, T.; Tantavisut, S.; Adisakwattana, S.; Tanavalee, A.; Yuktanandana, P.; Anomasiri, W.; Deepaisarnsakul, B.; Honsawek, S. Oxidative stress, vitamin e, and antioxidant capacity in knee osteoarthritis. J. Clin. Diagn. Res. 2013, 7, 1855–1859. [Google Scholar] [CrossRef] [PubMed]
- Abramson, S.B. Osteoarthritis and nitric oxide. Osteoarthr. Cartil. 2008, 16, S15–S20. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.G.; Yang, J.H. PCB126 induces apoptosis of chondrocytes via ROS-dependent pathways. Osteoarthr. Cartil. 2012, 20, 1179–1185. [Google Scholar] [CrossRef] [Green Version]
- Pons, M.; Cousins, S.W.; Csaky, K.G.; Striker, G.; Marin-Castaño, M.E. Cigarette smoke-related hydroquinone induces filamentous actin reorganization and heat shock protein 27 phosphorylation through p38 and extracellular signal-regulated kinase 1/2 in retinal pigment epithelium: Implication for age-related macular degeneration. Am. J. Pathol. 2010, 177, 1198–1213. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Arthur, D.; Liu, F.; Lee, J.; Xia, Q.; Lavin, M.F.; Ng, J.C. Genotoxicity of hydroquinone in A549 cells. Cell Biol. Toxicol. 2013, 29, 213–227. [Google Scholar] [CrossRef]
- Julliard, W.; Fechner, J.H.; Mezrich, J.D. The aryl hydrocarbon receptor meets immunology: Friend or foe? A little of both. Front. Immunol. 2014, 5, 458. [Google Scholar] [CrossRef] [Green Version]
- Denison, M.S.; Nagy, S.R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogeneous chemicals. Ann. Rev. Pharmacol. Toxicol. 2003, 43, 309–334. [Google Scholar] [CrossRef] [PubMed]
- Nakahama, T.; Kimura, A.; Nguyen, N.T.; Chinen, I.; Hanieh, H.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor deficiency in T cells suppresses the development of collagen-induced arthritis. Proc. Natl. Acad. Sci. USA 2011, 108, 14222–14227. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.T.; Nakahama, T.; Nguyen, C.H.; Tran, T.T.; Le, V.S.; Chu, H.H.; Kishimoto, T. Aryl hydrocarbon receptor antagonism and its role in rheumatoid arthritis. J. Exp. Pharmacol. 2015, 7, 29–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, S.; Okamoto, H.; Iwamoto, T.; Toyama, Y.; Tomatsu, T.; Yamanaka, H.; Momohara, S. A role for the aryl hydrocarbon receptor and the dioxin TCDD in rheumatoid arthritis. Rheumatology 2008, 47, 1317–1322. [Google Scholar] [CrossRef] [Green Version]
- Talbot, J.; Peres, R.S.; Pinto, L.G.; Oliveira, R.D.R.; Lima, K.A.; Donate, P.B.; Silva, J.R.; Ryffel, B.; Cunha, T.M.; Alves-Filho, J.C.; et al. Smoking-induced aggravation of experimental arthritis is dependent of aryl hydrocarbon receptor activation in Th17 cells. Arthritis Res. Ther. 2018, 20, 119. [Google Scholar] [CrossRef] [Green Version]
- Widerak, M.; Ghoneim, C.; Dumontier, M.-F.; Quesne, M.; Corvol, M.T.; Savouret, J.-F. The aryl hydrocarbon receptor activates the retinoic acid receptor α through SMRT antagonism. Biochimie 2006, 88, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Gargaro, M.; Scalisi, G.; Manni, G.; Mondanelli, G.; Grohmann, U.; Fallarino, F. The Landscape of AhR regulators and coregulators to fine-tune AhR functions. Int. J. Mol. Sci. 2021, 22, 757. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sense Primer | Anti-Sense Primer | Annealing Temperature (°C) |
|---|---|---|---|
| β-actin | AGCAGTCGGTTGGATCGAGCA | GGGAAGGCAAAGGACTTCCTGTAAC | 55 |
| SOX-9 | GCTCTGGGCAAGCTCTGGAGACT | GGCGCGGCTGGTACTTGTAGTCC | 60 |
| Col2a1 | ACGTCCAGATGACCTTCCTG | GGATGAGCAGAGCCTTCTTG | 55 |
| COL-X | AAAGGTCTAAGTGGCCCCTTTTGTC | GAGGTTCATGACAAAAGCACCTTGC | 55 |
| ADAMTS5 | ACGGGACCGTCATGAACTAC | CTTTTGGAGCCGACTTCTTG | 55 |
| MMP-3 | TGTGTGTCTTGCCCACTAGC | TGCCTGTTGCAGAATGCTAA | 55 |
| AhR | AGCAGCGCTAACATCACCTA | GGGACTGGCTTGACAGTTTTC | 60 |
| ARNT | CAGGTGCACCCAGATGATGT | AGATCCAGGATACGCCCTGT | 60 |
| Cyp1a1 | ACCTTGATCACTAACGGCCA | GCCTCCTTGTTCACATGCTCT | 60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heluany, C.S.; De Palma, A.; Day, N.J.; Farsky, S.H.P.; Nalesso, G. Hydroquinone, an Environmental Pollutant, Affects Cartilage Homeostasis through the Activation of the Aryl Hydrocarbon Receptor Pathway. Cells 2023, 12, 690. https://doi.org/10.3390/cells12050690
Heluany CS, De Palma A, Day NJ, Farsky SHP, Nalesso G. Hydroquinone, an Environmental Pollutant, Affects Cartilage Homeostasis through the Activation of the Aryl Hydrocarbon Receptor Pathway. Cells. 2023; 12(5):690. https://doi.org/10.3390/cells12050690
Chicago/Turabian StyleHeluany, Cintia Scucuglia, Anna De Palma, Nicholas James Day, Sandra Helena Poliselli Farsky, and Giovanna Nalesso. 2023. "Hydroquinone, an Environmental Pollutant, Affects Cartilage Homeostasis through the Activation of the Aryl Hydrocarbon Receptor Pathway" Cells 12, no. 5: 690. https://doi.org/10.3390/cells12050690