
CRISPR/dCas9-KRAB-Mediated Suppression of S100b Restores p53-Mediated Apoptosis in Melanoma Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. TCGA Patient Cohorts
2.2. Proteomics Data Set from Cases of Melanoma Treated with Immune Checkpoint Inhibitors (ICIs)
2.3. Identification of DNase Hypersensitive Sites (DHSs)
2.4. Prediction of Homology between Human and Murine Proteins
2.5. Prediction of Transcription Factor (TF) Binding
2.6. Development of CRISPR/dCas9-KRAB Tool
2.7. Cell Lines and Transduction
2.8. Quantitation of Expression with Quantitative Real-Time PCR (qPCR)
2.9. Western Blot Analysis of Protein Levels
2.10. Apoptosis and Cell Death Assay
2.11. Cell Proliferation and Chemotherapeutic Sensitivity Assay
2.12. Statistical Analysis
3. Results
3.1. Overexpression of S100B Is Linked to Copy-Number Changes and Epigenetic Alterations of the Gene
3.2. S100b Expression Is Suppressed by CRISPR/dCAS9-KRAB
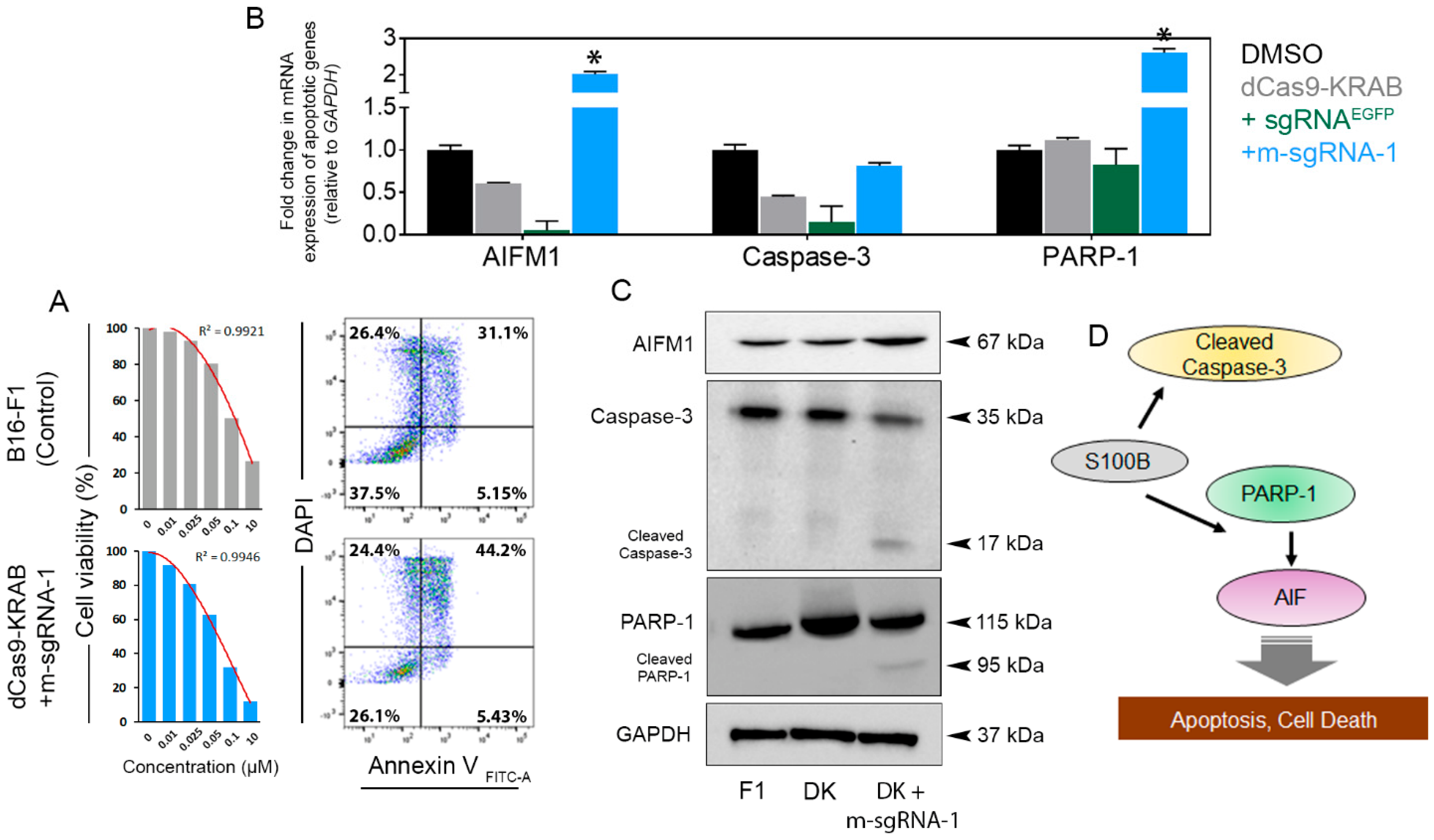
3.3. S100b Suppression Restores WT-p53 Level and Activates Apoptotic Signaling
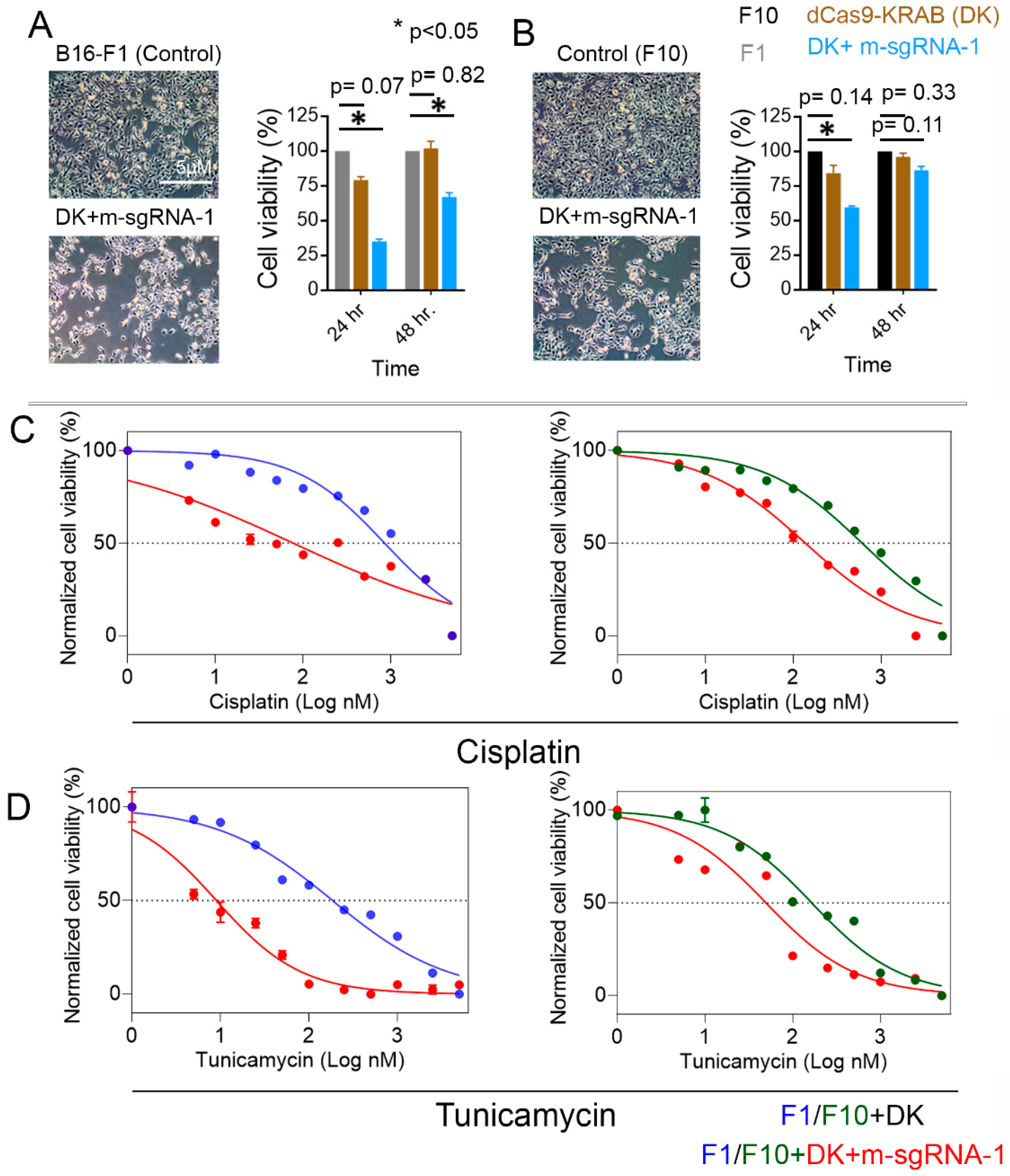
3.4. S100b Suppression in Melanoma Cells Decreases Their Viability and Increases Their Susceptibility to Chemotherapeutics
3.5. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guy, G.P., Jr.; Thomas, C.C.; Thompson, T.; Watson, M.; Massetti, G.M.; Richardson, L.C. Vital signs: Melanoma incidence and mortality trends and projections—United States, 1982–2030. Morb. Mortal. Wkly. Rep. 2015, 64, 591. [Google Scholar]
- Frauchiger, A.L.; Dummer, R.; Mangana, J. Serum S100B Levels in Melanoma. Methods Mol. Biol. 2019, 1929, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Tarhini, A.A.; Stuckert, J.; Lee, S.; Sander, C.; Kirkwood, J.M. Prognostic significance of serum S100B protein in high-risk surgically resected melanoma patients participating in Intergroup Trial ECOG 1694. J. Clin. Oncol. 2009, 27, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauschild, A.; Engel, G.; Brenner, W.; Gläser, R.; Mönig, H.; Henze, E.; Christophers, E. S100B Protein Detection in Serum Is a Significant Prognostic Factor in Metastatic Melanoma. Oncology 1999, 56, 338–344. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, L.; Zhang, I.Y.; Chen, X.; Da Fonseca, A.; Wu, S.; Ren, H.; Badie, S.; Sadeghi, S.; Ouyang, M.; et al. S100B promotes glioma growth through chemoattraction of myeloid-derived macrophages. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 3764–3775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclerc, E. Brain Metastases from Primary Tumors; Hayat, M.A., Ed.; Academic Press: Cambridge, MA, USA, 2016; Volume 3, pp. 119–132. [Google Scholar]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 Proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttunen, H.J.; Fages, C.; Kuja-Panula, J.; Ridley, A.J.; Rauvala, H. Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res. 2002, 62, 4805–4811. [Google Scholar] [PubMed]
- Baudier, J.; Delphin, C.; Grunwald, D.; Khochbin, S.; Lawrence, J.J. Characterization of the tumor suppressor protein p53 as a protein kinase C substrate and a S100b-binding protein. Proc. Natl. Acad. Sci. USA 1992, 89, 11627–11631. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Yang, Q.; Wilder, P.T.; Carrier, F.; Weber, D.J. The calcium-binding protein S100B down-regulates p53 and apoptosis in malignant melanoma. J. Biol. Chem. 2010, 285, 27487–27498. [Google Scholar] [CrossRef] [Green Version]
- Wilder, P.T.; Lin, J.; Bair, C.L.; Charpentier, T.H.; Yang, D.; Liriano, M.; Varney, K.M.; Lee, A.; Oppenheim, A.B.; Adhya, S.; et al. Recognition of the tumor suppressor protein p53 and other protein targets by the calcium-binding protein S100B. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2006, 1763, 1284–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, K.G.; McKnight, L.E.; Liriano, M.A.; Weber, D.J. The evolution of S100B inhibitors for the treatment of malignant melanoma. Future Med. Chem. 2013, 5, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsi, K.M.; Hennessy, E.; Kearns, N.; Maehr, R. Eukaryotic Transcriptional and Post-Transcriptional Gene Expression Regulation; Springer: Berlin, Germany, 2017; pp. 221–233. [Google Scholar]
- Thakore, P.I.; D’ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 2015, 12, 1143–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, E.M.; Koss, B.; Washam, C.L.; Shalin, S.C.; Barreto, J.C.; Graw, S.; Byrum, S.D.; Mackintosh, S.G.; Edmondson, R.D.; Tackett, A. Proteomic Signatures of Innate Immunity in Immunotherapy Naïve Melanomas Correlate with Anti-CTLA-4 Immunotherapy Responsiveness. 2020. Available online: https://assets.researchsquare.com/files/rs-69890/v1/29d1641d-4889-436d-9b7d-10ab50f9c4d1.pdf?c=1631854762 (accessed on 19 February 2023).
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Farré, D.; Roset, R.; Huerta, M.; Adsuara, J.E.; Roselló, L.; Albà, M.M.; Messeguer, X. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res. 2003, 31, 3651–3653. [Google Scholar] [CrossRef] [Green Version]
- Messeguer, X.; Escudero, R.; Farré, D.; Nuñez, O.; Martínez, J.; Albà, M.M. PROMO: Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002, 18, 333–334. [Google Scholar] [CrossRef] [Green Version]
- Sanson, K.R.; Hanna, R.E.; Hegde, M.; Donovan, K.F.; Strand, C.; Sullender, M.E.; Vaimberg, E.W.; Goodale, A.; Root, D.E.; Piccioni, F.; et al. Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nat. Commun. 2018, 9, 5416. [Google Scholar] [CrossRef] [Green Version]
- Garbe, C.; Leiter, U.; Ellwanger, U.; Blaheta, H.J.; Meier, F.; Rassner, G.; Schittek, B. Diagnostic value and prognostic significance of protein S-100beta, melanoma-inhibitory activity, and tyrosinase/MART-1 reverse transcription-polymerase chain reaction in the follow-up of high-risk melanoma patients. Cancer 2003, 97, 1737–1745. [Google Scholar] [CrossRef]
- Dunleavey, J.M.; Xiao, L.; Thompson, J.; Kim, M.M.; Shields, J.M.; Shelton, S.E.; Irvin, D.M.; Brings, V.E.; Ollila, D.W.; Brekken, R.A.; et al. Vascular channels formed by subpopulations of PECAM1+ melanoma cells. Nat. Commun. 2014, 5, 5200. [Google Scholar] [CrossRef] [Green Version]
- Vasilevska, J.; De Souza, G.A.; Stensland, M.; Skrastina, D.; Zhulenvovs, D.; Paplausks, R.; Kurena, B.; Kozlovska, T.; Zajakina, A. Comparative protein profiling of B16 mouse melanoma cells susceptible and non-susceptible to alphavirus infection: Effect of the tumor microenvironment. Cancer Biol. Ther. 2016, 17, 1035–1050. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.M.; Sun, G.G.; Han, L.N.; Liu, L.H.; Ren, F.Z.; Li, L.; Ma, M.; Shan, B.E. P-Hydroxycinnamaldehyde Induces B16-F1 Melanoma Cell Differentiation via the RhoA-MAPK Signaling Pathway. Cell. Physiol. Biochem. 2016, 38, 2247–2260. [Google Scholar] [CrossRef]
- Labun, K.; Montague, T.G.; Krause, M.; Cleuren, Y.N.T.; Tjeldnes, H.; Valen, E. CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019, 47, W171–W174. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liu, H.; Wang, Q.; Zhou, F.; Liu, Y.; Zhang, Y.; Ding, H.; Yuan, M.; Li, F.; Chen, Y. Involvement of c-Fos in cell proliferation, migration, and invasion in osteosarcoma cells accompanied by altered expression of Wnt2 and Fzd9. PLoS ONE 2017, 12, e0180558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, R.; Janz, M.; Galson, D.L.; Gries, M.; Li, S.; Jöhrens, K.; Anagnostopoulos, I.; Dörken, B.; Mapara, M.Y.; Borghesi, L.; et al. C/EBPbeta regulates transcription factors critical for proliferation and survival of multiple myeloma cells. Blood 2009, 114, 3890–3898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chikhirzhina, G.I.; Al’-Shekhadat, R.I.; Chikhirzhina, E.V. Transcription factors of the nuclear factor 1 (NF1) family. Role in chromatin remodelation. Mol. Biol. 2008, 42, 388–404. [Google Scholar] [CrossRef]
- Scotto, C.; Deloulme, J.C.; Rousseau, D.; Chambaz, E.; Baudier, J. Calcium and S100B Regulation of p53-Dependent Cell Growth Arrest and Apoptosis. Mol. Cell. Biol. 1998, 18, 4272–4281. [Google Scholar] [CrossRef] [Green Version]
- Tsoporis, J.N.; Mohammadzadeh, F.; Parker, T.G. Intracellular and Extracellular Effects of S100B in the Cardiovascular Response to Disease. Cardiovasc. Psychiatry Neurol. 2010, 2010, 206073. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Wang, X.; Deinum, J.; Huang, Z.; Gao, J.; Modjtahedi, N.; Neagu, M.R.; Nilsson, M.; Eriksson, P.S.; Hagberg, H.; et al. Cyclophilin A participates in the nuclear translocation of apoptosis-inducing factor in neurons after cerebral hypoxia-ischemia. J. Exp. Med. 2007, 204, 1741–1748. [Google Scholar] [CrossRef]
- Saelens, X.; Festjens, N.; Walle, L.V.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Debatin, K.M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.C.; Chen, L.H.; Gillespie, S.; Kiejda, K.A.; Mhaidat, N.; Wang, Y.F.; Thorne, R.; Zhang, X.D.; Hersey, P. Tunicamycin sensitizes human melanoma cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by up-regulation of TRAIL-R2 via the unfolded protein response. Cancer Res. 2007, 67, 5880–5888. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.; Tykodi, S.S.; Thompson, J.A. Treatment of metastatic melanoma: An overview. Oncology 2009, 23, 488–496. [Google Scholar] [PubMed]
- Choudhury, S.R.; Ashby, C.; Tytarenko, R.; Bauer, M.; Wang, Y.; Deshpande, S.; Den, J.; Schinke, C.; Zangari, M.; Thanendrarajan, S.; et al. The functional epigenetic landscape of aberrant gene expression in molecular subgroups of newly diagnosed multiple myeloma. J. Hematol. Oncol. 2020, 13, 108. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Cheng, J.; You, J.; Yan, B.; Liu, H.; Li, F. S100B promotes chemoresistance in ovarian cancer stem cells by regulating p53. Oncol. Rep. 2018, 40, 1574–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rust, R.R.; Baldisseri, D.M.; Weber, D.J. Structure of the negative regulatory domain of p53 bound to S100B(ββ). Nat. Struct. Biol. 2000, 7, 570–574. [Google Scholar] [CrossRef]
- Lin, J.; Blake, M.; Tang, C.; Zimmer, D.; Rustandi, R.R.; Weber, D.J.; Carrier, F. Inhibition of p53 transcriptional activity by the S100B calcium-binding protein. J. Biol. Chem. 2001, 276, 35037–35041. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, A.J.; Meyers, R.M.; Weir, B.A.; Vazquez, F.; Zhang, C.Z.; Ben-David, U.; Cook, A.; Ha, G.; Harrington, W.F.; Doshi, M.B.; et al. Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov. 2016, 6, 914–929. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Yoshikawa, N.; Yamaguchi, Y.; Kagota, S.; Shinozuka, K.; Kunitomo, M. Characterization of mouse melanoma cell lines by their mortal malignancy using an experimental metastatic model. Life Sci. 2002, 70, 791–798. [Google Scholar] [CrossRef]
- O’Geen, H.; Bates, S.L.; Carter, S.S.; Nisson, K.A.; Halmai, J.; Fink, K.D.; Rhie, S.K.; Farnham, P.J.; Segal, D.J. Ezh2-dCas9 and KRAB-dCas9 enable engineering of epigenetic memory in a context-dependent manner. Epigenet. Chromatin 2019, 12, 26. [Google Scholar] [CrossRef] [Green Version]
- Yeo, N.C.; Chavez, A.; Lance-Byrne, A.; Chan, Y.; Menn, D.; Milanova, D.; Kuo, C.-C.; Guo, X.; Sharma, S.; Tung, A.; et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat. Methods 2018, 15, 611–616. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy Choudhury, S.; Heflin, B.; Taylor, E.; Koss, B.; Avaritt, N.L.; Tackett, A.J. CRISPR/dCas9-KRAB-Mediated Suppression of S100b Restores p53-Mediated Apoptosis in Melanoma Cells. Cells 2023, 12, 730. https://doi.org/10.3390/cells12050730
Roy Choudhury S, Heflin B, Taylor E, Koss B, Avaritt NL, Tackett AJ. CRISPR/dCas9-KRAB-Mediated Suppression of S100b Restores p53-Mediated Apoptosis in Melanoma Cells. Cells. 2023; 12(5):730. https://doi.org/10.3390/cells12050730
Chicago/Turabian StyleRoy Choudhury, Samrat, Billie Heflin, Erin Taylor, Brian Koss, Nathan L. Avaritt, and Alan J. Tackett. 2023. "CRISPR/dCas9-KRAB-Mediated Suppression of S100b Restores p53-Mediated Apoptosis in Melanoma Cells" Cells 12, no. 5: 730. https://doi.org/10.3390/cells12050730