
PGRMC1 Ablation Protects from Energy-Starved Heart Failure by Promoting Fatty Acid/Pyruvate Oxidation
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Gene Expression Omnibus (GEO) Datasets
2.3. Comprehensive Laboratory Animal Monitoring System (CLAMS)
2.4. RNA Isolation, Reverse Transcription, and Quantitative Reverse Transcription–Polymerase Chain Reaction (qRT-PCR)
2.5. Western Blotting
2.6. Blood and Plasma Measurements
2.7. Cell Culture
2.8. Cardiac Fibrosis Measurement
2.9. Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End-Labeling (TUNEL) Staining and Immunostaining
2.10. Statistical Analysis
3. Results
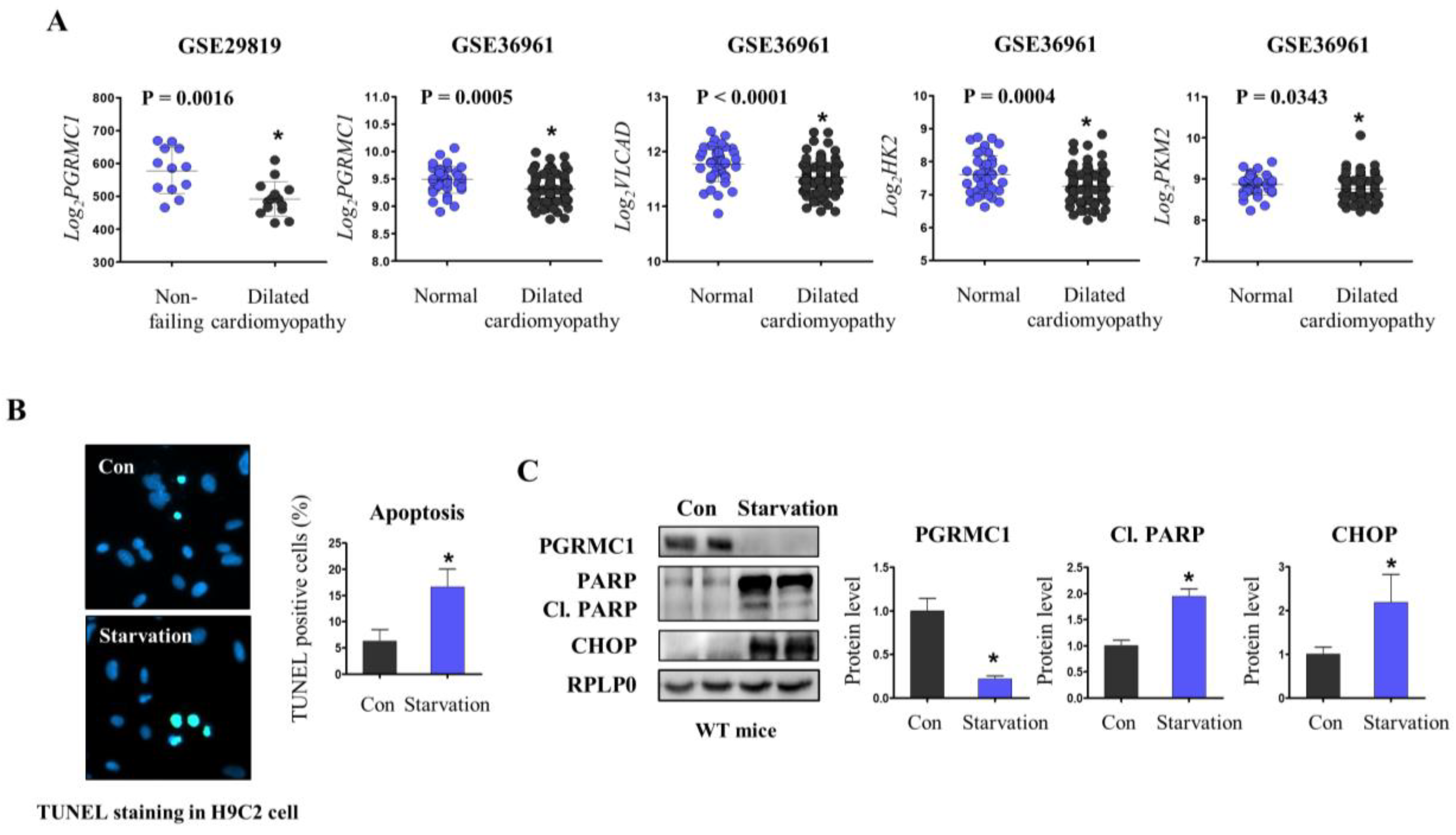
3.1. PGRMC1 Expression Is Associated with Energy-Starved Cardiomyopathy
3.2. Loss of PGRMC1 Maintains the Whole-Body Metabolism during Starvation
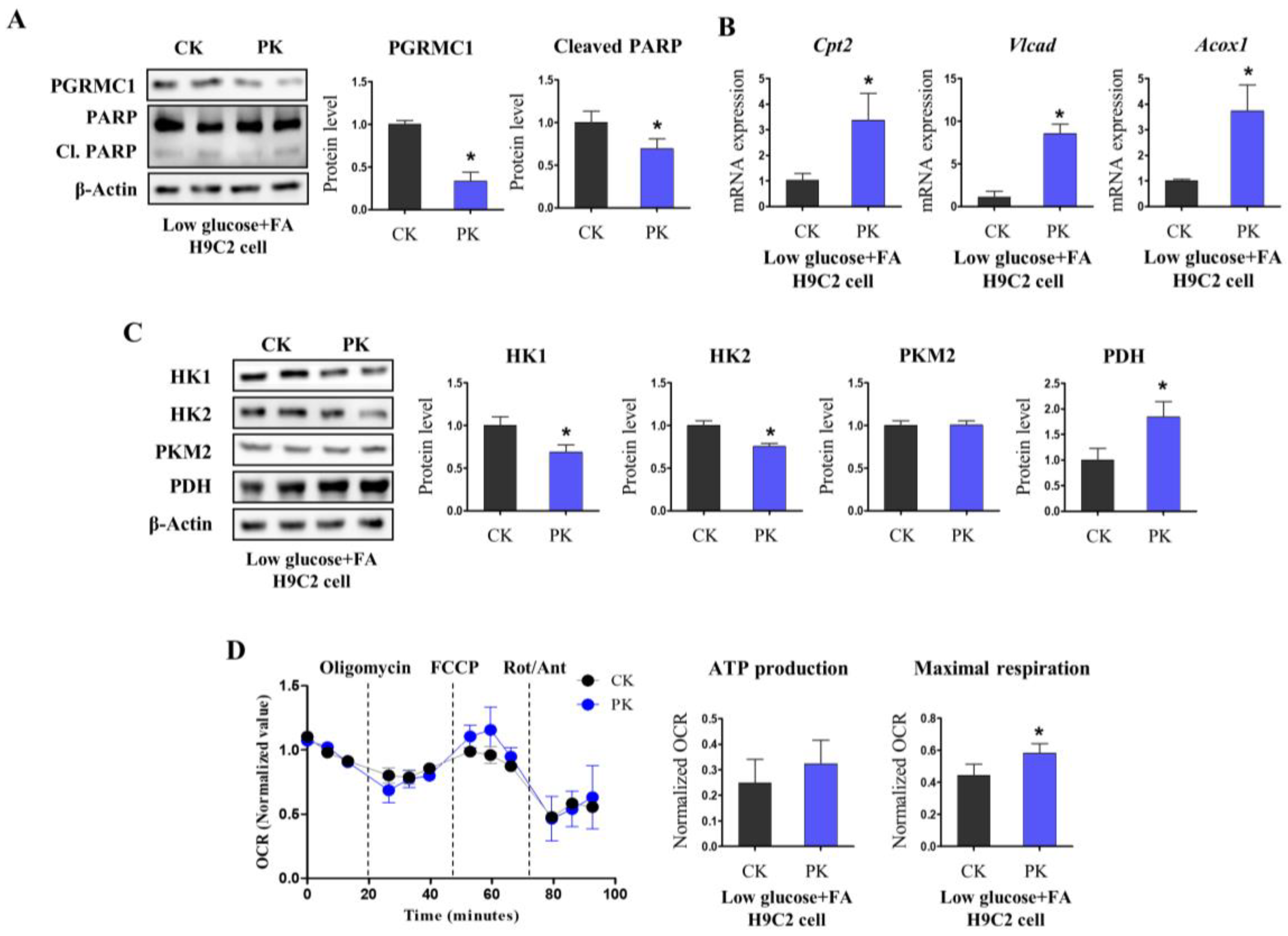
3.3. Pgrmc1 Loss Increases Fatty Acid/Pyruvate Oxidation and Decreases Starvation-Induced Cardiac Injury
3.4. AMPK Activation Is Associated with Pgrmc1-Induced Metabolic Alteration in the Heart
3.5. Pgrmc1 Ablation Protects the Heart from Isoproterenol-Induced Damage
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Groenewegen, A.; Rutten, F.H.; Mosterd, A.; Hoes, A.W. Epidemiology of Heart Failure. Eur. J. Heart Fail. 2020, 22, 1342–1356. [Google Scholar] [CrossRef]
- Roger, V.L. Epidemiology of Heart Failure: A Contemporary Perspective. Circ. Res. 2021, 128, 1421–1434. [Google Scholar] [CrossRef]
- Arikawa, R.; Kanda, D.; Ikeda, Y.; Tokushige, A.; Sonoda, T.; Anzaki, K.; Ohishi, M. Prognostic impact of malnutrition on cardiovascular events in coronary artery disease patients with myocardial damage. BMC Cardiovasc. Disord. 2021, 21, 479. [Google Scholar] [CrossRef]
- Webb, J.G.; Kiess, M.C.; Chan-Yan, C.C. Malnutrition and the heart. Can. Med. Assoc. J. 1986, 135, 753–758. [Google Scholar]
- Shah, A.; Gandhi, D.; Srivastava, S.; Shah, K.J.; Mansukhani, R. Heart Failure: A Class Review of Pharmacotherapy. P T Peer-Rev. J. Formul. Manag. 2017, 42, 464–472. [Google Scholar]
- Grynberg, A.; Demaison, L. Fatty Acid Oxidation in the Heart. J. Cardiovasc. Pharmacol. 1996, 28 (Suppl. S1), S11–S17. [Google Scholar]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Jaswal, J.S.; Keung, W.; Wang, W.; Ussher, J.R.; Lopaschuk, G.D. Targeting fatty acid and carbohydrate oxidation—A novel therapeutic intervention in the ischemic and failing heart. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2011, 1813, 1333–1350. [Google Scholar] [CrossRef] [Green Version]
- Heggermont, W.A.; Papageorgiou, A.P.; Heymans, S.; van Bilsen, M. Metabolic Support for the Heart: Complementary Therapy for Heart Failure? Eur. J. Heart Fail. 2016, 18, 1420–1429. [Google Scholar] [CrossRef] [Green Version]
- Lam, C.S.P.; Arnott, C.; Beale, A.L.; Chandramouli, C.; Hilfiker-Kleiner, D.; Kaye, D.M.; Ky, B.; Santema, B.T.; Sliwa, K.; Voors, A.A. Sex differences in heart failure. Eur. Heart J. 2019, 40, 3859–3868c. [Google Scholar] [CrossRef]
- Güder, G.; Allolio, B.; Angermann, C.E.; Störk, S. Androgen Deficiency in Heart Failure. Curr. Heart Fail. Rep. 2011, 8, 131–139. [Google Scholar] [CrossRef]
- Iorga, A.; Umar, S.; Ruffenach, G.; Aryan, L.; Li, J.; Sharma, S.; Motayagheni, N.; Nadadur, R.D.; Bopassa, J.C.; Eghbali, M. Estrogen rescues heart failure through estrogen receptor Beta activation. Biol. Sex Differ. 2018, 9, 48. [Google Scholar] [CrossRef]
- Hermsmeyer, R.K.; Thompson, T.L.; Pohost, G.M.; Kaski, J.C. Cardiovascular Effects of Medroxyprogesterone Acetate and Progesterone: A Case of Mistaken Identity? Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 387–395. [Google Scholar] [CrossRef]
- Lee, S.R.; Heo, J.H.; Jo, S.L.; Kim, G.; Kim, S.J.; Yoo, H.J.; Lee, K.-P.; Kwun, H.-J.; Shin, H.-J.; Baek, I.-J.; et al. Progesterone receptor membrane component 1 reduces cardiac steatosis and lipotoxicity via activation of fatty acid oxidation and mitochondrial respiration. Sci. Rep. 2021, 11, 8781. [Google Scholar] [CrossRef]
- Russo, N.; Kaplan-Kahn, E.A.; Wilson, J.; Criss, A.; Burack, J.A. Choices, challenges, and constraints: A pragmatic examination of the limits of mental age matching in empirical research. Dev. Psychopathol. 2021, 33, 727–738. [Google Scholar] [CrossRef]
- McCallum, M.L.; Pru, C.A.; Niikura, Y.; Yee, S.-P.; Lydon, J.P.; Peluso, J.J.; Pru, J.K. Conditional Ablation of Progesterone Receptor Membrane Component 1 Results in Subfertility in the Female and Development of Endometrial Cysts. Endocrinology 2016, 157, 3309–3319. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.R.; Choi, W.-Y.; Heo, J.H.; Huh, J.; Kim, G.; Lee, K.-P.; Kwun, H.-J.; Shin, H.-J.; Baek, I.-J.; Hong, E.-J. Progesterone increases blood glucose via hepatic progesterone receptor membrane component 1 under limited or impaired action of insulin. Sci. Rep. 2020, 10, 16316. [Google Scholar] [CrossRef]
- Furuhata, R.; Kabe, Y.; Kanai, A.; Sugiura, Y.; Tsugawa, H.; Sugiyama, E.; Hirai, M.; Yamamoto, T.; Koike, I.; Yoshikawa, N.; et al. Progesterone receptor membrane associated component 1 enhances obesity progression in mice by facilitating lipid accumulation in adipocytes. Commun. Biol. 2020, 3, 479. [Google Scholar] [CrossRef]
- Zhang, M.; Robitaille, M.; Showalter, A.D.; Huang, X.; Liu, Y.; Bhattacharjee, A.; Willard, F.S.; Han, J.; Froese, S.; Wei, L.; et al. Progesterone Receptor Membrane Component 1 Is a Functional Part of the Glucagon-Like Peptide-1 (Glp-1) Receptor Complex in Pancreatic Beta Cells. Mol. Cell Proteomics 2014, 13, 3049–3062. [Google Scholar] [CrossRef] [Green Version]
- Wanka, H.; Lutze, P.; Albers, A.; Golchert, J.; Staar, D.; Peters, J. Overexpression of Transcripts Coding for Renin-b but Not for Renin-a Reduce Oxidative Stress and Increase Cardiomyoblast Survival under Starvation Conditions. Cells 2021, 10, 1204. [Google Scholar] [CrossRef]
- Xu, X.; Pacheco, B.D.; Leng, L.; Bucala, R.; Ren, J. Macrophage migration inhibitory factor plays a permissive role in the maintenance of cardiac contractile function under starvation through regulation of autophagy. Cardiovasc. Res. 2013, 99, 412–421. [Google Scholar] [CrossRef] [Green Version]
- El-Demerdash, E.; Awad, A.S.; Taha, R.M.; El-Hady, A.M.; Sayed-Ahmed, M.M. Probucol attenuates oxidative stress and energy decline in isoproterenol-induced heart failure in rat. Pharmacol. Res. 2005, 51, 311–318. [Google Scholar] [CrossRef]
- Lee, S.R.; Kwon, S.W.; Kaya, P.; Lee, Y.H.; Lee, J.G.; Kim, G.; Lee, G.-S.; Baek, I.-J.; Hong, E.-J. Loss of progesterone receptor membrane component 1 promotes hepatic steatosis via the induced de novo lipogenesis. Sci. Rep. 2018, 8, 15711. [Google Scholar] [CrossRef] [Green Version]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac Metabolism in Heart Failure: Implications Beyond Atp Production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Lionetti, V.; Stanley, W.C.; Recchia, F.A. Modulating fatty acid oxidation in heart failure. Cardiovasc. Res. 2011, 90, 202–209. [Google Scholar] [CrossRef]
- Fillmore, N.; Levasseur, J.L.; Fukushima, A.; Wagg, C.S.; Wang, W.; Dyck, J.R.B.; Lopaschuk, G.D. Uncoupling of glycolysis from glucose oxidation accompanies the development of heart failure with preserved ejection fraction. Mol. Med. 2018, 24, 3. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Xu, J.; Qin, X.; Hou, Z.; Guo, Y.; Liu, Z.; Wu, J.; Zheng, H.; Zhang, X.; Gao, F. Glucose oxidation positively regulates glucose uptake and improves cardiac function recovery after myocardial reperfusion. Am. J. Physiol. Metab. 2017, 313, E577–E585. [Google Scholar] [CrossRef] [Green Version]
- Tran, D.H.; Wang, Z.V.; Heather, L.C.; Pates, K.M.; Atherton, H.J.; Cole, M.A.; Ball, D.R.; Evans, R.D.; Glatz, J.F.; Luiken, J.J.; et al. Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J. Am. Heart Assoc. 2019, 8, e012673. [Google Scholar] [CrossRef] [Green Version]
- Dorn, G.W. Mitochondrial Fission/Fusion and Cardiomyopathy. Curr. Opin. Genet. Dev. 2016, 38, 38–44. [Google Scholar]
- Zaha, V.G.; Young, L.H. AMP-Activated Protein Kinase Regulation and Biological Actions in the Heart. Circ. Res. 2012, 111, 800–814. [Google Scholar] [CrossRef] [Green Version]
- Brooks, W.W.; Conrad, C.H. Isoproterenol-induced myocardial injury and diastolic dysfunction in mice: Structural and functional correlates. Comp. Med. 2009, 59, 339–343. [Google Scholar] [PubMed]
- Grimm, D.; Elsner, D.; Schunkert, H.; Pfeifer, M.; Griese, D.; Bruckschlegel, G.; Muders, F.; Riegger, G.A.; Kromer, E.P. Development of Heart Failure Following Isoproterenol Administration in the Rat: Role of the Renin-Angiotensin System. Cardiovasc. Res. 1998, 37, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Nordlie, M.A.; Wold, L.E.; Kloner, R.A. Genetic contributors toward increased risk for ischemic heart disease. J. Mol. Cell. Cardiol. 2005, 39, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.; Hwang, K.-C.; Lee, K.-Y.; Kim, Y.-H. Ischemic heart diseases: Current treatments and future. J. Control Release 2009, 140, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Fillmore, N.; Mori, J.; Lopaschuk, G.D. Mitochondrial Fatty Acid Oxidation Alterations in Heart Failure, Ischaemic Heart Disease and Diabetic Cardiomyopathy. Br. J. Pharmacol. 2014, 171, 2080–2090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polak-Iwaniuk, A.; Harasim-Symbor, E.; Gołaszewska, K.; Chabowski, A. How Hypertension Affects Heart Metabolism. Front. Physiol. 2019, 10, 435. [Google Scholar] [CrossRef] [Green Version]
- Tuunanen, H.; Engblom, E.; Naum, A.; Någren, K.; Hesse, B.; Airaksinen, J.; Nuutila, P.; Iozzo, P.; Ukkonen, H.; Opie, L.H.; et al. Free Fatty Acid Depletion Acutely Decreases Cardiac Work and Efficiency in Cardiomyopathic Heart Failure. Circulation 2006, 114, 2130–2137. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Pan, D.A. Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem. Soc. Trans. 2002, 30, 1064–1070. [Google Scholar] [CrossRef]
- Dyck, J.R.; Lopaschuk, G.D. Ampk Alterations in Cardiac Physiology and Pathology: Enemy or Ally? J. Physiol. 2006, 574 Pt 1, 95–112. [Google Scholar] [CrossRef]
- Arad, M.; Seidman, C.E.; Seidman, J.G. Amp-Activated Protein Kinase in the Heart: Role during Health and Disease. Circ. Res. 2007, 100, 474–488. [Google Scholar] [CrossRef] [Green Version]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial Substrate Metabolism in the Normal and Failing Heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knottnerus, S.J.G.; Bleeker, J.C.; Wüst, R.C.I.; Ferdinandusse, S.; Ijlst, L.; Wijburg, F.A.; Wanders, R.J.A.; Visser, G.; Houtkooper, R.H. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev. Endocr. Metab. Disord. 2018, 19, 93–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Deng, S.; Wang, Y.; Li, P.; Tang, L.; Pang, Y. Specific Inhibition of Acyl-Coa Oxidase-1 by an Acetylenic Acid Improves Hepatic Lipid and Reactive Oxygen Species (Ros) Metabolism in Rats Fed a High Fat Diet. J. Biol. Chem. 2017, 292, 3800–3809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depre, C.; Rider, M.H.; Hue, L. Mechanisms of control of heart glycolysis. JBIC J. Biol. Inorg. Chem. 1998, 258, 277–290. [Google Scholar] [CrossRef]
- Allard, M.F.; Schonekess, B.O.; Henning, S.L.; English, D.R.; Lopaschuk, G.D. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol. Circ. Physiol. 1994, 267, H742–H750. [Google Scholar] [CrossRef]
- Fukushima, A.; Lopaschuk, G.D. Cardiac fatty acid oxidation in heart failure associated with obesity and diabetes. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2016, 1861, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Y. Ampk and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 85–108. [Google Scholar]
- Zhu, X.; Ji, M.; Han, Y.; Guo, Y.; Zhu, W.; Gao, F.; Yang, X.; Zhang, C. PGRMC1-dependent autophagy by hyperoside induces apoptosis and sensitizes ovarian cancer cells to cisplatin treatment. Int. J. Oncol. 2017, 50, 835–846. [Google Scholar] [CrossRef] [Green Version]
- Gustafsson, A.B.; Gottlieb, R.A. Autophagy in Ischemic Heart Disease. Circ. Res. 2009, 104, 150–158. [Google Scholar] [CrossRef] [Green Version]
- Saddik, M.; Lopaschuk, G.D. Myocardial triglyceride turnover and contribution to energy substrate utilization in isolated working rat hearts. J. Biol. Chem. 1991, 266, 8162–8170. [Google Scholar] [CrossRef]
- Borutaite, V.; Mildaziene, V.; Ivanoviene, L.; Kholodenko, B.; Toleikis, A.; Praskevicius, A. The Role of Long-Chain Acyl-Coa in the Damage of Oxidative Phosphorylation in Heart Mitochondria. FEBS Lett. 1989, 243, 264–266. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.X.; Hanley, P.J.; Ray, J.; Daut, J. Long-Chain Acyl-Coenzyme a Esters and Fatty Acids Directly Link Metabolism to K(Atp) Channels in the Heart. Circ. Res. 2001, 88, 918–924. [Google Scholar] [CrossRef] [Green Version]
- Ingwall, J.S.; Weiss, R.G. Is the Failing Heart Energy Starved? On Using Chemical Energy to Support Cardiac Function. Circ. Res. 2004, 95, 135–145. [Google Scholar] [CrossRef]
- Weiss, R.G.; Gerstenblith, G.; Bottomley, P.A. Atp Flux through Creatine Kinase in the Normal, Stressed, and Failing Human Heart. Proc. Natl. Acad. Sci. USA 2005, 102, 808–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hue, L.; Taegtmeyer, H. The Randle cycle revisited: A new head for an old hat. Am. J. Physiol. Metab. 2009, 297, E578–E591. [Google Scholar] [CrossRef] [Green Version]
- Bersin, R.M.; Wolfe, C.; Kwasman, M.; Lau, D.; Klinski, C.; Tanaka, K.; Khorrami, P.; Henderson, G.N.; de Marco, T.; Chatterjee, K. Improved hemodynamic function and mechanical efficiency in congestive heart failure with sodium dichloroacetate. J. Am. Coll. Cardiol. 1994, 23, 1617–1624. [Google Scholar] [CrossRef]
- Schmidt-Schweda, S.; Holubarsch, C. First Clinical Trial with Etomoxir in Patients with Chronic Congestive Heart Failure. Clin. Sci. 2000, 99, 27–35. [Google Scholar] [CrossRef]
- Schmitz, F.; Rösen, P.; Reinauer, H. Improvement of Myocardial Function and Metabolism in Diabetic Rats by the Carnitine Palmitoyl Transferase Inhibitor Etomoxir®. Horm. Metab. Res. 1995, 27, 515–522. [Google Scholar] [CrossRef]
- Holubarsch, C.J.F.; Rohrbach, M.; Karrasch, M.; Boehm, E.; Polonski, L.; Ponikowski, P.; Rhein, S. A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: The ERGO (etomoxir for the recovery of glucose oxidation) study. Clin. Sci. 2007, 113, 205–212. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, R.S.; Guo, L.; Ghassemi, S.; Snyder, N.W.; Worth, A.J.; Weng, L.; Kam, Y.; Philipson, B.; Trefely, S.; Nunez-Cruz, S.; et al. The CPT1a inhibitor, etomoxir induces severe oxidative stress at commonly used concentrations. Sci. Rep. 2018, 8, 6289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, T.; Takahashi, M.; Yamasaki, G.; Sugimoto, M.; Kuse, A.; Morichika, M.; Nakagawa, K.; Sakurada, M.; Asano, M.; Ueno, Y. Immunohistochemical analysis of CD31 expression in myocardial tissues from autopsies of patients with ischemic heart disease. Leg. Med. 2022, 59, 102127. [Google Scholar] [CrossRef] [PubMed]
- Woodfin, A.; Voisin, M.-B.; Nourshargh, S. PECAM-1: A Multi-Functional Molecule in Inflammation and Vascular Biology. Arter. Thromb. Vasc. Biol. 2007, 27, 2514–2523. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Ruan, X.; Ju, R.; Wang, Z.; Yang, Y.; Cheng, J.; Gu, M.; Mueck, A.O. Progesterone Receptor Membrane Component-1 May Promote Survival of Human Brain Microvascular Endothelial Cells in Alzheimer’s Disease. Am. J. Alzheimer’s Dis. Other Dement. 2022, 37, 15333175221109749. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Upper Primer (5′-3′) | Lower Primer (5′-3′) | Species |
|---|---|---|---|
| Cpt2 | CAG CAC AGC ATC GTA CCC A | TCC CAA TGC CGT TCT CAA AAT | Mouse |
| Vlcad | TAT CTC TGC CCA GCG ACT TT | TGG GTA TGG GAA CAC CTG AT | Mouse |
| Acox1 | TTG GAA ACC ACT GCC ACA TA | AGG CAT GTA ACC CGT AGC AC | Mouse |
| Tgfβ | GAC GTC ACT GGA GTT GTA CG | GGT TCA TGT CAT GGA TGG TG | Mouse |
| Anp | CCA TAT TGG AGC AAA TCC TGT G | CGG CAT CTT CTC CTC CAG GT | Mouse |
| Bnp | GGG AGA ACA CGG CAT CAT TG | ACA GCA CCT TCA GGA GAT CCA | Mouse |
| Mfn2 | GCC AGC TTC CTT GAA GAC AC | GCA GAA CTT TGT CCC AGA GC | Mouse |
| Drp1 | AGA AAA CTG TCT GCC CGA GA | GCT GCC CTA CCA GTT CAC TC | Mouse |
| Cpt2 | ACT AAG AGA TGC TCC GAG GC | GCA GAG CAT ACA AGT GTC GG | Rat |
| Vlcad | TGA CCC TGC CAA GAA TGA CT | GTC ATG CAT GCC CAC AAT CT | Rat |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.R.; Mukae, M.; Jeong, K.J.; Park, S.H.; Shin, H.J.; Kim, S.W.; Won, Y.S.; Kwun, H.-J.; Baek, I.-J.; Hong, E.-J. PGRMC1 Ablation Protects from Energy-Starved Heart Failure by Promoting Fatty Acid/Pyruvate Oxidation. Cells 2023, 12, 752. https://doi.org/10.3390/cells12050752
Lee SR, Mukae M, Jeong KJ, Park SH, Shin HJ, Kim SW, Won YS, Kwun H-J, Baek I-J, Hong E-J. PGRMC1 Ablation Protects from Energy-Starved Heart Failure by Promoting Fatty Acid/Pyruvate Oxidation. Cells. 2023; 12(5):752. https://doi.org/10.3390/cells12050752
Chicago/Turabian StyleLee, Sang R., Moeka Mukae, Kang Joo Jeong, Se Hee Park, Hi Jo Shin, Sang Woon Kim, Young Suk Won, Hyo-Jung Kwun, In-Jeoung Baek, and Eui-Ju Hong. 2023. "PGRMC1 Ablation Protects from Energy-Starved Heart Failure by Promoting Fatty Acid/Pyruvate Oxidation" Cells 12, no. 5: 752. https://doi.org/10.3390/cells12050752