

Blocking EREG/GPX4 Sensitizes Head and Neck Cancer to Cetuximab through Ferroptosis Induction
 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture, Transfection and Drugs
2.2. Sphere Evasion Assay
2.3. IncuCyte® Assay
2.4. Clonogenic Survival Assay
2.5. Metabolic Assays
2.6. Western Blot
2.7. Quantification of Intracellular Fe2+ Accumulation
2.8. Detection of the Accumulation of Lipid Peroxides
2.9. Tumoroids Culture
2.10. Immunohistochemistry on Tumoroids
2.11. Statistical Analysis
3. Results
3.1. Silencing EREG Prevents Survival and Growth and Sensitizes to CTX
3.2. Silencing EREG Promotes Mitochondrial Dysfunction and Inhibits Autophagy in Reponse to CTX
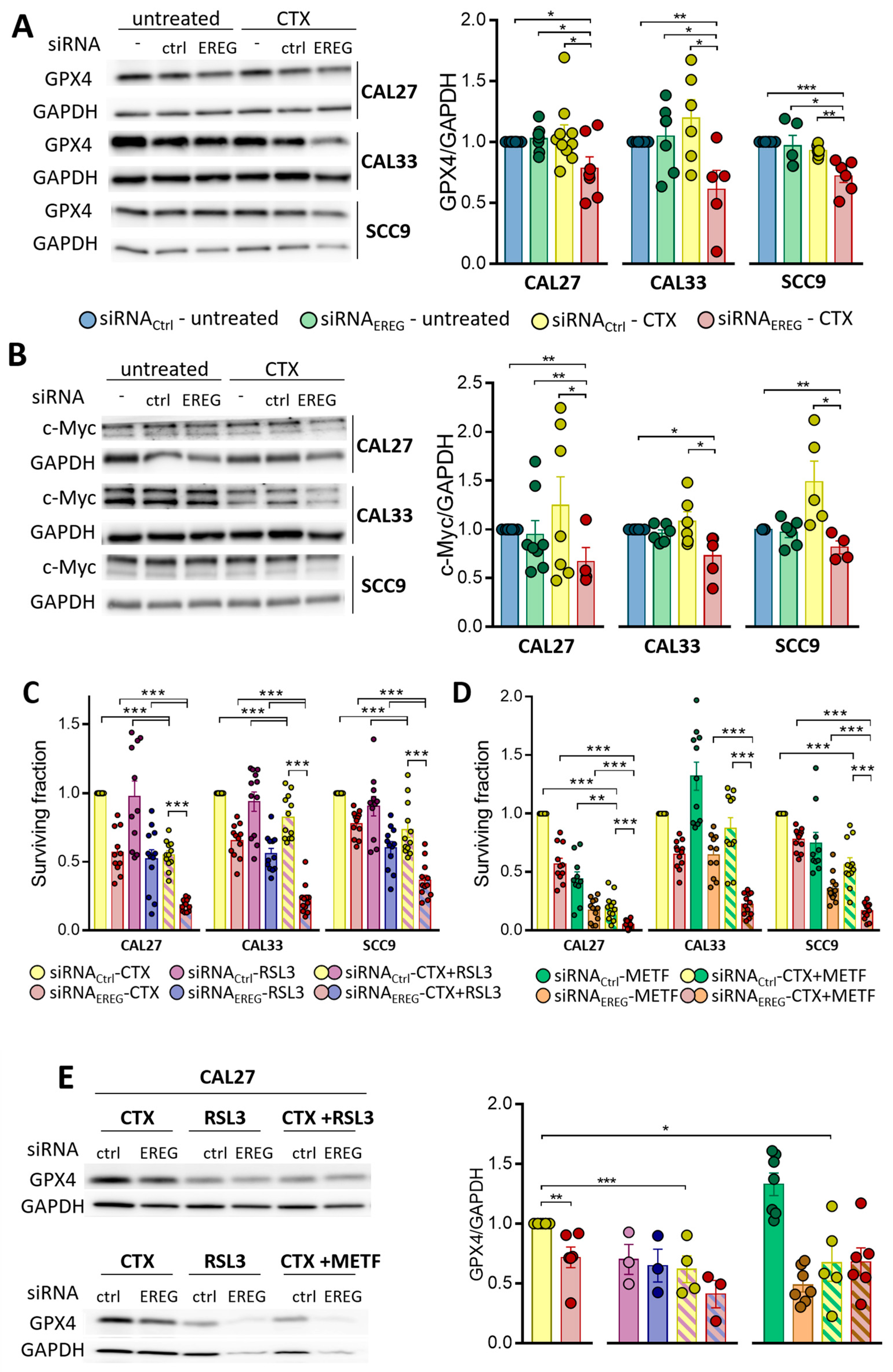
3.3. Silencing EREG Promotes Ferroptosis in Response to CTX
3.4. EREG-Silencing Uncovers the Vulnerability of Cells to GPX4 Inhibition
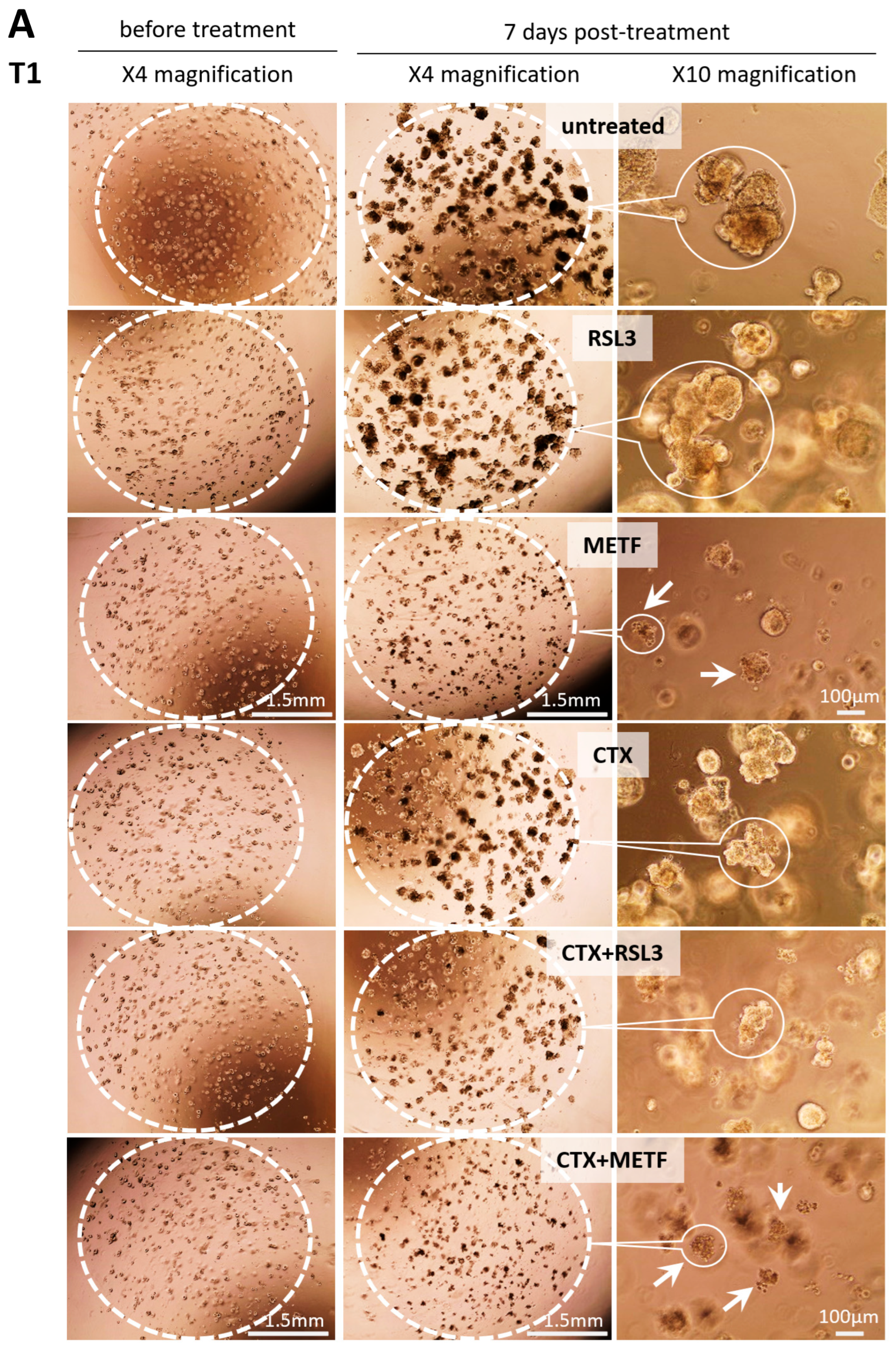
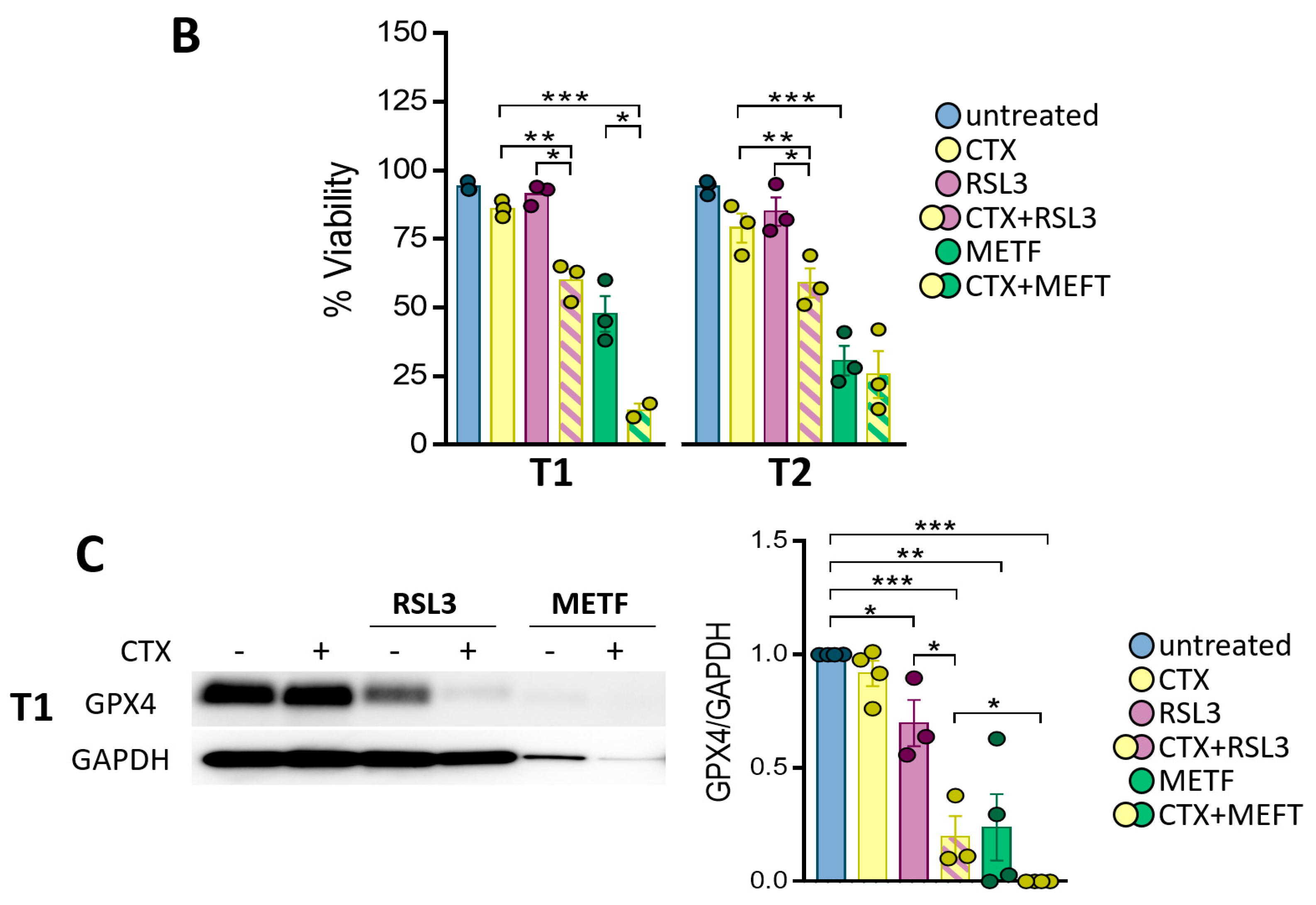
3.5. GPX4 Inhibition Sensitizes the Patient-Derived Tumoroid to CTX
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef] [Green Version]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Cohen, R.B.; Jones, C.U.; Sur, R.K.; Raben, D.; Baselga, J.; Spencer, S.A.; Zhu, J.; et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010, 11, 21–28. [Google Scholar] [CrossRef]
- Goldstein, N.I.; Prewett, M.; Zuklys, K.; Rockwell, P.; Mendelsohn, J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin. Cancer Res. 1995, 1, 1311–1318. [Google Scholar]
- Masuelli, L.; Budillon, A.; Marzocchella, L.; Mrozek, M.A.; Vitolo, D.; Di Gennaro, E.; Losito, S.; Sale, P.; Longo, F.; Ionna, F.; et al. Caveolin-1 overexpression is associated with simultaneous abnormal expression of the E-cadherin/alpha-beta catenins complex and multiple ErbB receptors and with lymph nodes metastasis in head and neck squamous cell carcinomas. J. Cell. Physiol. 2012, 227, 3344–3353. [Google Scholar] [CrossRef] [PubMed]
- Brand, T.M.; Iida, M.; Wheeler, D.L. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol. Ther. 2011, 11, 777–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciardiello, F.; Tortora, G. EGFR antagonists in cancer treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgy, M.; Jehl, A.; Conrad, O.; Foppolo, S.; Bruban, V.; Etienne-Selloum, N.; Jung, A.C.; Masson, M.; Macabre, C.; Ledrappier, S.; et al. Cav1/EREG/YAP Axis in the Treatment Resistance of Cav1-Expressing Head and Neck Squamous Cell Carcinoma. Cancers 2021, 13, 3038. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Weskamp, G.; Kelly, K.; Zhou, H.M.; Higashiyama, S.; Peschon, J.; Hartmann, D.; Saftig, P.; Blobel, C.P. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J. Cell Biol. 2004, 164, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Toyoda, H.; Komurasaki, T.; Uchida, D.; Takayama, Y.; Isobe, T.; Okuyama, T.; Hanada, K. Epiregulin. A novel epidermal growth factor with mitogenic activity for rat primary hepatocytes. J. Biol. Chem. 1995, 270, 7495–7500. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wang, Y.; Han, Y.; Xia, W.; Zhang, L.; Xu, S.; Ju, H.; Zhang, X.; Ren, G.; Liu, L.; et al. EREG-driven oncogenesis of Head and Neck Squamous Cell Carcinoma exhibits higher sensitivity to Erlotinib therapy. Theranostics 2020, 10, 10589–10605. [Google Scholar] [CrossRef]
- Komurasaki, T.; Toyoda, H.; Uchida, D.; Morimoto, S. Epiregulin binds to epidermal growth factor receptor and ErbB-4 and induces tyrosine phosphorylation of epidermal growth factor receptor, ErbB-2, ErbB-3 and ErbB-4. Oncogene 1997, 15, 2841–2848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roepstorff, K.; Grandal, M.V.; Henriksen, L.; Knudsen, S.L.; Lerdrup, M.; Grovdal, L.; Willumsen, B.M.; van Deurs, B. Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 2009, 10, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Freed, D.M.; Bessman, N.J.; Kiyatkin, A.; Salazar-Cavazos, E.; Byrne, P.O.; Moore, J.O.; Valley, C.C.; Ferguson, K.M.; Leahy, D.J.; Lidke, D.S.; et al. EGFR Ligands Differentially Stabilize Receptor Dimers to Specify Signaling Kinetics. Cell 2017, 171, 683–695.e18. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Jing, Y.; Ding, L.; Zhang, X.; Song, Y.; Chen, S.; Zhao, X.; Huang, X.; Pu, Y.; Wang, Z.; et al. Epiregulin reprograms cancer-associated fibroblasts and facilitates oral squamous cell carcinoma invasion via JAK2-STAT3 pathway. J. Exp. Clin. Cancer Res. 2019, 38, 274. [Google Scholar] [CrossRef] [Green Version]
- Sunaga, N.; Kaira, K. Epiregulin as a therapeutic target in non-small-cell lung cancer. Lung Cancer 2015, 6, 91–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.L.; Feng, P.H.; Lee, K.Y.; Chen, K.Y.; Sun, W.L.; Van Hiep, N.; Luo, C.S.; Wu, S.M. The Role of EREG/EGFR Pathway in Tumor Progression. Int. J. Mol. Sci. 2021, 22, 12828. [Google Scholar] [CrossRef]
- Shigeishi, H.; Higashikawa, K.; Hiraoka, M.; Fujimoto, S.; Mitani, Y.; Ohta, K.; Takechi, M.; Kamata, N. Expression of epiregulin, a novel epidermal growth factor ligand associated with prognosis in human oral squamous cell carcinomas. Oncol. Rep. 2008, 19, 1557–1564. [Google Scholar]
- Kogashiwa, Y.; Inoue, H.; Kuba, K.; Araki, R.; Yasuda, M.; Nakahira, M.; Sugasawa, M. Prognostic role of epiregulin/amphiregulin expression in recurrent/metastatic head and neck cancer treated with cetuximab. Head Neck 2018, 40, 2424–2431. [Google Scholar] [CrossRef]
- Hu, K.; Li, S.L.; Gan, Y.H.; Wang, C.Y.; Yu, G.Y. Epiregulin promotes migration and invasion of salivary adenoid cystic carcinoma cell line SACC-83 through activation of ERK and Akt. Oral Oncol. 2009, 45, 156–163. [Google Scholar] [CrossRef]
- Job, S.; Reynies, A.; Heller, B.; Weiss, A.; Guerin, E.; Macabre, C.; Ledrappier, S.; Bour, C.; Wasylyk, C.; Etienne-Selloum, N.; et al. Preferential Response of Basal-Like Head and Neck Squamous Cell Carcinoma Cell Lines to EGFR-Targeted Therapy Depending on EREG-Driven Oncogenic Addiction. Cancers 2019, 11, 795. [Google Scholar] [CrossRef] [Green Version]
- Driehuis, E.; Kretzschmar, K.; Clevers, H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat. Protoc. 2020, 15, 3380–3409. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino Acid transporters in cancer and their relevance to “glutamine addiction”: Novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.Y.; Xu, W.W.; Huan, X.K.; Wu, G.N.; Li, G.; Zhou, Y.H.; Najafi, M. Mechanisms of cancer cell killing by metformin: A review on different cell death pathways. Mol. Cell. Biochem. 2022, 478, 197–214. [Google Scholar] [CrossRef]
- Yang, J.; Zhou, Y.; Xie, S.; Wang, J.; Li, Z.; Chen, L.; Mao, M.; Chen, C.; Huang, A.; Chen, Y.; et al. Metformin induces Ferroptosis by inhibiting UFMylation of SLC7A11 in breast cancer. J. Exp. Clin. Cancer Res. 2021, 40, 206. [Google Scholar] [CrossRef] [PubMed]
- Oliveras-Ferraros, C.; Cufi, S.; Queralt, B.; Vazquez-Martin, A.; Martin-Castillo, B.; de Llorens, R.; Bosch-Barrera, J.; Brunet, J.; Menendez, J.A. Cross-suppression of EGFR ligands amphiregulin and epiregulin and de-repression of FGFR3 signalling contribute to cetuximab resistance in wild-type KRAS tumour cells. Br. J. Cancer 2012, 106, 1406–1414. [Google Scholar] [CrossRef] [Green Version]
- Sloan-Lancaster, J.; Raddad, E.; Deeg, M.A.; Eli, M.; Flynt, A.; Tumlin, J. Evaluation of the Safety, Pharmacokinetics, Pharmacodynamics, and Efficacy after Single and Multiple Dosings of LY3016859 in Healthy Subjects and Patients with Diabetic Nephropathy. Clin. Pharmacol. Drug Dev. 2018, 7, 759–772. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, L.; Ren, Y.; Dai, W.; Chen, T.; Luo, L.; Zeng, J.; Mi, K.; Lang, J.; Cao, B. Epiregulin confers EGFR-TKI resistance via EGFR/ErbB2 heterodimer in non-small cell lung cancer. Oncogene 2021, 40, 2596–2609. [Google Scholar] [CrossRef]
- Poliakova, M.; Aebersold, D.M.; Zimmer, Y.; Medova, M. The relevance of tyrosine kinase inhibitors for global metabolic pathways in cancer. Mol. Cancer 2018, 17, 27. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Jin, Q.; Chen, C.; Liu, Y.; Ye, X.; Jiang, Y.; Ji, F.; Qian, H.; Gan, D.; Yue, S.; et al. The miR-186-3p/EREG axis orchestrates tamoxifen resistance and aerobic glycolysis in breast cancer cells. Oncogene 2019, 38, 5551–5565. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, R.; Shuai, Y.; Huang, Y.; Jin, R.; Wang, X.; Luo, J. ASCT2 (SLC1A5)-dependent glutamine uptake is involved in the progression of head and neck squamous cell carcinoma. Br. J. Cancer 2020, 122, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Li, X.; Lu, Y.; Qiu, S.; Fan, Z. ASCT2 (SLC1A5) is an EGFR-associated protein that can be co-targeted by cetuximab to sensitize cancer cells to ROS-induced apoptosis. Cancer Lett. 2016, 381, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, X.; Lu, Y.; Qiu, S.; Wang, Y.; Qin, J.; Fan, Z. AP1G1 is involved in cetuximab-mediated downregulation of ASCT2-EGFR complex and sensitization of human head and neck squamous cell carcinoma cells to ROS-induced apoptosis. Cancer Lett. 2017, 408, 33–42. [Google Scholar] [CrossRef]
- Yang, J.; Mo, J.; Dai, J.; Ye, C.; Cen, W.; Zheng, X.; Jiang, L.; Ye, L. Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer. Cell Death Dis. 2021, 12, 1079. [Google Scholar] [CrossRef]
- Zhou, R.P.; Chen, Y.; Wei, X.; Yu, B.; Xiong, Z.G.; Lu, C.; Hu, W. Novel insights into ferroptosis: Implications for age-related diseases. Theranostics 2020, 10, 11976–11997. [Google Scholar] [CrossRef]
- Roh, J.L.; Kim, E.H.; Jang, H.J.; Park, J.Y.; Shin, D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 2016, 381, 96–103. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, C.; Yang, Z.; Bai, Y.; Shukuya, T.; Poh, M.E.; Ekman, S.; Li, J.; Xu, Y.; Deng, S. Identification of GPX4 as a therapeutic target for lung adenocarcinoma after EGFR-TKI resistance. Transl. Lung Cancer Res. 2022, 11, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Vilaseca, I.; Fuster, G.; Aviles-Jurado, F.X. The impact of diabetes in head and neck cancer. Curr. Opin. Otolaryngol. Head Neck Surg. 2020, 28, 107–111. [Google Scholar] [CrossRef]
- Figueiredo, R.A.; Weiderpass, E.; Tajara, E.H.; Strom, P.; Carvalho, A.L.; de Carvalho, M.B.; Kanda, J.L.; Moyses, R.A.; Wunsch-Filho, V. Diabetes mellitus, metformin and head and neck cancer. Oral Oncol. 2016, 61, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Ogunsakin, A.; Infield, J.; Zuber, J.; Solomon, S.S. Metformin Associated with Improved Outcomes in Diabetic Patients with Laryngeal and Oropharyngeal Carcinoma. Am. J. Med. Sci. 2018, 356, 574–575. [Google Scholar] [CrossRef] [PubMed]
- Tsou, Y.A.; Chang, W.C.; Lin, C.D.; Chang, R.L.; Tsai, M.H.; Shih, L.C.; Staniczek, T.; Wu, T.F.; Hsu, H.Y.; Chang, W.D.; et al. Metformin Increases Survival in Hypopharyngeal Cancer Patients with Diabetes Mellitus: Retrospective Cohort Study and Cell-Based Analysis. Pharmaceuticals 2021, 14, 191. [Google Scholar] [CrossRef] [PubMed]
- Pernicova, I.; Korbonits, M. Metformin-mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Qin, C.; Zhou, Y.; Chen, Y.; Mao, M.; Yang, J. Metformin may induce ferroptosis by inhibiting autophagy via lncRNA H19 in breast cancer. FEBS Open Bio 2022, 12, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016, 18, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Wensink, G.E.; Elias, S.G.; Mullenders, J.; Koopman, M.; Boj, S.F.; Kranenburg, O.W.; Roodhart, J.M. Patient-derived organoids as a predictive biomarker for treatment response in cancer patients. NPJ Precis. Oncol. 2021, 5, 30. [Google Scholar] [CrossRef]
- Driehuis, E.; Kolders, S.; Spelier, S.; Lõhmussaar, K.; Willems, S.M.; Devriese, L.A.; de Bree, R.; de Ruiter, E.J.; Korving, J.; Begthel, H.; et al. Oral Mucosal Organoids as a Potential Platform for Personalized Cancer Therapy. Cancer Discov. 2019, 9, 852–871. [Google Scholar] [CrossRef]
- Tanaka, N.; Osman, A.A.; Takahashi, Y.; Lindemann, A.; Patel, A.A.; Zhao, M.; Takahashi, H.; Myers, J.N. Head and neck cancer organoids established by modification of the CTOS method can be used to predict in vivo drug sensitivity. Oral Oncol. 2018, 87, 49–57. [Google Scholar] [CrossRef]
- Wang, W.M.; Yang, S.S.; Shao, S.H.; Nie, H.Q.; Zhang, J.; Su, T. Metformin Downregulates the Expression of Epidermal Growth Factor Receptor Independent of Lowering Blood Glucose in Oral Squamous Cell Carcinoma. Front. Endocrinol. 2022, 13, 828608. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jehl, A.; Conrad, O.; Burgy, M.; Foppolo, S.; Vauchelles, R.; Ronzani, C.; Etienne-Selloum, N.; Chenard, M.-P.; Danic, A.; Dourlhes, T.; et al. Blocking EREG/GPX4 Sensitizes Head and Neck Cancer to Cetuximab through Ferroptosis Induction. Cells 2023, 12, 733. https://doi.org/10.3390/cells12050733
Jehl A, Conrad O, Burgy M, Foppolo S, Vauchelles R, Ronzani C, Etienne-Selloum N, Chenard M-P, Danic A, Dourlhes T, et al. Blocking EREG/GPX4 Sensitizes Head and Neck Cancer to Cetuximab through Ferroptosis Induction. Cells. 2023; 12(5):733. https://doi.org/10.3390/cells12050733
Chicago/Turabian StyleJehl, Aude, Ombline Conrad, Mickaël Burgy, Sophie Foppolo, Romain Vauchelles, Carole Ronzani, Nelly Etienne-Selloum, Marie-Pierre Chenard, Aurélien Danic, Thomas Dourlhes, and et al. 2023. "Blocking EREG/GPX4 Sensitizes Head and Neck Cancer to Cetuximab through Ferroptosis Induction" Cells 12, no. 5: 733. https://doi.org/10.3390/cells12050733