
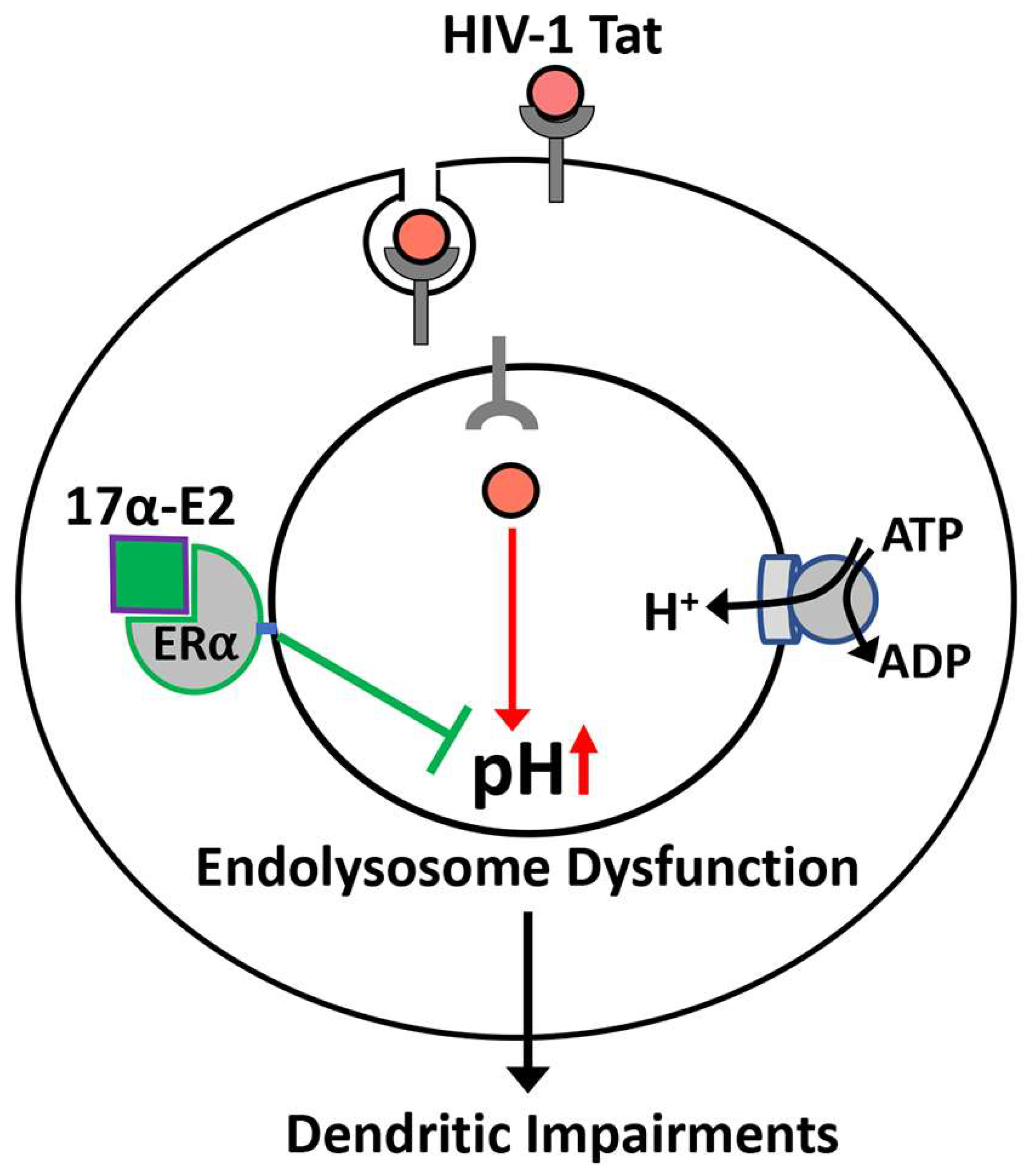
17⍺-Estradiol Protects against HIV-1 Tat-Induced Endolysosome Dysfunction and Dendritic Impairments in Neurons



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
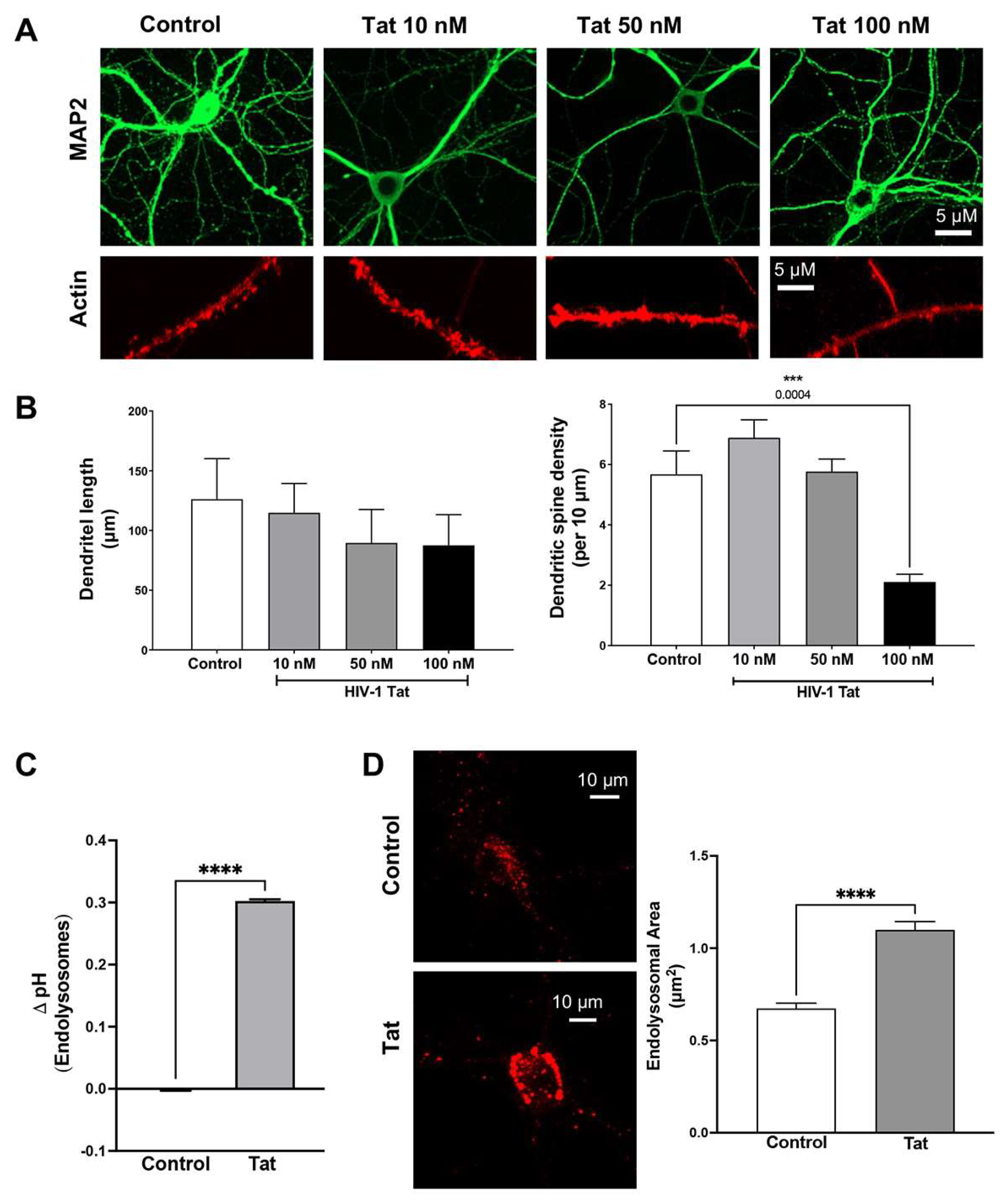
3.1. Tat Induces Dendritic Spine Impairment and Endolysosome Dysfunction
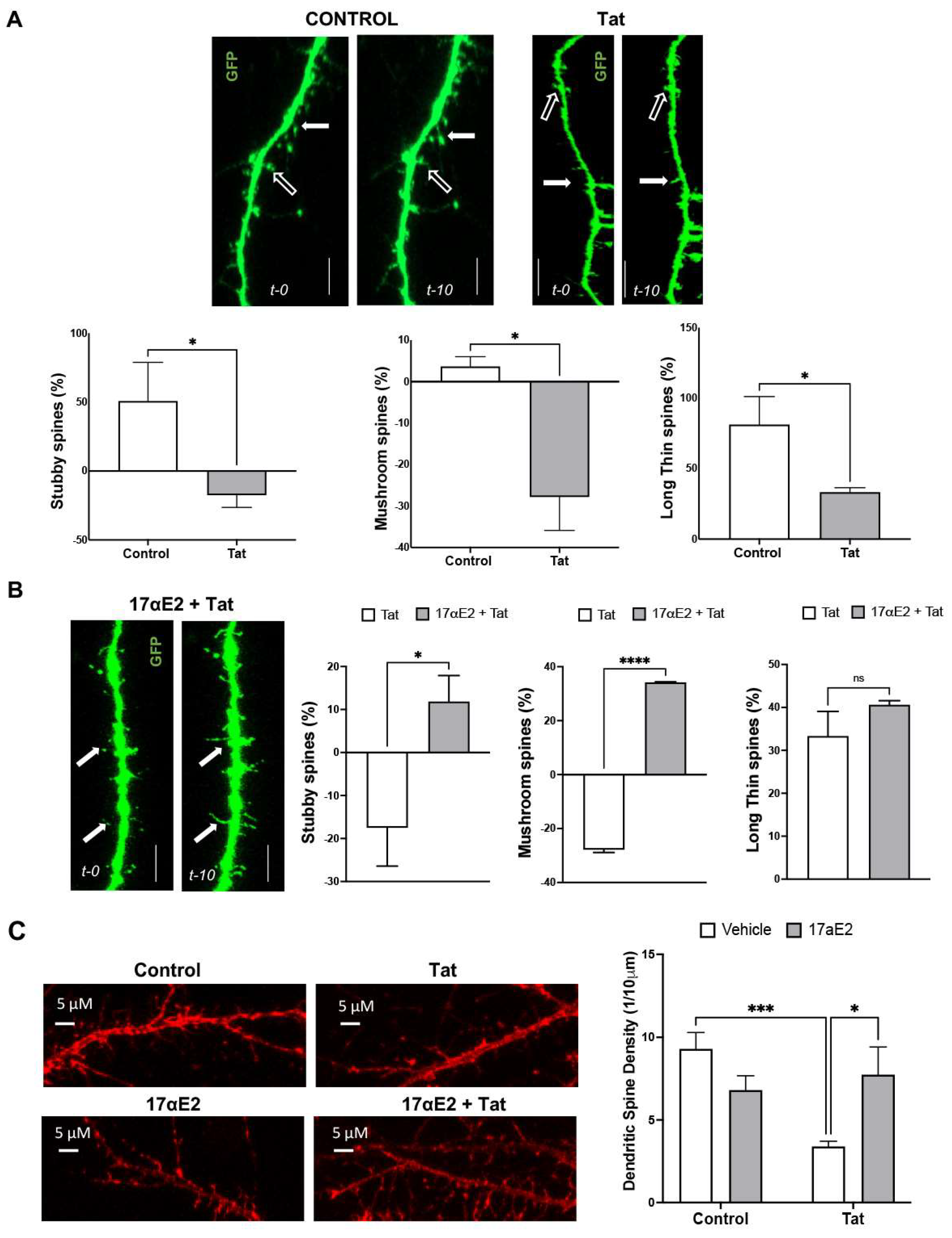
3.2. 17αE2 Prevents Tat-Induced Dendritic Damage
3.3. 17αE2 Prevents Tat-Induced Endolysosome Dysfunction
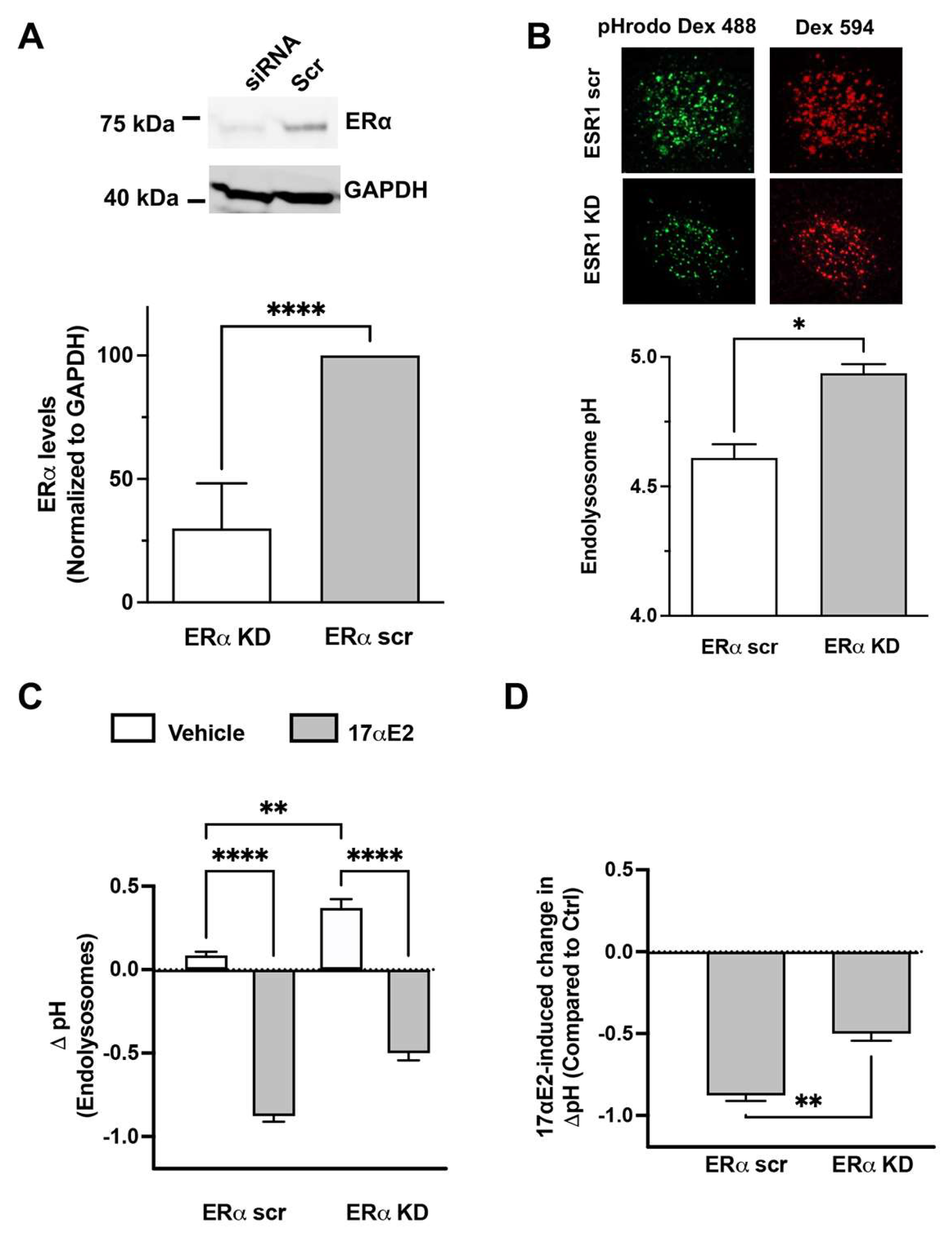
3.4. 17αE2 Enhances Endolysosome Function via ERα
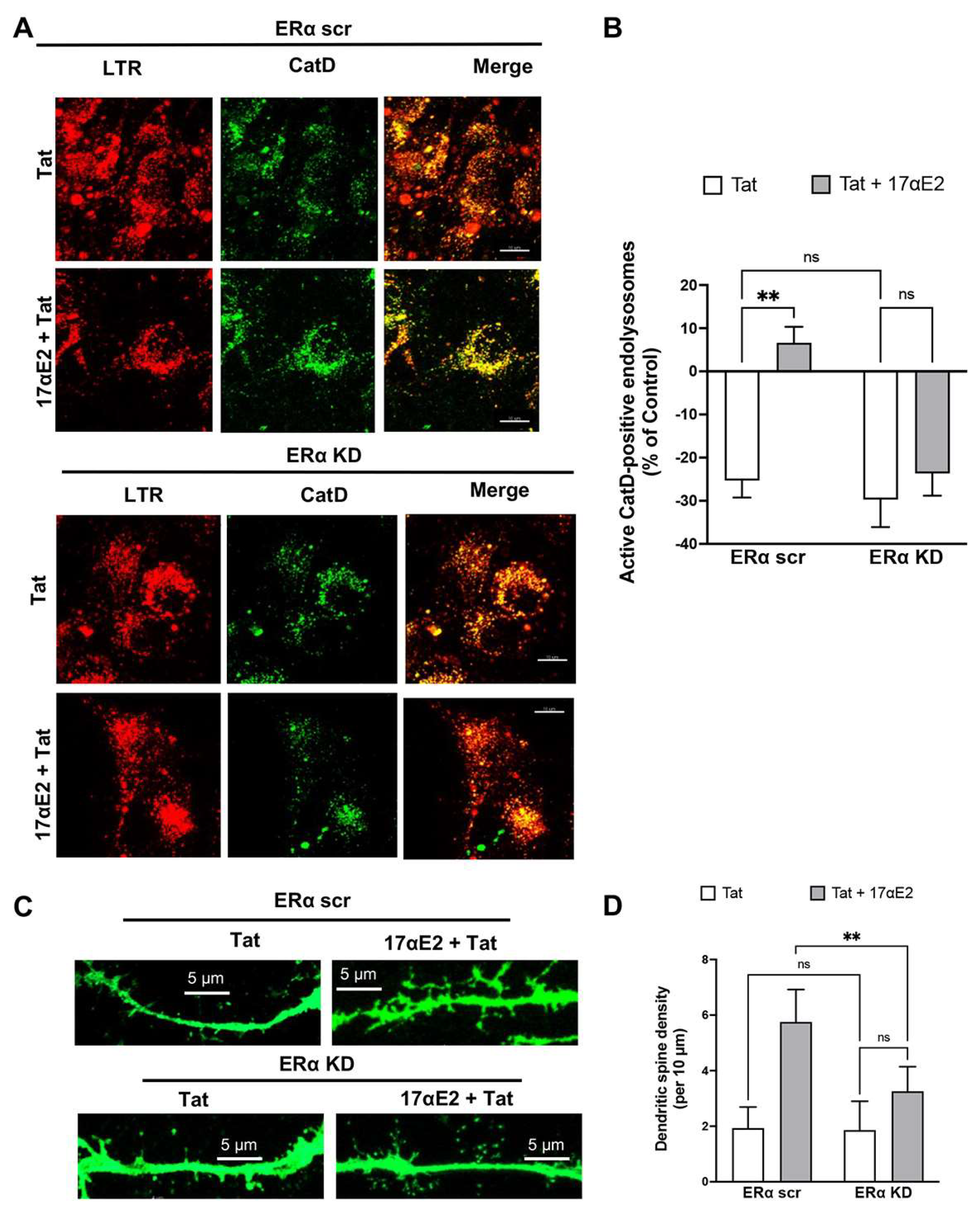
3.5. 17αE2 Protects against Tat-Induced Endolysosome Dysfunction and Impairment in Dendritic Spines via ERα
3.6. 17αE2 Protects against Tat-Induced Endolysosome Dysfunction and Impairment in Dendritic Spines via Endolysosome Localization of ERα
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, Y.; Liu, M.; Lu, Q.; Farrell, M.; Lappin, J.; Shi, J.; Lu, L.; Bao, Y. Data from: Global prevalence and burden of HIV-associated neurocognitive disorder: A meta-analysis. Neurology 2020, 95, e2610–e2621. [Google Scholar] [CrossRef]
- Sacktor, N.; Skolasky, R.L.; Seaberg, E.; Munro, C.; Becker, J.T.; Martin, E.; Ragin, A.; Levine, A.; Miller, E. Prevalence of HIV-associated neurocognitive disorders in the Multicenter AIDS Cohort Study. Neurology 2016, 86, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maschke, M.; Kastrup, O.; Esser, S.; Ross, B.; Hengge, U.; Hufnagel, A. Incidence and prevalence of neurological disorders associated with HIV since the introduction of highly active antiretroviral therapy (HAART). J. Neurol. Neurosurg. Psychiatry 2000, 69, 376–380. [Google Scholar] [CrossRef] [Green Version]
- Crews, L.; Patrick, C.; Achim, C.L.; Everall, I.P.; Masliah, E. Molecular Pathology of Neuro-AIDS (CNS-HIV). Int. J. Mol. Sci. 2009, 10, 1045–1063. [Google Scholar] [CrossRef] [Green Version]
- McArthur, J.C. HIV dementia: An evolving disease. J. Neuroimmunol. 2004, 157, 3–10. [Google Scholar] [CrossRef]
- Masliah, E.; Ellis, R.J.; Bs, M.M.; Heaton, R.K.; Marcotte, T.D.; Ba, J.A.N.; Grant, I.; Atkinson, J.H.; Wiley, C.A.; Achim, C.L.; et al. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann. Neurol. 1997, 42, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Albright, A.V.; Soldan, S.S.; Gonzalez-Scarano, F. Pathogenesis of human immunodeficiency virus-induced neurological disease. J. Neurovirol. 2003, 9, 222–227. [Google Scholar] [CrossRef]
- Archibald, S.L.; Masliah, E.; Fennema-Notestine, C.; Marcotte, T.D.; Ellis, R.; McCutchan, J.A.; Heaton, R.K.; Grant, I.; Mallory, M.; Miller, A.; et al. Correlation of In Vivo Neuroimaging Abnormalities With Postmortem Human Immunodeficiency Virus Encephalitis and Dendritic Loss. Arch. Neurol. 2004, 61, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Everall, I.P.; Heaton, R.K.; Marcotte, T.D.; Ellis, R.; McCutchan, J.A.; Atkinson, J.H.; Grant, I.; Mallory, M.; Masliah, E.; HNRC Group. Cortical Synaptic Density is Reduced in Mild to Moderate Human Immunodeficiency Virus Neurocognitive Disorder. Brain Pathol. 2006, 9, 209–217. [Google Scholar] [CrossRef]
- Sá, M.J.; Madeira, M.D.; Ruela, C.; Volk, B.; Mota-Miranda, A.; Paula-Barbosa, M.M. Dendritic changes in the hippocampal formation of AIDS patients: A quantitative Golgi study. Acta Neuropathol. 2004, 107, 97–110. [Google Scholar] [CrossRef]
- Ellis, R.; Langford, D.; Masliah, E. HIV and antiretroviral therapy in the brain: Neuronal injury and repair. Nat. Rev. Neurosci. 2007, 8, 33–44. [Google Scholar] [CrossRef]
- Marban, C.; Forouzanfar, F.; Ait-Ammar, A.; Fahmi, F.; El Mekdad, H.; Daouad, F.; Rohr, O.; Schwartz, C. Targeting the Brain Reservoirs: Toward an HIV Cure. Front. Immunol. 2016, 7, 397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajasin, D.; Eugenin, E.A. HIV-1 Tat: Role in Bystander Toxicity. Front. Cell. Infect. Microbiol. 2020, 10, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avdoshina, V.M.; Mocchetti, I. Recent Advances in the Molecular and Cellular Mechanisms of gp120-Mediated Neurotoxicity. Cells 2022, 11, 1599. [Google Scholar] [CrossRef] [PubMed]
- Sviridov, D.; Mukhamedova, N.; Makarov, A.A.; Adzhubei, A.; Bukrinsky, M. Comorbidities of HIV infection: Role of Nef-induced impairment of cholesterol metabolism and lipid raft functionality. AIDS 2020, 34, 1–13. [Google Scholar] [CrossRef]
- Li, G.; Makar, T.; Gerzanich, V.; Kalakonda, S.; Ivanova, S.; Pereira, E.F.R.; Andharvarapu, S.; Zhang, J.; Simard, J.M.; Zhao, R.Y. HIV-1 Vpr-Induced Proinflammatory Response and Apoptosis Are Mediated through the Sur1-Trpm4 Channel in Astrocytes. Mbio 2020, 11, e02939-20. [Google Scholar] [CrossRef] [PubMed]
- Underwood, J.; Robertson, K.R.; Winston, A. Could antiretroviral neurotoxicity play a role in the pathogenesis of cognitive impairment in treated HIV disease? AIDS 2015, 29, 253–261. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, M.; Atluri, V.S.R.; Nair, M. Drugs of Abuse in HIV infection and neurotoxicity. Front. Microbiol. 2015, 6, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitting, S.; Xu, R.; Bull, C.; Buch, S.K.; El-Hage, N.; Nath, A.; Knapp, P.E.; Hauser, K.F. Interactive Comorbidity between Opioid Drug Abuse and HIV-1 Tat: Chronic Exposure Augments Spine Loss and Sublethal Dendritic Pathology in Striatal Neurons. Am. J. Pathol. 2010, 177, 1397–1410. [Google Scholar] [CrossRef]
- Soontornniyomkij, V.; Translational Methamphetamine AIDS Research Center (TMARC) Group; Kesby, J.P.; Morgan, E.E.; Bischoff-Grethe, A.; Minassian, A.; Brown, G.G.; Grant, I. Effects of HIV and Methamphetamine on Brain and Behavior: Evidence from Human Studies and Animal Models. J. Neuroimmune Pharmacol. 2016, 11, 495–510. [Google Scholar] [CrossRef] [Green Version]
- Irollo, E.; Luchetta, J.; Ho, C.; Nash, B.; Meucci, O. Mechanisms of neuronal dysfunction in HIV-associated neurocognitive disorders. Cell. Mol. Life Sci. 2021, 78, 4283–4303. [Google Scholar] [CrossRef]
- Saylor, D.; Dickens, A.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder—pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef]
- Heaton, R.K.; Franklin, D.R.; Deutsch, R.; Letendre, S.; Ellis, R.J.; Casaletto, K.B.; Marquine, M.J.; Woods, S.P.; Vaida, F.; Atkinson, J.H.; et al. Neurocognitive Change in the Era of HIV Combination Antiretroviral Therapy: The Longitudinal CHARTER Study. Clin. Infect. Dis. 2014, 60, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Ru, W.; Tang, S.J. HIV-associated synaptic degeneration. Mol. Brain 2017, 10, 40. [Google Scholar] [CrossRef] [Green Version]
- Bandera, A.; Taramasso, L.; Bozzi, G.; Muscatello, A.; Robinson, J.A.; Burdo, T.H.; Gori, A. HIV-Associated Neurocognitive Impairment in the Modern ART Era: Are We Close to Discovering Reliable Biomarkers in the Setting of Virological Suppression? Front. Aging Neurosci. 2019, 11, 187. [Google Scholar] [CrossRef] [Green Version]
- Gelman, B.B.; Soukup, V.M.; Holzer, C.E.; Fabian, R.H.; Schuenke, K.W.; Keherly, M.J.; Richey, F.J.; Lahart, C.J. Potential Role for White Matter Lysosome Expansion in HIV-Associated Dementia. J. Acquir. Immune Defic. Syndr. 2005, 39, 422–425. [Google Scholar] [CrossRef]
- Spector, S.A.; Zhou, D. Autophagy: An overlooked mechanism of HIV-1 pathogenesis and NeuroAIDS? Autophagy 2008, 4, 704–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Spector, S.A. Human immunodeficiency virus type-1 infection inhibits autophagy. AIDS 2008, 22, 695–699. [Google Scholar] [CrossRef] [Green Version]
- Cysique, L.A.; Hewitt, T.; Croitoru-Lamoury, J.; Taddei, K.; Martins, R.N.; Chew, C.S.; Davies, N.N.; Price, P.; Brew, B.J. APOE epsilon4 moderates abnormal CSF-abeta-42 levels, while neurocognitive impairment is associated with abnormal CSF tau levels in HIV+ individuals—A cross-sectional observational study. BMC Neurol. 2015, 15, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixon, R.A.; Cataldo, A.M. The endosomal-lysosomal system of neurons: New roles. Trends Neurosci. 1995, 18, 489–496. [Google Scholar] [CrossRef]
- Nixon, R.A.; Cataldo, A.M. Lysosomal system pathways: Genes to neurodegeneration in Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Goo, M.S.; Sancho, L.; Slepak, N.; Boassa, D.; Deerinck, T.J.; Ellisman, M.H.; Bloodgood, B.L.; Patrick, G.N. Activity-dependent trafficking of lysosomes in dendrites and dendritic spines. J. Cell Biol. 2017, 216, 2499–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padamsey, Z.; McGuinness, L.; Bardo, S.J.; Reinhart, M.; Tong, R.; Hedegaard, A.; Hart, M.L.; Emptage, N.J. Activity-Dependent Exocytosis of Lysosomes Regulates the Structural Plasticity of Dendritic Spines. Neuron 2017, 93, 132–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikoletopoulou, V.; Tavernarakis, N. Regulation and Roles of Autophagy at Synapses. Trends Cell Biol. 2018, 28, 646–661. [Google Scholar] [CrossRef]
- Yap, C.C.; Digilio, L.; McMahon, L.P.; Garcia, A.D.R.; Winckler, B. Degradation of dendritic cargos requires Rab7-dependent transport to somatic lysosomes. J. Cell Biol. 2018, 217, 3141–3159. [Google Scholar] [CrossRef] [Green Version]
- Farías, G.G.; Guardia, C.M.; Britt, D.J.; Guo, X.; Bonifacino, J.S. Sorting of Dendritic and Axonal Vesicles at the Pre-axonal Exclusion Zone. Cell Rep. 2015, 13, 1221–1232. [Google Scholar] [CrossRef] [Green Version]
- Farías, G.G.; Guardia, C.M.; De Pace, R.; Britt, D.J.; Bonifacino, J.S. BORC/kinesin-1 ensemble drives polarized transport of lysosomes into the axon. Proc. Natl. Acad. Sci. USA 2017, 114, E2955–E2964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, S.M. Axonal transport and maturation of lysosomes. Curr. Opin. Neurobiol. 2018, 51, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Agostini, S.; Ali, H.; Vardabasso, C.; Fittipaldi, A.; Tasciotti, E.; Cereseto, A.; Bugatti, A.; Rusnati, M.; Lusic, M.; Giacca, M. Inhibition of Non Canonical HIV-1 Tat Secretion Through the Cellular Na+, K+-ATPase Blocks HIV-1 Infection. EBioMedicine 2017, 21, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Ensoli, B.; Barillari, G.; Salahuddin, S.Z.; Gallo, R.C.; Wong-Staal, F. Tat protein of HIV-1 stimulates growth of cells derived from Kaposi’s sarcoma lesions of AIDS patients. Nature 1990, 345, 84–86. [Google Scholar] [CrossRef]
- Chang, H.C.; Samaniego, F.; Nair, B.C.; Buonaguro, L.; Ensoli, B. HIV-1 Tat protein exits from cells via a leaderless secretory pathway and binds to extracellular matrix-associated heparan sulfate proteoglycans through its basic region. AIDS 1997, 11, 1421–1431. [Google Scholar] [CrossRef]
- Rayne, F.; Debaisieux, S.; Yezid, H.; Lin, Y.-L.; Mettling, C.; Konate, K.; Chazal, N.; Arold, S.T.; Pugniere, M.; Sanchez, F.; et al. Phosphatidylinositol-(4,5)-bisphosphate enables efficient secretion of HIV-1 Tat by infected T-cells. EMBO J. 2010, 29, 1348–1362. [Google Scholar] [CrossRef]
- Mediouni, S.; Darque, A.; Baillat, G.; Ravaux, I.; Dhiver, C.; Tissot-Dupont, H.; Mokhtari, M.; Moreau, H.; Tamalet, C.; Brunet, C.; et al. Antiretroviral therapy does not block the secretion of the human immunodeficiency virus Tat protein. Infect. Disord. Drug Targets 2012, 12, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.P.; Patel, K.; Johnson, K.R.; Maric, D.; Calabresi, P.A.; Hasbun, R.; Nath, A. Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc. Natl. Acad. Sci. USA 2013, 110, 13588–13593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, L.J.; Johnson, T.P.; Smith, B.R.; Reoma, L.; Santamaria, U.A.; Bachani, M.; DeMarino, C.; Barclay, R.A.; Snow, J.; Sacktor, N.; et al. Presence of Tat and transactivation response element in spinal fluid despite antiretroviral therapy. AIDS 2019, 33 (Suppl. S2), S145–S157. [Google Scholar] [CrossRef]
- Chang, J.R.; Mukerjee, R.; Bagashev, A.; Del Valle, L.; Chabrashvili, T.; Hawkins, B.J.; He, J.J.; Sawaya, B.E. HIV-1 Tat Protein Promotes Neuronal Dysfunction through Disruption of MicroRNAs. J. Biol. Chem. 2011, 286, 41125–41134. [Google Scholar] [CrossRef] [Green Version]
- Donoso, M.; D’Amico, D.; Valdebenito, S.; Hernandez, C.A.; Prideaux, B.; Eugenin, E.A. Identification, Quantification, and Characterization of HIV-1 Reservoirs in the Human Brain. Cells 2022, 11, 2379. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Huang, Y.; Reid, R.; Steiner, J.; Malpica-Llanos, T.; Darden, T.A.; Shankar, S.K.; Mahadevan, A.; Satishchandra, P.; Nath, A. NMDA Receptor Activation by HIV-Tat Protein Is Clade Dependent. J. Neurosci. 2008, 28, 12190–12198. [Google Scholar] [CrossRef] [Green Version]
- Sabatier, J.M.; Vives, E.; Mabrouk, K.; Benjouad, A.; Rochat, H.; Duval, A.; Hue, B.; Bahraoui, E. Evidence for neurotoxic activity of Tat from human immunodeficiency virus type 1. J. Virol. 1991, 65, 961–967. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, S.J.; Mactutus, C.F.; Aksenova, M.V.; Espensen-Sturges, T.D.; Booze, R.M. Synaptodendritic recovery following HIV Tat exposure: Neurorestoration by phytoestrogens. J. Neurochem. 2014, 128, 140–151. [Google Scholar] [CrossRef] [Green Version]
- Fitting, S.; Ignatowska-Jankowska, B.M.; Bull, C.; Skoff, R.P.; Lichtman, A.H.; Wise, L.E.; Fox, M.A.; Su, J.; Medina, A.E.; Krahe, T.E.; et al. Synaptic Dysfunction in the Hippocampus Accompanies Learning and Memory Deficits in Human Immunodeficiency Virus Type-1 Tat Transgenic Mice. Biol. Psychiatry 2013, 73, 443–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hargus, N.J.; Thayer, S.A. Human Immunodeficiency Virus-1 Tat Protein Increases the Number of Inhibitory Synapses between Hippocampal Neurons in Culture. J. Neurosci. 2013, 33, 17908–17920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, A.; Steiner, J. Synaptodendritic injury with HIV-Tat protein: What is the therapeutic target? Exp. Neurol. 2014, 251, 112–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debaisieux, S.; Rayne, F.; Yezid, H.; Beaumelle, B. The ins and outs of HIV-1 Tat. Traffic 2012, 13, 355–363. [Google Scholar] [CrossRef]
- Tyagi, M.; Rusnati, M.; Presta, M.; Giacca, M. Internalization of HIV-1 Tat Requires Cell Surface Heparan Sulfate Proteoglycans. J. Biol. Chem. 2001, 276, 3254–3261. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Jones, M.; Hingtgen, C.M.; Bu, G.; Laribee, N.; Tanzi, R.E.; Moir, R.D.; Nath, A.; He, J.J. Uptake of HIV-1 Tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat. Med. 2000, 6, 1380–1387. [Google Scholar] [CrossRef]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the Tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.A.; Frankel, A.D. Endocytosis and targeting of exogenous HIV-1 Tat protein. EMBO J. 1991, 10, 1733–1739. [Google Scholar] [CrossRef]
- Gaskill, P.J.; Miller, D.R.; Gamble-George, J.; Yano, H.; Khoshbouei, H. HIV, Tat and dopamine transmission. Neurobiol. Dis. 2017, 105, 51–73. [Google Scholar] [CrossRef]
- Hui, L.; Chen, X.; Haughey, N.J.; Geiger, J.D. Role of Endolysosomes in HIV-1 Tat-Induced Neurotoxicity. ASN Neuro 2012, 4, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Hui, L.; Geiger, N.H.; Haughey, N.J.; Geiger, J.D. Endolysosome involvement in HIV-1 transactivator protein-induced neuronal amyloid beta production. Neurobiol. Aging 2013, 34, 2370–2378. [Google Scholar] [CrossRef] [Green Version]
- Datta, G.; Miller, N.N.; Du, W.; Geiger, J.D.; Chang, S.; Chen, X. Endolysosome Localization of ERalpha Is Involved in the Protective Effect of 17alpha-Estradiol against HIV-1 gp120-Induced Neuronal Injury. J. Neurosci. 2021, 41, 10365–10381. [Google Scholar] [CrossRef]
- Christensen, A.; Dewing, P.; Micevych, P. Membrane-Initiated Estradiol Signaling Induces Spinogenesis Required for Female Sexual Receptivity. J. Neurosci. 2011, 31, 17583–17589. [Google Scholar] [CrossRef] [Green Version]
- Nash, B.; Tarn, K.; Irollo, E.; Luchetta, J.; Festa, L.; Halcrow, P.; Datta, G.; Geiger, J.D.; Meucci, O. Morphine-Induced Modulation of Endolysosomal Iron Mediates Upregulation of Ferritin Heavy Chain in Cortical Neurons. Eneuro 2019, 6, ENEURO.0237-19.2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, A.; Turchan, J.; Pocernich, C.; Bruce-Keller, A.; Roth, S.; Butterfield, D.A.; Major, E.O.; Nath, A. Intracellular Human Immunodeficiency Virus Tat Expression in Astrocytes Promotes Astrocyte Survival but Induces Potent Neurotoxicity at Distant Sites via Axonal Transport. J. Biol. Chem. 2003, 278, 13512–13519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Knight, A.G.; Gupta, S.; Knapp, P.E.; Hauser, K.F.; Keller, J.N.; Bruce-Keller, A.J. HIV-Tat elicits microglial glutamate release: Role of NAPDH oxidase and the cystine-glutamate antiporter. Neurosci. Lett. 2010, 485, 233–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.; Halcrow, P.W.; Afghah, Z.; Baral, A.; Geiger, J.D.; Chen, X. HIV-1 Tat endocytosis and retention in endolysosomes affects HIV-1 Tat-induced LTR transactivation in astrocytes. FASEB J. 2022, 36, e22184. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Bucci, C.; Weis, J.; Kruttgen, A. The Small GTPase Rab7 Controls the Endosomal Trafficking and Neuritogenic Signaling of the Nerve Growth Factor Receptor TrkA. J. Neurosci. 2005, 25, 10930–10940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, T.; Makino, Y.; Yamada, M.K. 17α-Estradiol is generated locally in the male rat brain and can regulate GAD65 expression and anxiety. Neuropharmacology 2015, 90, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Toran-Allerand, C.D.; Tinnikov, A.A.; Singh, R.J.; Nethrapalli, I.S. 17alpha-estradiol: A brain-active estrogen? Endocrinology 2005, 146, 3843–3850. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, D.C.; Lantz, C.L.; Rumi, M.A.K.; Quinlan, E.M. 17α Estradiol promotes plasticity of spared inputs in the adult amblyopic visual cortex. Sci. Rep. 2019, 9, 19040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heron, P.M.; Turchan-Cholewo, J.; Bruce-Keller, A.J.; Wilson, M.E. Estrogen Receptor Alpha Inhibits the Estrogen-Mediated Suppression of HIV Transcription in Astrocytes: Implications for Estrogen Neuroprotection in HIV Dementia. AIDS Res. Hum. Retroviruses 2009, 25, 1071–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.P.; Zeng, M.; Hu, X.-Y.; Xu, H.; Swaab, D.; Ravid, R.; Zhou, J.-N. Estrogen receptor alpha-immunoreactive astrocytes are increased in the hippocampus in Alzheimer’s disease. Exp. Neurol. 2003, 183, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Milner, T.A.; Ayoola, K.; Drake, C.T.; Herrick, S.P.; Tabori, N.E.; McEwen, B.S.; Warrier, S.; Alves, S.E. Ultrastructural localization of estrogen receptor beta immunoreactivity in the rat hippocampal formation. J. Comp. Neurol. 2005, 491, 81–95. [Google Scholar] [CrossRef]
- Milner, T.A.; McEwen, B.S.; Hayashi, S.; Li, C.J.; Reagan, L.P.; Alves, S.E. Ultrastructural evidence that hippocampal alpha estrogen receptors are located at extranuclear sites. J. Comp. Neurol. 2001, 429, 355–371. [Google Scholar] [CrossRef]
- Lai, Y.J.; Yu, D.; Zhang, J.H.; Chen, G.-J. Cooperation of Genomic and Rapid Nongenomic Actions of Estrogens in Synaptic Plasticity. Mol. Neurobiol. 2016, 54, 4113–4126. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, D.P.; Waters, E.M.; Mermelstein, P.G.; Kramár, E.A.; Shors, T.J.; Liu, F. Rapid Estrogen Signaling in the Brain: Implications for the Fine-Tuning of Neuronal Circuitry. J. Neurosci. 2011, 31, 16056–16063. [Google Scholar] [CrossRef] [Green Version]
- Frick, K.M. Molecular mechanisms underlying the memory-enhancing effects of estradiol. Horm. Behav. 2015, 74, 4–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukai, H.; Tsurugizawa, T.; Murakami, G.; Kominami, S.; Ishii, H.; Ogiue-Ikeda, M.; Takata, N.; Tanabe, N.; Furukawa, A.; Hojo, Y.; et al. Rapid modulation of long-term depression and spinogenesis via synaptic estrogen receptors in hippocampal principal neurons. J. Neurochem. 2007, 100, 950–967. [Google Scholar] [CrossRef]
- Hojo, Y.; Murakami, G.; Mukai, H.; Higo, S.; Hatanaka, Y.; Ogiue-Ikeda, M.; Ishii, H.; Kimoto, T.; Kawato, S. Estrogen synthesis in the brain—Role in synaptic plasticity and memory. Mol. Cell. Endocrinol. 2008, 290, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, P.A.S.; Choleris, E.; Galea, L.A.M. Structural plasticity of the hippocampus in response to estrogens in female rodents. Mol. Brain 2019, 12, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampayo, R.G.; Toscani, A.M.; Rubashkin, M.G.; Thi, K.; Masullo, L.A.; Violi, I.L.; Lakins, J.N.; Cáceres, A.; Hines, W.C.; Leskow, F.C.; et al. Fibronectin rescues estrogen receptor α from lysosomal degradation in breast cancer cells. J. Cell Biol. 2018, 217, 2777–2798. [Google Scholar] [CrossRef] [Green Version]
- Liao, T.L.; Tzeng, C.-R.; Yu, C.-L.; Wang, Y.-P.; Kao, S.-H. Estrogen receptor-beta in mitochondria: Implications for mitochondrial bioenergetics and tumorigenesis. Ann. N. Y. Acad Sci. 2015, 1350, 52–60. [Google Scholar] [CrossRef]
- Yang, S.H.; Liu, R.; Perez, E.J.; Wen, Y.; Stevens, S.M., Jr.; Valencia, T.; Brun-Zinkernagel, A.-M.; Prokai, L.; Will, Y.; Dykens, J.; et al. Mitochondrial localization of estrogen receptor beta. Proc. Natl. Acad. Sci. USA 2004, 101, 4130–4135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [Green Version]
- Azcoitia, I.; Mendez, P.; Garcia-Segura, L.M. Aromatase in the Human Brain. Androg. Clin. Res. Ther. 2021, 2, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Kretz, O.; Fester, L.; Wehrenberg, U.; Zhou, L.; Brauckmann, S.; Zhao, S.; Prange-Kiel, J.; Naumann, T.; Jarry, H.; Frotscher, M.; et al. Hippocampal Synapses Depend on Hippocampal Estrogen Synthesis. J. Neurosci. 2004, 24, 5913–5921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acconcia, F.; Ascenzi, P.; Bocedi, A.; Spisni, E.; Tomasi, V.; Trentalance, A.; Visca, P.; Marino, M. Palmitoylation-dependent estrogen receptor alpha membrane localization: Regulation by 17beta-estradiol. Mol. Biol. Cell 2005, 16, 231–237. [Google Scholar] [CrossRef] [Green Version]
- Meitzen, J.; Luoma, J.I.; Boulware, M.I.; Hedges, V.L.; Peterson, B.M.; Tuomela, K.; Britson, K.A.; Mermelstein, P.G. Palmitoylation of Estrogen Receptors Is Essential for Neuronal Membrane Signaling. Endocrinology 2013, 154, 4293–4304. [Google Scholar] [CrossRef] [Green Version]
- Adlanmerini, M.; Solinhac, R.; Abot, A.; Fabre, A.; Raymond-Letron, I.; Guihot, A.-L.; Boudou, F.; Sautier, L.; Vessières, E.; Kim, S.H.; et al. Mutation of the palmitoylation site of estrogen receptor α in vivo reveals tissue-specific roles for membrane versus nuclear actions. Proc. Natl. Acad. Sci. USA 2014, 111, E283–E290. [Google Scholar] [CrossRef] [Green Version]
- Mindell, J.A. Lysosomal Acidification Mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86. [Google Scholar] [CrossRef] [Green Version]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- McGuire, C.; Stransky, L.; Cotter, K.; Forgac, M. Regulation of V-ATPase activity. Front. Biosci.-Landmark 2017, 22, 609–622. [Google Scholar]
- Prasad, H.; Rao, R. Histone deacetylase–mediated regulation of endolysosomal pH. J. Biol. Chem. 2018, 293, 6721–6735. [Google Scholar] [CrossRef] [Green Version]
- de Duve, C. The lysosome turns fifty. Nat. Cell Biol. 2005. 7, 847–849. [CrossRef]
- Paroutis, P.; Touret, N.; Grinstein, S. The pH of the Secretory Pathway: Measurement, Determinants, and Regulation. Physiology 2004, 19, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Kellokumpu, S. Golgi pH, Ion and Redox Homeostasis: How Much Do They Really Matter? Front. Cell Dev. Biol. 2019, 7, 93. [Google Scholar] [CrossRef] [Green Version]
- Weisz, O.A. Organelle Acidification and Disease. Traffic 2003, 4, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Winckler, B.; Faundez, V.; Maday, S.; Cai, Q.; Almeida, C.G.; Zhang, H. The Endolysosomal System and Proteostasis: From Development to Degeneration. J. Neurosci. 2018, 38, 9364–9374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Sato, Y.; Nixon, R.A. Lysosomal proteolysis inhibition selectively disrupts axonal transport of degradative organelles and causes an Alzheimer’s-like axonal dystrophy. J. Neurosci. 2011, 31, 7817–7830. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.J.; Attwell, D. The Energetics of CNS White Matter. J. Neurosci. 2012, 32, 356–371. [Google Scholar] [CrossRef] [Green Version]
- Price, J.C.; Guan, S.; Burlingame, A.; Prusiner, S.B.; Ghaemmaghami, S. Analysis of proteome dynamics in the mouse brain. Proc. Natl. Acad. Sci. USA 2010, 107, 14508–14513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, S.; Diering, G.H.; Na, C.H.; Nirujogi, R.S.; Bachman, J.L.; Pandey, A.; Huganir, R.L. Identification of long-lived synaptic proteins by proteomic analysis of synaptosome protein turnover. Proc. Natl. Acad. Sci. USA 2018, 115, E3827–E3836. [Google Scholar] [CrossRef] [Green Version]
- Nikoletopoulou, V.; Papandreou, M.-E.; Tavernarakis, N. Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ. 2015, 22, 398–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Root, J.; Merino, P.; Nuckols, A.; Johnson, M.; Kukar, T. Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 154, 105360. [Google Scholar] [CrossRef]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef] [Green Version]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. The aging lysosome: An essential catalyst for late-onset neurodegenerative diseases. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140443. [Google Scholar] [CrossRef]
- Cinti, A.; Le Sage, V.; Milev, M.P.; Valiente-Echeverría, F.; Crossie, C.; Miron, M.-J.; Panté, N.; Olivier, M.; Mouland, A.J. HIV-1 enhances mTORC1 activity and repositions lysosomes to the periphery by co-opting Rag GTPases. Sci. Rep. 2017, 7, 5515. [Google Scholar] [CrossRef] [Green Version]
- Moorjani, H.; Craddock, B.P.; Morrison, S.A.; Steigbigel, R.T. Impairment of Phagosome-Lysosome Fusion in HIV-1-Infected Macrophages. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1996, 13, 18–22. [Google Scholar] [CrossRef]
- Fields, J.; Dumaop, W.; Elueteri, S.; Campos, S.; Serger, E.; Trejo, M.; Kosberg, K.; Adame, A.; Spencer, B.; Rockenstein, E.; et al. HIV-1 Tat Alters Neuronal Autophagy by Modulating Autophagosome Fusion to the Lysosome: Implications for HIV-Associated Neurocognitive Disorders. J. Neurosci. 2015, 35, 1921–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, M.; Patel, N.; Xu, H.; Lee, M.; Tominaga-Yamanaka, K.; Nath, A.; Geiger, J.; Gorospe, M.; Mattson, M.P.; Haughey, N.J. Activation of TRPML1 clears intraneuronal Abeta in preclinical models of HIV infection. J. Neurosci. 2014, 34, 11485–11503. [Google Scholar] [CrossRef] [Green Version]
- Datta, G.; Miller, N.M.; Afghah, Z.; Geiger, J.D.; Chen, X. HIV-1 gp120 Promotes Lysosomal Exocytosis in Human Schwann Cells. Front. Cell. Neurosci. 2019, 13, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halcrow, P.W.; Lakpa, K.L.; Khan, N.; Afghah, Z.; Miller, N.; Datta, G.; Chen, X.; Geiger, J.D. HIV-1 gp120-Induced Endolysosome de-Acidification Leads to Efflux of Endolysosome Iron, and Increases in Mitochondrial Iron and Reactive Oxygen Species. J. Neuroimmune Pharmacol. 2021, 17, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Rawat, P.; Bruckman, R.S.; Spector, S.A. Human Immunodeficiency Virus Type 1 Nef Inhibits Autophagy through Transcription Factor EB Sequestration. PLoS Pathog. 2015, 11, e1005018. [Google Scholar] [CrossRef] [Green Version]
- Santerre, M.; Arjona, S.P.; Allen, C.N.; Buch, S.; E Sawaya, B. HIV-1 Vpr protein impairs lysosome clearance causing SNCA/alpha-synuclein accumulation in neurons. Autophagy 2021, 17, 1768–1782. [Google Scholar] [CrossRef]
- Hui, L.; Ye, Y.; Soliman, M.L.; Lakpa, K.L.; Miller, N.M.; Afghah, Z.; Geiger, J.D.; Chen, X. Antiretroviral Drugs Promote Amyloidogenesis by De-Acidifying Endolysosomes. J. Neuroimmune Pharmacol. 2021, 16, 159–168. [Google Scholar] [CrossRef]
- Cubells, J.; Rayport, S.; Rajendran, G.; Sulzer, D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J. Neurosci. 1994, 14, 2260–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nara, A.; Aki, T.; Funakoshi, T.; Unuma, K.; Uemura, K. Hyperstimulation of macropinocytosis leads to lysosomal dysfunction during exposure to methamphetamine in SH-SY5Y cells. Brain Res. 2012, 1466, 1–14. [Google Scholar] [CrossRef]
- Kawai, A.; Uchiyama, H.; Takano, S.; Nakamura, N.; Ohkuma, S. Autophagosome-Lysosome Fusion Depends on the pH in Acidic Compartments in CHO Cells. Autophagy 2007, 3, 154–157. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.E.; Ostrowski, P.; Jaumouillé, V.; Grinstein, S. The position of lysosomes within the cell determines their luminal pH. J. Cell Biol. 2016, 212, 677–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuruta, F.; Dolmetsch, R.E. PIKfyve mediates the motility of late endosomes and lysosomes in neuronal dendrites. Neurosci. Lett. 2015, 605, 18–23. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Datta, G.; Miller, N.M.; Chen, X. 17⍺-Estradiol Protects against HIV-1 Tat-Induced Endolysosome Dysfunction and Dendritic Impairments in Neurons. Cells 2023, 12, 813. https://doi.org/10.3390/cells12050813
Datta G, Miller NM, Chen X. 17⍺-Estradiol Protects against HIV-1 Tat-Induced Endolysosome Dysfunction and Dendritic Impairments in Neurons. Cells. 2023; 12(5):813. https://doi.org/10.3390/cells12050813
Chicago/Turabian StyleDatta, Gaurav, Nicole M. Miller, and Xuesong Chen. 2023. "17⍺-Estradiol Protects against HIV-1 Tat-Induced Endolysosome Dysfunction and Dendritic Impairments in Neurons" Cells 12, no. 5: 813. https://doi.org/10.3390/cells12050813
APA StyleDatta, G., Miller, N. M., & Chen, X. (2023). 17⍺-Estradiol Protects against HIV-1 Tat-Induced Endolysosome Dysfunction and Dendritic Impairments in Neurons. Cells, 12(5), 813. https://doi.org/10.3390/cells12050813