

Chimeric Antigen Receptor T Cell Therapy for Pancreatic Cancer: A Review of Current Evidence


Abstract
:1. Introduction
2. Immunotherapy Challenges in Pancreatic Cancer
3. CAR-T Cells
3.1. Structure and Evolution of CAR-T Cells
3.2. Antitumor Mechanisms of CAR-T Cells
4. CAR-T Cell Therapy in Pancreatic Cancer
4.1. Studies Performed in Cell Culture and Animal Models
4.2. Studies Performed in Humans
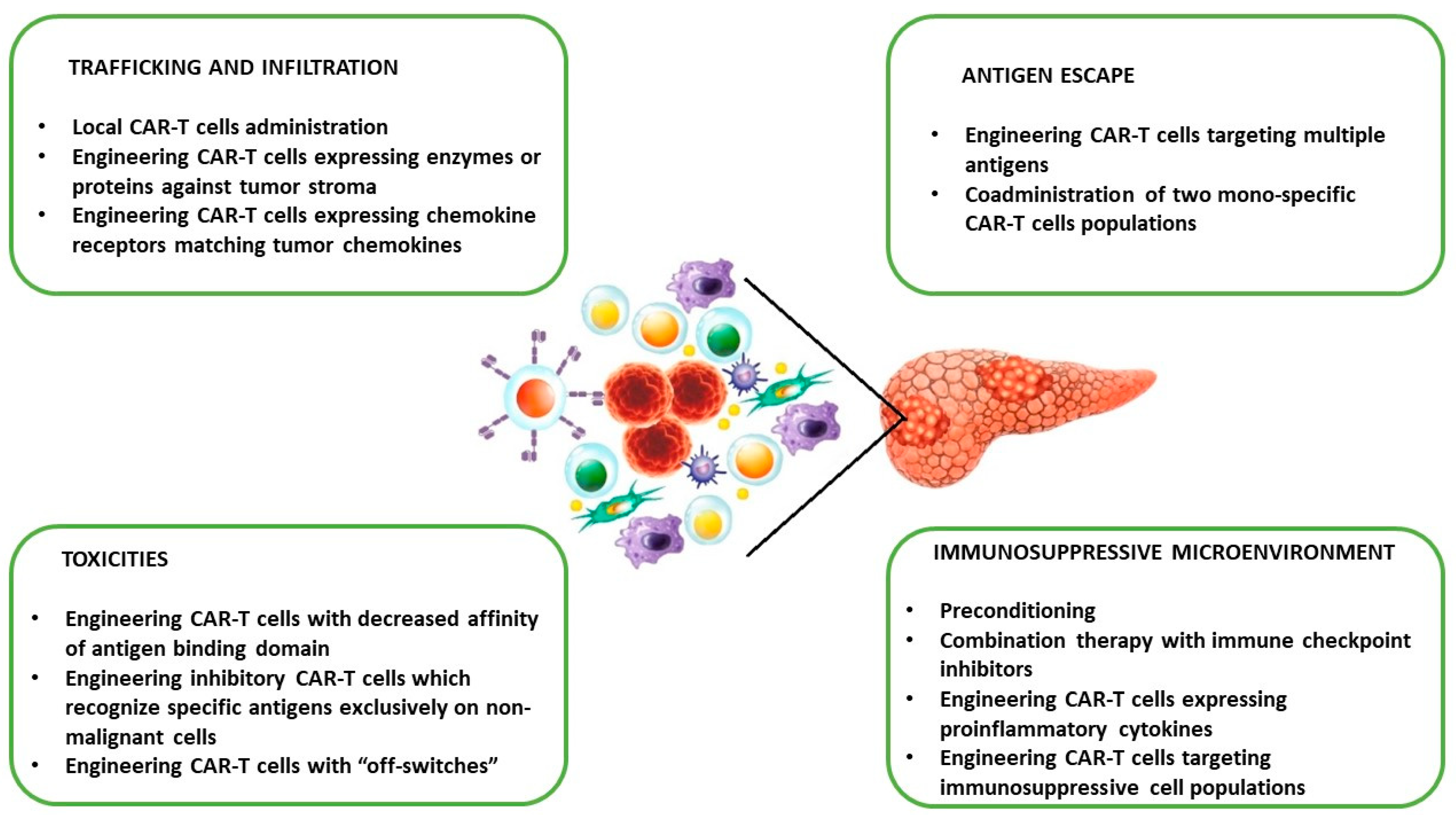
4.3. Challenges of PC CAR-T Cell Therapy in Clinical Translation and Potential Strategies to Overcome Limitations
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today. Lyon, France: International Agency for Research on Cancer. 2018. Available online: https://gco.iarc.fr/today (accessed on 16 February 2019).
- Stoffel, E.M.; Brand, R.E.; Goggins, M. Pancreatic Cancer: Changing Epidemiology and New Approaches to Risk Assessment, Early Detection, and Prevention. Gastroenterology 2023, 164, 752–765. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute. Surveillance, Epidemiology and End Results Program. Available online: www.seer.cancer.gov (accessed on 10 February 2023).
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921, Erratum in Cancer Res. 2014, 74, 4006. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.P. Pancreatic cancer epidemiology: Understanding the role of lifestyle and inherited risk factors. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Kleeff, J.; Michl, P.; Costello, E.; Greenhalf, W.; Palmer, D.H. Therapeutic developments in pancreatic cancer: Current and future perspectives. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 333–348. [Google Scholar] [CrossRef]
- Fudalej, M.; Kwaśniewska, D.; Nurzyński, P.; Badowska-Kozakiewicz, A.; Mękal, D.; Czerw, A.; Sygit, K.; Deptała, A. New Treatment Options in Metastatic Pancreatic Cancer. Cancers 2023, 15, 2327. [Google Scholar] [CrossRef]
- Nagrial, A.M.; Chin, V.T.; Sjoquist, K.M.; Pajic, M.; Horvath, L.G.; Biankin, A.V.; Yip, D. Second-line treatment in inoperable pancreatic adenocarcinoma: A systematic review and synthesis of all clinical trials. Crit. Rev. Oncol. Hematol. 2015, 96, 483–497. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, H.; Liu, C.; Wang, Z.; Wu, W.; Zhang, N.; Zhang, L.; Hu, J.; Luo, P.; Zhang, J.; et al. Immune checkpoint modulators in cancer immunotherapy: Recent advances and emerging concepts. J. Hematol. Oncol. 2022, 15, 111. [Google Scholar] [CrossRef]
- Mucciolo, G.; Roux, C.; Scagliotti, A.; Brugiapaglia, S.; Novelli, F.; Cappello, P. The dark side of immunotherapy: Pancreatic cancer. Cancer Drug. Resist. 2020, 3, 491–520. [Google Scholar] [CrossRef]
- Li, T.J.; Wang, W.Q.; Yu, X.J.; Liu, L. Killing the “BAD”: Challenges for immunotherapy in pancreatic cancer. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188384. [Google Scholar] [CrossRef]
- Das, M.; Zhou, X.; Liu, Y.; Das, A.; Vincent, B.G.; Li, J.; Liu, R.; Huang, L. Tumor neoantigen heterogeneity impacts bystander immune inhibition of pancreatic cancer growth. Transl. Oncol. 2020, 13, 100856. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kunisada, Y.; Miyamura, N.; Eikawa, S.; Hurtado de Mendoza, T.; Mose, E.S.; Lu, C.; Kuroda, Y.; Ruoslahti, E.; Lowy, A.M.; et al. Tumor-resident regulatory T cells in pancreatic cancer express the αvβ5 integrin as a targetable activation marker. bioRxiv 2023. [Google Scholar] [CrossRef]
- Murakami, T.; Hiroshima, Y.; Matsuyama, R.; Homma, Y.; Hoffman, R.M.; Endo, I. Role of the tumor microenvironment in pancreatic cancer. Ann. Gastroenterol. Surg. 2019, 3, 130–137. [Google Scholar] [CrossRef]
- Griesmann, H.; Drexel, C.; Milosevic, N.; Sipos, B.; Rosendahl, J.; Gress, T.M.; Michl, P. Pharmacological macrophage inhibition decreases metastasis formation in a genetic model of pancreatic cancer. Gut 2017, 66, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Xie, K. Pancreatic cancer stromal biology and therapy. Genes Dis. 2015, 2, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Liebl, M.C.; Hofmann, T.G. Identification of responders to immune checkpoint therapy: Which biomarkers have the highest value? J. Eur. Acad. Dermatol. Venereol. 2019, 33 (Suppl. 8), 52–56. [Google Scholar] [CrossRef] [PubMed]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O′Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 247. [Google Scholar] [CrossRef]
- Iranzo, P.; Callejo, A.; Assaf, J.D.; Molina, G.; Lopez, D.E.; Garcia-Illescas, D.; Pardo, N.; Navarro, A.; Martinez-Marti, A.; Cedres, S.; et al. Overview of Checkpoint Inhibitors Mechanism of Action: Role of Immune-Related Adverse Events and Their Treatment on Progression of Underlying Cancer. Front. Med. (Lausanne) 2022, 9, 875974. [Google Scholar] [CrossRef]
- Laface, C.; Memeo, R.; Maselli, F.M.; Santoro, A.N.; Iaia, M.L.; Ambrogio, F.; Laterza, M.; Cazzato, G.; Guarini, C.; De Santis, P.; et al. Immunotherapy and Pancreatic Cancer: A Lost Challenge? Life 2023, 13, 1482. [Google Scholar] [CrossRef]
- Gebauer, F.; Wicklein, D.; Horst, J.; Sundermann, P.; Maar, H.; Streichert, T.; Tachezy, M.; Izbicki, J.R.; Bockhorn, M.; Schumacher, U. Carcinoembryonic antigen-related cell adhesion molecules (CEACAM) 1, 5 and 6 as biomarkers in pancreatic cancer. PLoS ONE 2014, 9, e113023. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Tsang, Y.; Yang, Y. Identification of CEACAM5 as a stemness-related inhibitory immune checkpoint in pancreatic cancer. BMC Cancer 2022, 22, 1291. [Google Scholar] [CrossRef] [PubMed]
- Kankeu Fonkoua, L.A.; Sirpilla, O.; Sakemura, R.; Siegler, E.L.; Kenderian, S.S. CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol. Ther. Oncolytics 2022, 25, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Lesch, S.; Blumenberg, V.; Stoiber, S.; Gottschlich, A.; Ogonek, J.; Cadilha, B.L.; Dantes, Z.; Rataj, F.; Dorman, K.; Lutz, J.; et al. T cells armed with C-X-C chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours. Nat. Biomed. Eng. 2021, 5, 1246–1260. [Google Scholar] [CrossRef] [PubMed]
- Cadilha, B.L.; Benmebarek, M.R.; Dorman, K.; Oner, A.; Lorenzini, T.; Obeck, H.; Vänttinen, M.; Di Pilato, M.; Pruessmann, J.N.; Stoiber, S.; et al. Combined tumor-directed recruitment and protection from immune suppression enable CAR T cell efficacy in solid tumors. Sci. Adv. 2021, 7, eabi5781. [Google Scholar] [CrossRef] [PubMed]
- Mellor, A.L.; Munn, D.H. IDO expression by dendritic cells: Tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef]
- Maalej, K.M.; Merhi, M.; Inchakalody, V.P.; Mestiri, S.; Alam, M.; Maccalli, C.; Cherif, H.; Uddin, S.; Steinhoff, M.; Marincola, F.M.; et al. CAR-cell therapy in the era of solid tumor treatment: Current challenges and emerging therapeutic advances. Mol. Cancer 2023, 22, 20. [Google Scholar] [CrossRef]
- Tang, H.K.C.; Wang, B.; Tan, H.X.; Sarwar, M.A.; Baraka, B.; Shafiq, T.; Rao, A.R. CAR T-Cell Therapy for Cancer: Latest Updates and Challenges, with a Focus on B-Lymphoid Malignancies and Selected Solid Tumours. Cells 2023, 12, 1586. [Google Scholar] [CrossRef]
- Yan, T.; Zhu, L.; Chen, J. Current advances and challenges in CAR T-Cell therapy for solid tumors: Tumor-associated antigens and the tumor microenvironment. Exp. Hematol. Oncol. 2023, 12, 14. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Śledź, M.; Wojciechowska, A.; Zagożdżon, R.; Kaleta, B. In Situ Programming of CAR-T Cells: A Pressing Need in Modern Immunotherapy. Arch. Immunol. Ther. Exp. 2023, 71, 18, Erratum in Arch. Immunol. Ther. Exp. 2023, 71, 22. [Google Scholar] [CrossRef]
- Guedan, S.; Calderon, H.; Posey, A.D., Jr.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. Methods Clin. Dev. 2018, 12, 145–156. [Google Scholar] [CrossRef]
- Jayaraman, J.; Mellody, M.P.; Hou, A.J.; Desai, R.P.; Fung, A.W.; Pham, A.H.T.; Chen, Y.Y.; Zhao, W. CAR-T design: Elements and their synergistic function. EBioMedicine 2020, 58, 102931. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef]
- Brocker, T. Chimeric Fv-zeta or Fv-epsilon receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood 2000, 96, 1999–2001. [Google Scholar] [CrossRef]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef]
- Schubert, M.L.; Schmitt, A.; Neuber, B.; Hückelhoven-Krauss, A.; Kunz, A.; Wang, L.; Gern, U.; Michels, B.; Sellner, L.; Hofmann, S.; et al. Third-generation CAR T cells targeting CD19 are associated with an excellent safety profile and might improve persistence of CAR T cells in treated patients. Blood 2019, 134, 51. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef]
- Moghanloo, E.; Mollanoori, H.; Talebi, M.; Pashangzadeh, S.; Faraji, F.; Hadjilooei, F.; Mahmoodzadeh, H. Remote controlling of CAR-T cells and toxicity management: Molecular switches and next generation CARs. Transl. Oncol. 2021, 14, 101070. [Google Scholar] [CrossRef]
- Benmebarek, M.R.; Karches, C.H.; Cadilha, B.L.; Lesch, S.; Endres, S.; Kobold, S. Killing Mechanisms of Chimeric Antigen Receptor (CAR) T Cells. Int. J. Mol. Sci. 2019, 20, 1283. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, B.; Mestre, D.; Martínez-Martín, N. The immunological synapse: A cause or consequence of T-cell receptor triggering? Immunology 2011, 133, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Davenport, A.J.; Jenkins, M.R. Programming a serial killer: CAR T cells form non-classical immune synapses. Oncoscience 2018, 5, 69–70. [Google Scholar] [CrossRef] [PubMed]
- Davenport, A.J.; Cross, R.S.; Watson, K.A.; Liao, Y.; Shi, W.; Prince, H.M.; Beavis, P.A.; Trapani, J.A.; Kershaw, M.H.; Ritchie, D.S.; et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl. Acad. Sci. USA 2019, 115, E2068–E2076, Erratum in Proc. Natl. Acad. Sci. USA 2019, 116, 11075–11076. [Google Scholar] [CrossRef] [PubMed]
- Trapani, J.A.; Smyth, M.J. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2002, 2, 735–747. [Google Scholar] [CrossRef]
- Cullen, S.P.; Martin, S.J. Mechanisms of granule-dependent killing. Cell Death Differ. 2008, 15, 251–262. [Google Scholar] [CrossRef]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef]
- Volpe, E.; Sambucci, M.; Battistini, L.; Borsellino, G. Fas-Fas Ligand: Checkpoint of T Cell Functions in Multiple Sclerosis. Front. Immunol. 2016, 7, 382. [Google Scholar] [CrossRef]
- O’Brien, D.I.; Nally, K.; Kelly, R.G.; O′Connor, T.M.; Shanahan, F.; O′Connell, J. Targeting the Fas/Fas ligand pathway in cancer. Expert Opin. Ther. Targets 2005, 9, 1031–1044. [Google Scholar] [CrossRef]
- Textor, A.; Listopad, J.J.; Wührmann, L.L.; Perez, C.; Kruschinski, A.; Chmielewski, M.; Abken, H.; Blankenstein, T.; Charo, J. Efficacy of CAR T-cell therapy in large tumors relies upon stromal targeting by IFNγ. Cancer Res. 2014, 74, 6796–6805. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, Y.; Francisco, N.M.; Zhang, Y.; Wu, M. The application of CAR-T cell therapy in hematological malignancies: Advantages and challenges. Acta Pharm. Sin. B 2018, 8, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Haslauer, T.; Greil, R.; Zaborsky, N.; Geisberger, R. CAR T-Cell Therapy in Hematological Malignancies. Int. J. Mol. Sci. 2021, 22, 8996. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Tao, C.; Li, J.; Tang, J.C.; Chan, A.S.; Zhou, Y. Chimeric antigen receptor T cells applied to solid tumors. Front. Immunol. 2022, 13, 984864. [Google Scholar] [CrossRef] [PubMed]
- Daei Sorkhabi, A.; Mohamed Khosroshahi, L.; Sarkesh, A.; Mardi, A.; Aghebati-Maleki, A.; Aghebati-Maleki, L.; Baradaran, B. The current landscape of CAR T-cell therapy for solid tumors: Mechanisms, research progress, challenges, and counterstrategies. Front. Immunol. 2023, 14, 1113882. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.; Yang, P.; Gu, J.; Wu, H.; Chi, X.; Liu, C.; Wang, Y.; Xue, J.; Qi, W.; Sun, Q.; et al. Recombination of a dual-CAR-modified T lymphocyte to accurately eliminate pancreatic malignancy. J. Hematol. Oncol. 2018, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Tomar, S.; Zhang, J.; Khanal, M.; Hong, J.; Venugopalan, A.; Jiang, Q.; Sengupta, M.; Miettinen, M.; Li, N.; Pastan, I.; et al. Development of Highly Effective Anti-Mesothelin hYP218 Chimeric Antigen Receptor T Cells With Increased Tumor Infiltration and Persistence for Treating Solid Tumors. Mol. Cancer Ther. 2022, 21, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bethigh FoxO1low Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef]
- Inaguma, S.; Lasota, J.; Czapiewski, P.; Langfort, R.; Rys, J.; Szpor, J.; Waloszczyk, P.; Okoń, K.; Biernat, W.; Schrump, D.S.; et al. CD70 expression correlates with a worse prognosis in malignant pleural mesothelioma patients via immune evasion and enhanced invasiveness. J. Pathol. 2020, 250, 205–216. [Google Scholar] [CrossRef]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W.; et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 2019, 10, 4016. [Google Scholar] [CrossRef]
- Kremer, V.; Ligtenberg, M.A.; Zendehdel, R.; Seitz, C.; Duivenvoorden, A.; Wennerberg, E.; Colón, E.; Scherman-Plogell, A.H.; Lundqvist, A. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J. Immunother. Cancer 2017, 5, 73, Erratum in J. Immunother. Cancer 2017, 5, 88. [Google Scholar] [CrossRef]
- Lachota, M.; Zielniok, K.; Palacios, D.; Kanaya, M.; Peena, L.; Hoel, H.J.; Wiiger, M.T.; Kveberg, L.; Hautz, W.; Zagożdżon, R.; et al. Mapping the chemotactic landscape in NK cells reveals subset-specific synergistic migratory responses to dual chemokine receptor ligation. EBioMedicine 2023, 96, 104811. [Google Scholar] [CrossRef]
- Shvartsur, A.; Bonavida, B. Trop2 and its overexpression in cancers: Regulation and clinical/therapeutic implications. Genes Cancer 2015, 6, 84–105. [Google Scholar] [CrossRef] [PubMed]
- Mas, L.; Cros, J.; Svrcek, M.; Van Laethem, J.L.; Emile, J.F.; Rebours, V.; Nicolle, R.; Bachet, J.B. Trop-2 is a ubiquitous and promising target in pancreatic adenocarcinoma. Clin. Res. Hepatol. Gastroenterol. 2023, 47, 102108. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Fang, X.; Tuhin, I.J.; Tan, J.; Ye, J.; Jia, Y.; Xu, N.; Kang, L.; Li, M.; Lou, X.; et al. CAR T cells equipped with a fully human scFv targeting Trop2 can be used to treat pancreatic cancer. J. Cancer Res. Clin. Oncol. 2022, 148, 2261–2274. [Google Scholar] [CrossRef] [PubMed]
- Parriott, G.; Deal, K.; Crean, S.; Richardson, E.; Nylen, E.; Barber, A. T-cells expressing a chimeric-PD1-Dap10-CD3zeta receptor reduce tumour burden in multiple murine syngeneic models of solid cancer. Immunology 2020, 160, 280–294. [Google Scholar] [CrossRef] [PubMed]
- El-Gazzar, A.; Groh, V.; Spies, T. Immunobiology and conflicting roles of the human NKG2D lymphocyte receptor and its ligands in cancer. J. Immunol. 2013, 191, 1509–1515. [Google Scholar] [CrossRef]
- Gao, Y.; Lin, H.; Guo, D.; Cheng, S.; Zhou, Y.; Zhang, L.; Yao, J.; Farooq, M.A.; Ajmal, I.; Duan, Y.; et al. Suppression of 4.1R enhances the potency of NKG2D-CAR T cells against pancreatic carcinoma via activating ERK signaling pathway. Oncogenesis 2021, 10, 62. [Google Scholar] [CrossRef]
- Si, C.; Yuan, L.; Chen, C.; Wang, T.; Kang, Q. The Role of Cytoskeleton Protein 4.1 in Immunotherapy. Int. J. Mol. Sci. 2023, 24, 3777. [Google Scholar] [CrossRef]
- Pietrobon, V.; Todd, L.A.; Goswami, A.; Stefanson, O.; Yang, Z.; Marincola, F. Improving CAR T-Cell Persistence. Int. J. Mol. Sci. 2021, 22, 10828. [Google Scholar] [CrossRef]
- van der Stegen, S.J.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Chen, X.; Madar, A.; Carpenito, C.; McGettigan, S.E.; Frigault, M.J.; Lee, J.; Posey, A.D., Jr.; Scholler, J.; Scholler, N.; et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood 2014, 124, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef] [PubMed]
- Luu, M.; Riester, Z.; Baldrich, A.; Reichardt, N.; Yuille, S.; Busetti, A.; Klein, M.; Wempe, A.; Leister, H.; Raifer, H.; et al. Microbial short-chain fatty acids modulate CD8+ T cell responses and improve adoptive immunotherapy for cancer. Nat. Commun. 2021, 12, 4077. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Sharbeen, G.; Phillips, P.; Australian Pancreatic Cancer Genome Initiative; Ford, C.E. ROR1 and ROR2 expression in pancreatic cancer. BMC Cancer 2021, 21, 1199. [Google Scholar] [CrossRef] [PubMed]
- Sato, O.; Tsuchikawa, T.; Kato, T.; Amaishi, Y.; Okamoto, S.; Mineno, J.; Takeuchi, Y.; Sasaki, K.; Nakamura, T.; Umemoto, K.; et al. Tumor Growth Suppression of Pancreatic Cancer Orthotopic Xenograft Model by CEA-Targeting CAR-T Cells. Cancers 2023, 15, 601. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, D.; Tomiuk, S.; Küster, L.N.; Rawashdeh, W.A.; Henze, J.; Tischler-Höhle, G.; Agorku, D.J.; Brauner, J.; Linnartz, C.; Lock, D.; et al. Identification of CD318, TSPAN8 and CD66c as target candidates for CAR T cell based immunotherapy of pancreatic adenocarcinoma. Nat. Commun. 2021, 12, 1453. [Google Scholar] [CrossRef] [PubMed]
- Raj, D.; Nikolaidi, M.; Garces, I.; Lorizio, D.; Castro, N.M.; Caiafa, S.G.; Moore, K.; Brown, N.F.; Kocher, H.M.; Duan, X.; et al. CEACAM7 Is an Effective Target for CAR T-cell Therapy of Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2021, 27, 1538–1552. [Google Scholar] [CrossRef]
- Jancewicz, I.; Śmiech, M.; Winiarska, M.; Zagozdzon, R.; Wisniewski, P. New CEACAM-targeting 2A3 single-domain antibody-based chimeric antigen receptor T-cells produce anticancer effects in vitro and in vivo. Cancer Immunol. Immunother. 2024, in press.
- Zhou, Y.; Farooq, M.A.; Ajmal, I.; He, C.; Gao, Y.; Guo, D.; Duan, Y.; Jiang, W. Co-expression of IL-4/IL-15-based inverted cytokine receptor in CAR-T cells overcomes IL-4 signaling in immunosuppressive pancreatic tumor microenvironment. Biomed. Pharmacother. 2023, 168, 115740. [Google Scholar] [CrossRef]
- Haas, A.R.; Tanyi, J.L.; O′Hara, M.H.; Gladney, W.L.; Lacey, S.F.; Torigian, D.A.; Soulen, M.C.; Tian, L.; McGarvey, M.; Nelson, A.M.; et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol. Ther. 2019, 27, 1919–1929. [Google Scholar] [CrossRef]
- Beatty, G.L.; O′Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Guo, Y.; Wu, Z.; Feng, K.; Tong, C.; Wang, Y.; Dai, H.; Shi, F.; Yang, Q.; Han, W. Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: A phase I clinical trial. Cytotherapy 2020, 22, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Liu, Y.; Guo, Y.; Qiu, J.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Han, W. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell. 2018, 9, 838–847. [Google Scholar] [CrossRef]
- Botta, G.P.; Becerra, C.R.; Jin, Z.; Kim, D.W.; Zhao, D.; Lenz, H.-J.; Ma, H.; Ween, A.; Acha, P.; Li, Z.; et al. Multicenter phase Ib trial in the U.S. of salvage CT041 CLDN18.2-specific chimeric antigen receptor T-cell therapy for patients with advanced gastric and pancreatic adenocarcinoma. J. Clin. Oncol. 2022, 40, 2538. [Google Scholar] [CrossRef]
- Qi, C.; Gong, J.; Li, J.; Liu, D.; Qin, Y.; Ge, S.; Zhang, M.; Peng, Z.; Zhou, J.; Cao, Y.; et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: Phase 1 trial interim results. Nat. Med. 2022, 28, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; Liu, Y.; Huang, J.; Lv, H.; Luo, C.; et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology 2018, 7, e1440169. [Google Scholar] [CrossRef] [PubMed]
- Yeo, D.; Giardina, C.; Saxena, P.; Rasko, J.E.J. The next wave of cellular immunotherapies in pancreatic cancer. Mol. Ther. Oncolytics 2022, 24, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.H.; Jordan, A.C.; Tooker, E.; Lacey, S.F.; Chang, R.B.; Li, Y.; Venook, A.P.; Tempero, M.; Damon, L.; Fong, L.; et al. Dual Targeting of Mesothelin and CD19 with Chimeric Antigen Receptor-Modified T Cells in Patients with Metastatic Pancreatic Cancer. Mol. Ther. 2020, 28, 2367–2378. [Google Scholar] [CrossRef]
- Xu, J.; Wang, Y.; Shi, J.; Liu, J.; Li, Q.; Chen, L. Combination therapy: A feasibility strategy for CAR-T cell therapy in the treatment of solid tumors. Oncol. Lett. 2018, 16, 2063–2070. [Google Scholar] [CrossRef]
- Yang, C.Y.; Fan, M.H.; Miao, C.H.; Liao, Y.J.; Yuan, R.H.; Liu, C.L. Engineering Chimeric Antigen Receptor T Cells against Immune Checkpoint Inhibitors PD-1/PD-L1 for Treating Pancreatic Cancer. Mol. Ther. Oncolytics 2020, 17, 571–585. [Google Scholar] [CrossRef]
- Mohammed, S.; Sukumaran, S.; Bajgain, P.; Watanabe, N.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Fisher, W.E.; Leen, A.M.; Vera, J.F. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol. Ther. 2017, 25, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Schüßler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 2017, 9, 1183–1197. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Model | Targeted Tumor Antigen | Type of CAR-T Cells | Main Outcomes | Reference |
|---|---|---|---|---|
| In vitro and in vivo | CEA and MSLN | Dual receptor (anti-CEA and anti-MSLN) CAR-T cells | High cytotoxic activity against target cell line (AsPC-1). Inhibition of tumor growth in a mouse model. High IL-2, IL-6, TNF-α, and IFN-γ secretion in mouse model. | Zhang et al. [58] |
| In vitro and in vivo | MSLN | Anti-MSLN CAR-T cells | Cytotoxic activity against target cell lines (NCI-Meso29 and NCI-Meso63). High IL-2, TNF-α, and IFN-γ secretion by NCI-Meso63 cell line. Significant tumor regression in a mouse model. | Tomar et al. [59] |
| In vivo | CEA | IL-18-secreting CAR-T cells | High expression of granzyme and perforin. Increased number of M2 macrophages and NKG2D+ Treg cells. Significant tumor regression. | Chmielewski and Abken [61] |
| In vitro and in vivo | CD70 | Anti-CD70 CAR-T cells expressing CXCR1 and CXCR2 | Cytotoxic activity against target cell line (PANC-1). Decreased expression of exhaustion markers on T cells and enhanced migration of T cells in the tumor in mice inoculated with PANC-1.i720 tumor cells. High granzyme secretion and reduced tumor size. | Jin et al. [62] |
| In vitro and in vivo | Trop2 | Anti-Trop2 CAR-T cells | Cytotoxic activity against target cell lines (ASPC-1, CFPAC-1, BxPC-3). Upregulated IL-17A, IL-2, TNF-α, and IFN- γ production by BxPC-3 cells. Complete tumor regression and increased IFN-γ in mice inoculated with BxPC-3 tumor cells. | Zhu et al. [67] |
| In vitro and in vivo | PD-1 | PD1-Dap10-CD3zeta CAR-T cells | Cytotoxic activity against target cell lines (Pan02 and TGP49). Increased synthesis of IL-2, IL-17, IL-21, TNF-α, IFN-γ, and GM-CSF. Reduced tumor burden in mice inoculated with Pan02 tumor cells. | Parriott et al. [68] |
| In vitro and in vivo | NKG2D | NKG2D CAR-T cells with deleted 4.1R protein | 4.1R deletion in NKG2D CAR-T cells resulted in higher cytotoxicity against target cell lines (SW1990, CAPAN2, and PANC28). Significant tumor regression in mice inoculated with PANC28 tumor cells. | Gao et al. [70] |
| In vitro and in vivo | MSLN | CAR-T cells with ICOS | Increased synthesis of IL-17A, IL-17F, IFN-γ, and IL-22 in vitro. Stronger antitumor response in mice inoculated with Capan-2 tumor cells. Enhanced persistence compared with CD28- or 4-1BB-based CAR-T cells. | Guedan et al. [74,75] |
| In vitro and in vivo | ROR1 | SCFAs—modified CAR-T cells | Increased production of CD25, IFN-γ, and TNF-α in vitro. Significant tumor regression in mice inoculated with ROR1+ Pan02 tumor cells. | Luu et al. [76] |
| Trial Number | Phase | Target | Number of Patients/Treatment | Efficacy | Reference |
|---|---|---|---|---|---|
| NCT02159716 | I | MSLN | 6 PDAC patients with or without cyclophosphamide preconditioning/single CAR-T cell infusion | 3/5 evaluable patients with PD; 2/5 patients with SD | Haas et al. [83] |
| NCT01897415 | I | MSLN | 6 PDAC patients (information about preconditioning not available)/3 CAR-T cells infusion cycles 3 times/week for 3 weeks | 2/3 evaluable patients with SD; 1/3 patients with DP | Beatty et al. [84] |
| NCT01869166 | I | EGFR | 16 PDAC patients with nab-paclitaxel and cyclophosphamide preconditioning/single CAR-T cells infusion | 4/14 evaluable patients with PR; 8/14 patients with SD; 2/14 patients with DP | Liu et al. [85] |
| NCT01935843 | I | HER2 | 11 PDAC patients with nab-paclitaxel and cyclophosphamide preconditioning/1–2 CAR-T cells infusion cycles for 3–5 days | 2/11 evaluable patients with SD | Feng et al. [86] |
| NCT04404595 | Ib | CLDN18.2 | 6 PDAC patients with fludarbine, nab-paclitaxel, and cyclophosphamide preconditioning/single CAR-T cells infusion | 2/5 evaluable patients with SD | Botta et al. [87] |
| NCT03874897 | I | CLDN18.2 | 5 PDAC patients: 3 with fludarbine, nab-paclitaxel, and cyclophosphamide preconditioning, 2 with fludarbine, gemcitabine, and cyclophosphamide preconditioning/single CAR-T cells infusion | 1/5 evaluable patients with PD; 3/5 patients with SD | Oi et al. [88] |
| NCT02541370 | I | CD133 | 23 PDAC patients with nab-paclitaxel and cyclophosphamide preconditioning/2–4 CAR-T cells infusion cycles | 2/7 evaluable patients with PD; 3/7 patients with SD; 2/7 patients with PR | Wang et al. [89] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czaplicka, A.; Lachota, M.; Pączek, L.; Zagożdżon, R.; Kaleta, B. Chimeric Antigen Receptor T Cell Therapy for Pancreatic Cancer: A Review of Current Evidence. Cells 2024, 13, 101. https://doi.org/10.3390/cells13010101
Czaplicka A, Lachota M, Pączek L, Zagożdżon R, Kaleta B. Chimeric Antigen Receptor T Cell Therapy for Pancreatic Cancer: A Review of Current Evidence. Cells. 2024; 13(1):101. https://doi.org/10.3390/cells13010101
Chicago/Turabian StyleCzaplicka, Agata, Mieszko Lachota, Leszek Pączek, Radosław Zagożdżon, and Beata Kaleta. 2024. "Chimeric Antigen Receptor T Cell Therapy for Pancreatic Cancer: A Review of Current Evidence" Cells 13, no. 1: 101. https://doi.org/10.3390/cells13010101
APA StyleCzaplicka, A., Lachota, M., Pączek, L., Zagożdżon, R., & Kaleta, B. (2024). Chimeric Antigen Receptor T Cell Therapy for Pancreatic Cancer: A Review of Current Evidence. Cells, 13(1), 101. https://doi.org/10.3390/cells13010101