
CREB Is Indispensable to KIT Function in Human Skin Mast Cells—A Positive Feedback Loop between CREB and KIT Orchestrates Skin Mast Cell Fate

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Treatments
2.2. Immunoblot Analysis
2.3. BrdU Incorporation
2.4. Flow Cytometry
2.5. Caspase-3 Activity
2.6. Reverse Transcription-Quantitative PCR (RT-qPCR)
2.7. Accell®-Mediated RNA Interference
2.8. Statistics
3. Results
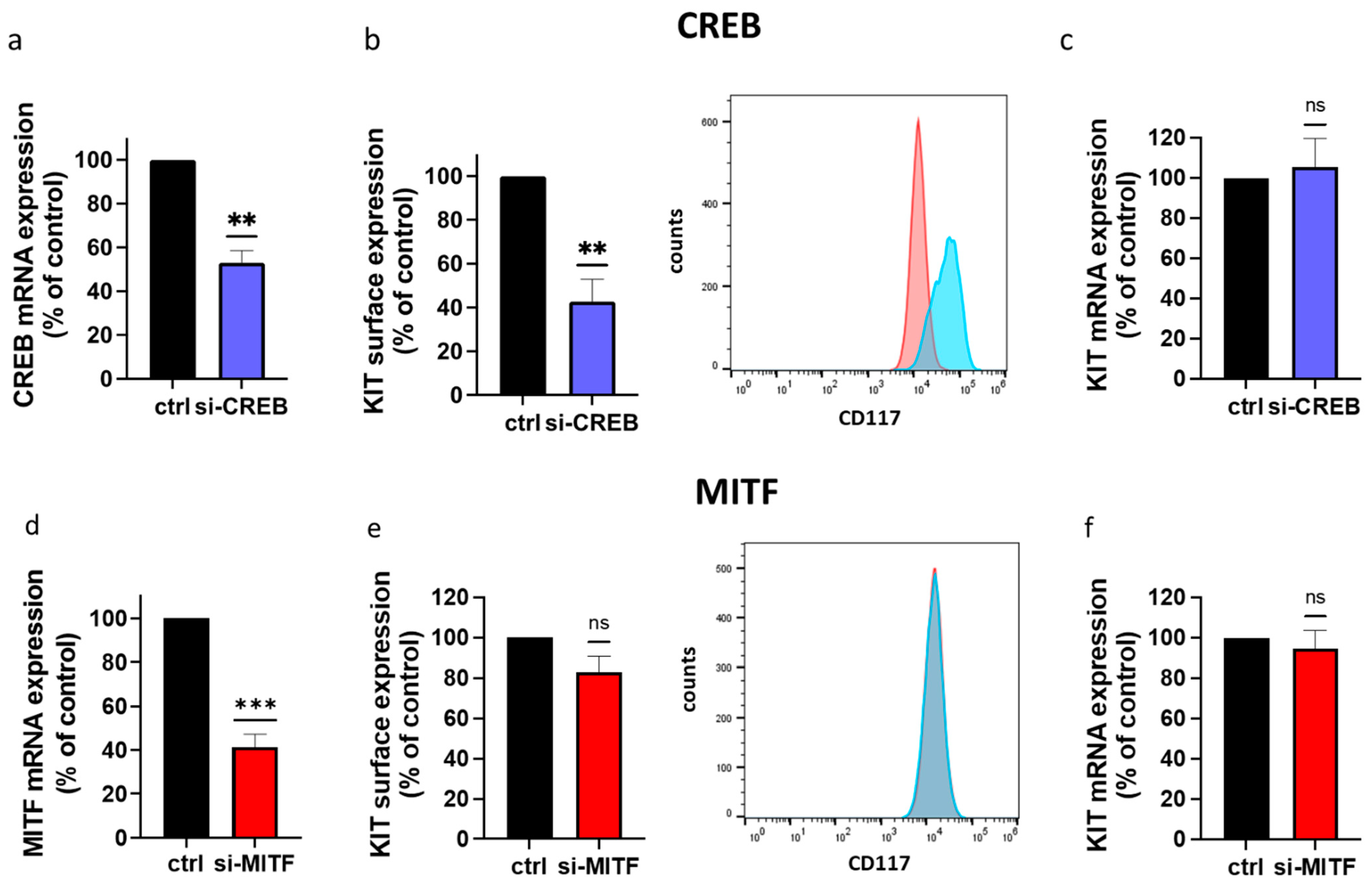
3.1. CREB Is Essential to KIT Expression in Skin MCs
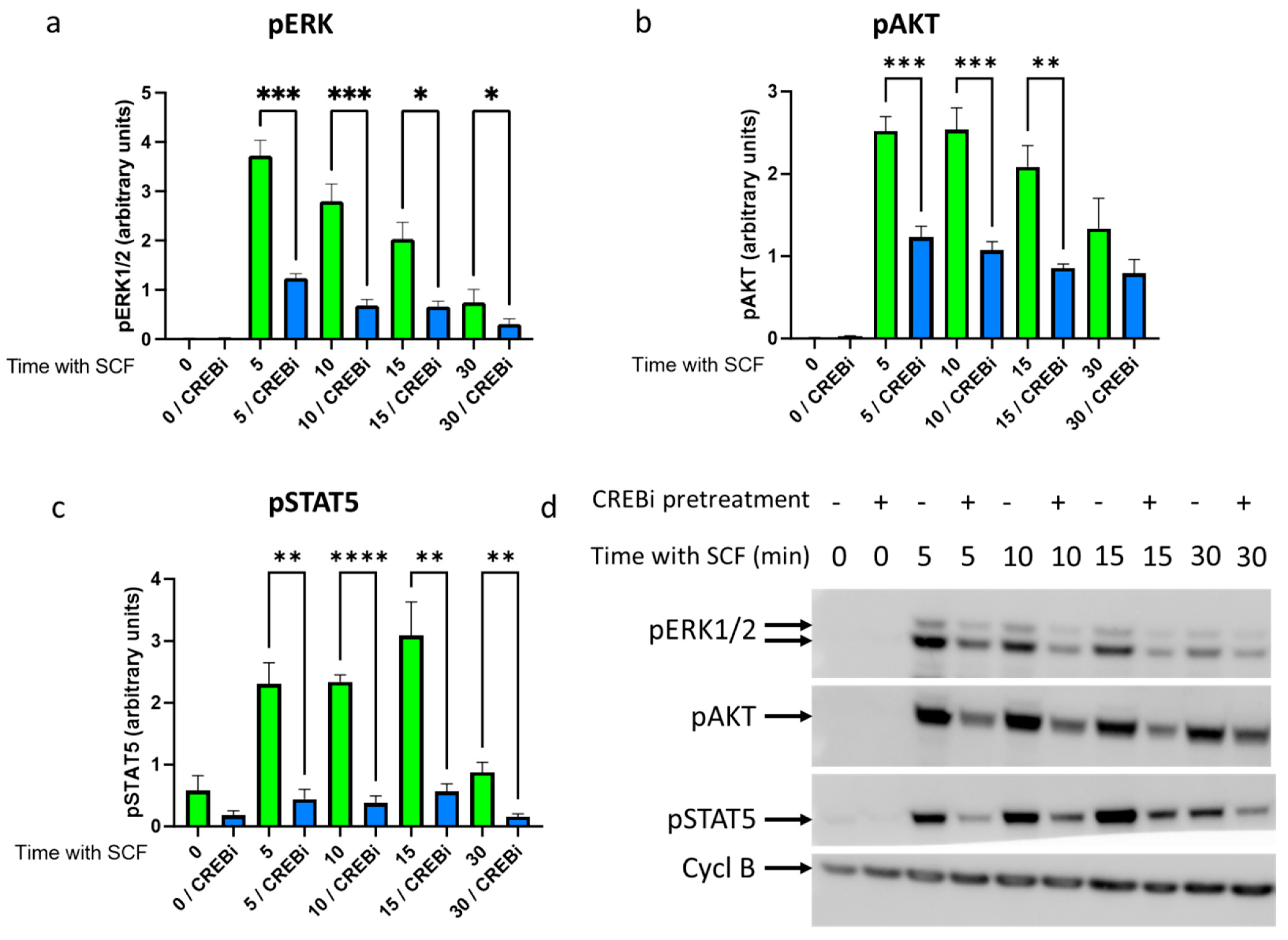
3.2. CREB Maintains KIT Functionality–Pretreatment with CREB Inhibitor Compromises KIT Signaling
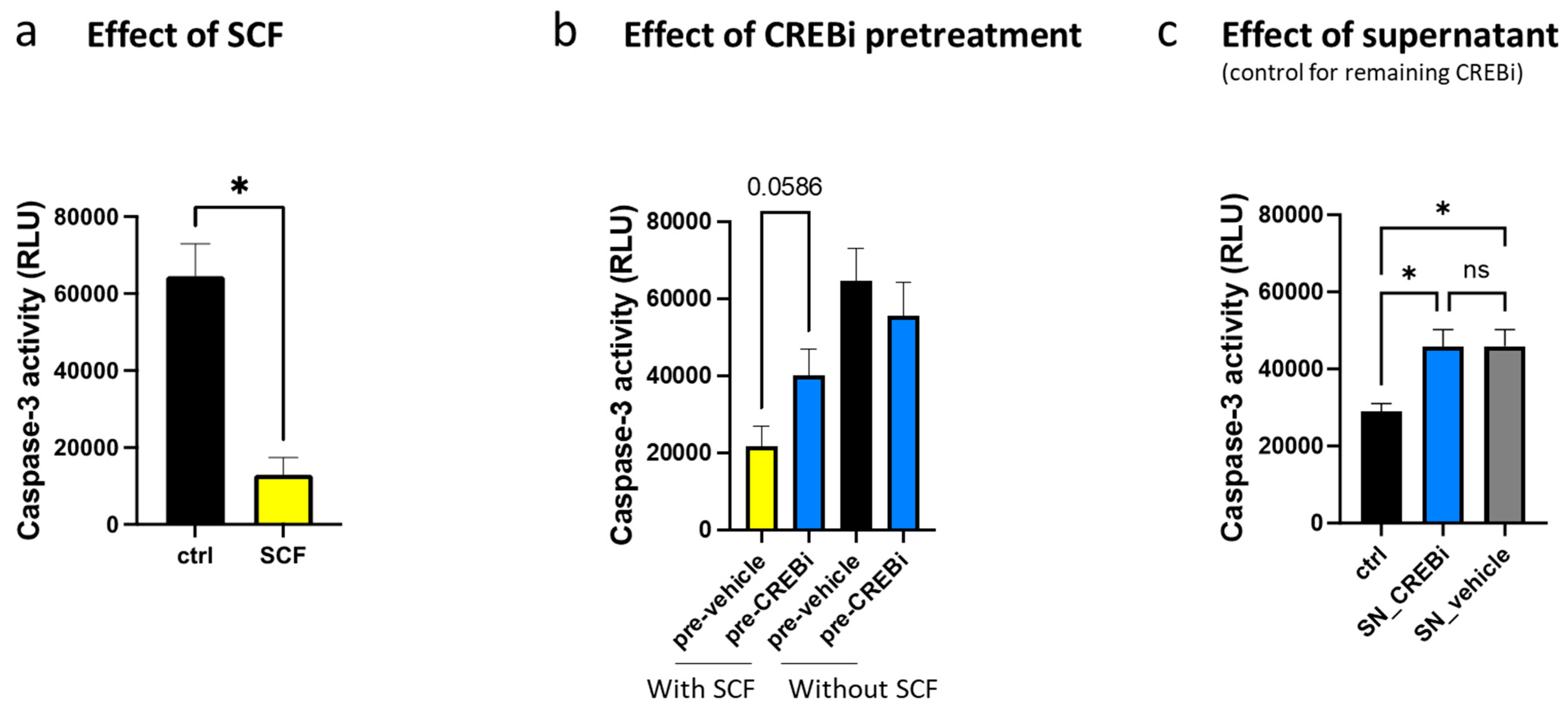
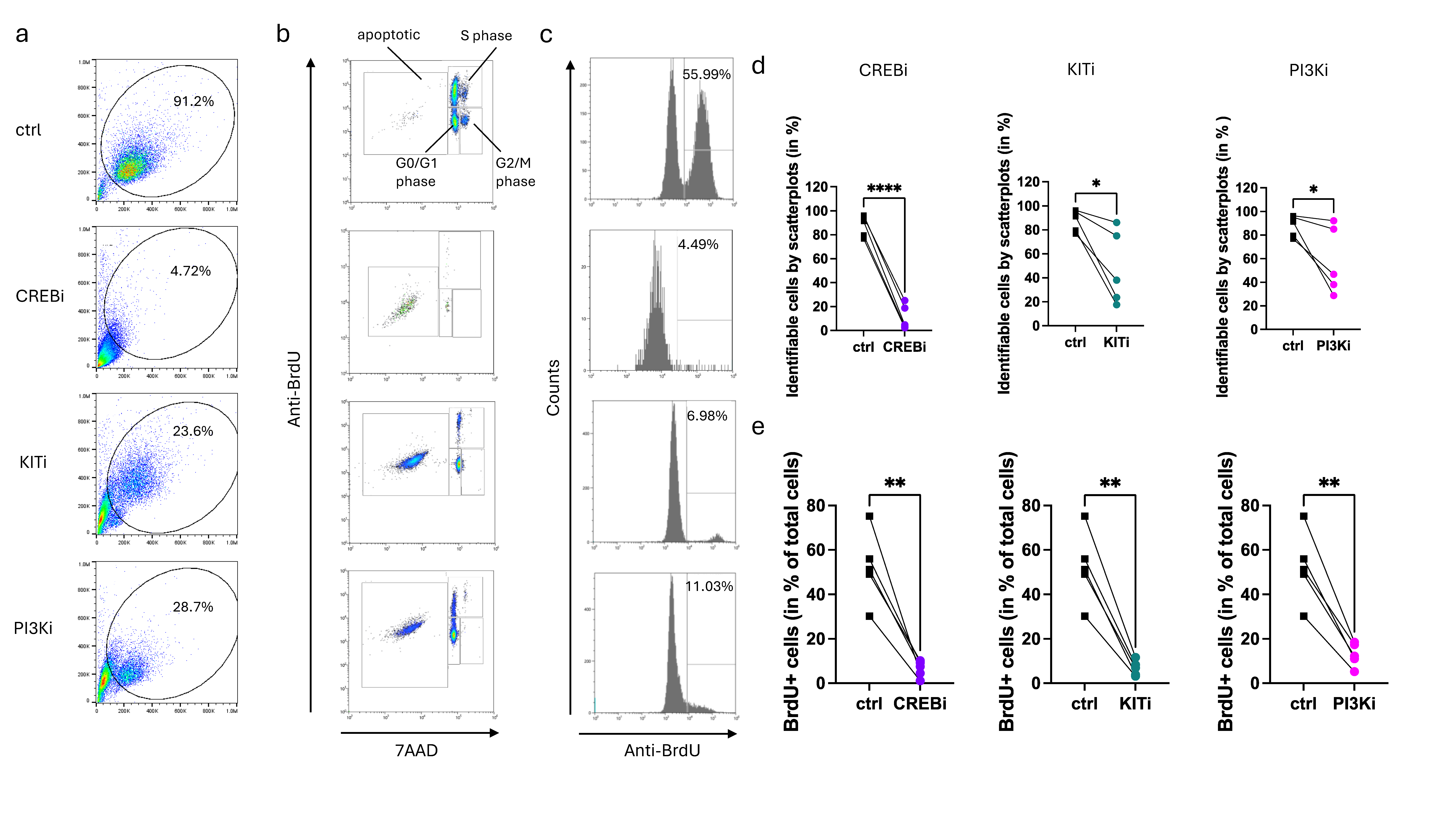
3.3. CREB Is Essential for KIT-Dependent Anti-Apoptosis and Cell Cycle Progression
3.4. Prolonged Inhibition of CREB Leads to MC Elimination, in a Way Exceeding the Inhibition of KIT Itself
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Metcalfe, D.D. Mast cells and mastocytosis. Blood 2008, 112, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Okayama, Y.; Kawakami, T. Development, migration, and survival of mast cells. Immunol. Res. 2006, 34, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Akin, C.; Metcalfe, D.D. The biology of Kit in disease and the application of pharmacogenetics. J. Allergy Clin. Immunol. 2004, 114, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Lennartsson, J.; Ronnstrand, L. Stem cell factor receptor/c-Kit: From basic science to clinical implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef] [PubMed]
- Cruse, G.; Metcalfe, D.D.; Olivera, A. Functional deregulation of KIT: Link to mast cell proliferative diseases and other neoplasms. Immunol. Allergy Clin. N. Am. 2014, 34, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, A.; Bahri, R.; Bulfone-Paus, S.; Arkwright, P.D. Mast cell disorders: From infancy to maturity. Allergy 2019, 74, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Lennartsson, J.; Ronnstrand, L. The stem cell factor receptor/c-Kit as a drug target in cancer. Curr. Cancer Drug Targets 2006, 6, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Franke, K.; Kirchner, M.; Mertins, P.; Zuberbier, T.; Babina, M. The SCF/KIT axis in human mast cells: Capicua acts as potent KIT repressor and ERK predominates PI3K. Allergy 2022, 77, 3337–3349. [Google Scholar] [CrossRef]
- Zhang, X.; Odom, D.T.; Koo, S.H.; Conkright, M.D.; Canettieri, G.; Best, J.; Chen, H.; Jenner, R.; Herbolsheimer, E.; Jacobsen, E.; et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 4459–4464. [Google Scholar] [CrossRef]
- Mora-Garcia, P.; Cheng, J.; Crans-Vargas, H.N.; Countouriotis, A.; Shankar, D.; Sakamoto, K.M. Transcriptional regulators and myelopoiesis: The role of serum response factor and CREB as targets of cytokine signaling. Stem Cells 2003, 21, 123–130. [Google Scholar] [CrossRef]
- Lonze, B.E.; Ginty, D.D. Function and regulation of CREB family transcription factors in the nervous system. Neuron 2002, 35, 605–623. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, M.; Delghandi, M.P.; Moens, U. What turns CREB on? Cell. Signal. 2004, 16, 1211–1227. [Google Scholar] [CrossRef] [PubMed]
- Franke, K.; Bal, G.; Li, Z.; Zuberbier, T.; Babina, M. CREB Is Activated by the SCF/KIT Axis in a Partially ERK-Dependent Manner and Orchestrates Survival and the Induction of Immediate Early Genes in Human Skin Mast Cells. Int. J. Mol. Sci. 2023, 24, 4135. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; d’Amati, A. Hematopoiesis and Mast Cell Development. Int. J. Mol. Sci. 2023, 24, 10679. [Google Scholar] [CrossRef] [PubMed]
- St John, A.L.; Rathore, A.P.S.; Ginhoux, F. New perspectives on the origins and heterogeneity of mast cells. Nat. Rev. Immunol. 2023, 23, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Hamey, F.K.; Lau, W.W.Y.; Kucinski, I.; Wang, X.; Diamanti, E.; Wilson, N.K.; Gottgens, B.; Dahlin, J.S. Single-cell molecular profiling provides a high-resolution map of basophil and mast cell development. Allergy 2021, 76, 1731–1742. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Oboki, K.; Ito, A. Development of mast cells. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2007, 83, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Nishiyama, C.; Ogawa, H.; Okumura, K. GATA2 and Sp1 positively regulate the c-kit promoter in mast cells. J. Immunol. 2010, 185, 4252–4260. [Google Scholar] [CrossRef]
- Debaize, L.; Jakobczyk, H.; Avner, S.; Gaudichon, J.; Rio, A.G.; Serandour, A.A.; Dorsheimer, L.; Chalmel, F.; Carroll, J.S.; Zornig, M.; et al. Interplay between transcription regulators RUNX1 and FUBP1 activates an enhancer of the oncogene c-KIT and amplifies cell proliferation. Nucleic Acids Res. 2018, 46, 11214–11228. [Google Scholar] [CrossRef]
- Lecuyer, E.; Herblot, S.; Saint-Denis, M.; Martin, R.; Begley, C.G.; Porcher, C.; Orkin, S.H.; Hoang, T. The SCL complex regulates c-kit expression in hematopoietic cells through functional interaction with Sp1. Blood 2002, 100, 2430–2440. [Google Scholar] [CrossRef]
- Da Ros, S.; Nicoletto, G.; Rigo, R.; Ceschi, S.; Zorzan, E.; Dacasto, M.; Giantin, M.; Sissi, C. G-Quadruplex Modulation of SP1 Functional Binding Sites at the KIT Proximal Promoter. Int. J. Mol. Sci. 2020, 22, 329. [Google Scholar] [CrossRef] [PubMed]
- Calero-Nieto, F.J.; Ng, F.S.; Wilson, N.K.; Hannah, R.; Moignard, V.; Leal-Cervantes, A.I.; Jimenez-Madrid, I.; Diamanti, E.; Wernisch, L.; Gottgens, B. Key regulators control distinct transcriptional programmes in blood progenitor and mast cells. EMBO J. 2014, 33, 1212–1226. [Google Scholar] [CrossRef]
- Poliseno, L.; Tuccoli, A.; Mariani, L.; Evangelista, M.; Citti, L.; Woods, K.; Mercatanti, A.; Hammond, S.; Rainaldi, G. MicroRNAs modulate the angiogenic properties of HUVECs. Blood 2006, 108, 3068–3071. [Google Scholar] [CrossRef] [PubMed]
- Babina, M.; Rex, C.; Guhl, S.; Thienemann, F.; Artuc, M.; Henz, B.M.; Zuberbier, T. Baseline and stimulated turnover of cell surface c-Kit expression in different types of human mast cells. Exp. Dermatol. 2006, 15, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Bayle, J.; Letard, S.; Frank, R.; Dubreuil, P.; De Sepulveda, P. Suppressor of cytokine signaling 6 associates with KIT and regulates KIT receptor signaling. J. Biol. Chem. 2004, 279, 12249–12259. [Google Scholar] [CrossRef]
- Zadjali, F.; Pike, A.C.; Vesterlund, M.; Sun, J.; Wu, C.; Li, S.S.; Ronnstrand, L.; Knapp, S.; Bullock, A.N.; Flores-Morales, A. Structural basis for c-KIT inhibition by the suppressor of cytokine signaling 6 (SOCS6) ubiquitin ligase. J. Biol. Chem. 2011, 286, 480–490. [Google Scholar] [CrossRef]
- Franke, K.; Bal, G.; Li, Z.; Zuberbier, T.; Babina, M. Clorfl86/RHEX Is a Negative Regulator of SCF/KIT Signaling in Human Skin Mast Cells. Cells 2023, 12, 1306. [Google Scholar] [CrossRef]
- Espinosa-Riquer, Z.P.; Segura-Villalobos, D.; Ramirez-Moreno, I.G.; Perez Rodriguez, M.J.; Lamas, M.; Gonzalez-Espinosa, C. Signal Transduction Pathways Activated by Innate Immunity in Mast Cells: Translating Sensing of Changes into Specific Responses. Cells 2020, 9, 2411. [Google Scholar] [CrossRef]
- Katsoulis-Dimitriou, K.; Kotrba, J.; Voss, M.; Dudeck, J.; Dudeck, A. Mast Cell Functions Linking Innate Sensing to Adaptive Immunity. Cells 2020, 9, 2538. [Google Scholar] [CrossRef]
- Dudeck, A.; Koberle, M.; Goldmann, O.; Meyer, N.; Dudeck, J.; Lemmens, S.; Rohde, M.; Roldan, N.G.; Dietze-Schwonberg, K.; Orinska, Z.; et al. Mast cells as protectors of health. J. Allergy Clin. Immunol. 2019, 144, S4–S18. [Google Scholar] [CrossRef]
- Galli, S.J.; Metz, M.; Starkl, P.; Marichal, T.; Tsai, M. Mast cells and IgE in defense against lethality of venoms: Possible “benefit” of allergy. Allergo J. Int. 2020, 29, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Eyerich, S.; Metz, M.; Bossios, A.; Eyerich, K. New biological treatments for asthma and skin allergies. Allergy 2020, 75, 546–560. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Ando, T.; Kimura, M.; Wilson, B.S.; Kawakami, Y. Mast cells in atopic dermatitis. Curr. Opin. Immunol. 2009, 21, 666–678. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Nograles, K.E.; Krueger, J.G. Contrasting pathogenesis of atopic dermatitis and psoriasis--part II: Immune cell subsets and therapeutic concepts. J. Allergy Clin. Immunol. 2011, 127, 1420–1432. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Babina, M. MRGPRX2 signals its importance in cutaneous mast cell biology: Does MRGPRX2 connect mast cells and atopic dermatitis? Exp. Dermatol. 2020, 29, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Y.; Chen, K.; Zhang, J.A. Mast cells as important regulators in the development of psoriasis. Front. Immunol. 2022, 13, 1022986. [Google Scholar] [CrossRef] [PubMed]
- Gaudenzio, N.; Marichal, T.; Galli, S.J.; Reber, L.L. Genetic and Imaging Approaches Reveal Pro-Inflammatory and Immunoregulatory Roles of Mast Cells in Contact Hypersensitivity. Front. Immunol. 2018, 9, 1275. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.J.; Hao, D.; Wen, X.; Du, D.; He, G.; Jiang, X. The Theranostics Role of Mast Cells in the Pathophysiology of Rosacea. Front. Med. 2019, 6, 324. [Google Scholar] [CrossRef]
- Numata, T.; Harada, K.; Nakae, S. Roles of Mast Cells in Cutaneous Diseases. Front. Immunol. 2022, 13, 923495. [Google Scholar] [CrossRef]
- Parente, R.; Giudice, V.; Cardamone, C.; Serio, B.; Selleri, C.; Triggiani, M. Secretory and Membrane-Associated Biomarkers of Mast Cell Activation and Proliferation. Int. J. Mol. Sci. 2023, 24, 7071. [Google Scholar] [CrossRef]
- Afrin, L.B.; Ackerley, M.B.; Bluestein, L.S.; Brewer, J.H.; Brook, J.B.; Buchanan, A.D.; Cuni, J.R.; Davey, W.P.; Dempsey, T.T.; Dorff, S.R.; et al. Diagnosis of mast cell activation syndrome: A global “consensus-2”. Diagnosis 2021, 8, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Steinhoff, M.; Neisius, U.; Ikoma, A.; Fartasch, M.; Heyer, G.; Skov, P.S.; Luger, T.A.; Schmelz, M. Proteinase-activated receptor-2 mediates itch: A novel pathway for pruritus in human skin. J. Neurosci. 2003, 23, 6176–6180. [Google Scholar] [CrossRef] [PubMed]
- Tey, H.L.; Yosipovitch, G. Targeted treatment of pruritus: A look into the future. Br. J. Dermatol. 2011, 165, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Corbiere, A.; Loste, A.; Gaudenzio, N. MRGPRX2 sensing of cationic compounds—A bridge between nociception and skin diseases? Exp. Dermatol. 2021, 30, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Aich, A.; Afrin, L.B.; Gupta, K. Mast Cell-Mediated Mechanisms of Nociception. Int. J. Mol. Sci. 2015, 16, 29069–29092. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, H.; Kolkhir, P.; Babina, M.; Dull, M.; Frischbutter, S.; Fok, J.S.; Jiao, Q.; Metz, M.; Scheffel, J.; Wolf, K.; et al. Mas-related G protein-coupled receptor X2 and its activators in dermatologic allergies. J. Allergy Clin. Immunol. 2021, 147, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Falduto, G.H.; Pfeiffer, A.; Luker, A.; Metcalfe, D.D.; Olivera, A. Emerging mechanisms contributing to mast cell-mediated pathophysiology with therapeutic implications. Pharmacol. Ther. 2021, 220, 107718. [Google Scholar] [CrossRef]
- Voss, M.; Kotrba, J.; Gaffal, E.; Katsoulis-Dimitriou, K.; Dudeck, A. Mast Cells in the Skin: Defenders of Integrity or Offenders in Inflammation? Int. J. Mol. Sci. 2021, 22, 4589. [Google Scholar] [CrossRef]
- Babina, M. The pseudo-allergic/neurogenic route of mast cell activation via MRGPRX2: Discovery, functional programs, regulation, relevance to disease, and relation with allergic stimulation. Itch 2020, 5, e32. [Google Scholar] [CrossRef]
- Terhorst-Molawi, D.; Hawro, T.; Grekowitz, E.; Kiefer, L.; Merchant, K.; Alvarado, D.; Thomas, L.J.; Hawthorne, T.; Crowley, E.; Heath-Chiozzi, M.; et al. Anti-KIT antibody, barzolvolimab, reduces skin mast cells and disease activity in chronic inducible urticaria. Allergy 2023, 78, 1269–1279. [Google Scholar] [CrossRef]
- Costa, J.J.; Demetri, G.D.; Harrist, T.J.; Dvorak, A.M.; Hayes, D.F.; Merica, E.A.; Menchaca, D.M.; Gringeri, A.J.; Schwartz, L.B.; Galli, S.J. Recombinant human stem cell factor (kit ligand) promotes human mast cell and melanocyte hyperplasia and functional activation in vivo. J. Exp. Med. 1996, 183, 2681–2686. [Google Scholar] [CrossRef] [PubMed]
- Franke, K.; Wang, Z.; Zuberbier, T.; Babina, M. Cytokines Stimulated by IL-33 in Human Skin Mast Cells: Involvement of NF-kappaB and p38 at Distinct Levels and Potent Co-Operation with FcepsilonRI and MRGPRX2. Int. J. Mol. Sci. 2021, 22, 3580. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Guhl, S.; Franke, K.; Artuc, M.; Zuberbier, T.; Babina, M. IL-33 and MRGPRX2-Triggered Activation of Human Skin Mast Cells-Elimination of Receptor Expression on Chronic Exposure, but Reinforced Degranulation on Acute Priming. Cells 2019, 8, 341. [Google Scholar] [CrossRef]
- Babina, M.; Wang, Z.; Franke, K.; Guhl, S.; Artuc, M.; Zuberbier, T. Yin-Yang of IL-33 in Human Skin Mast Cells: Reduced Degranulation, but Augmented Histamine Synthesis through p38 Activation. J. Investig. Dermatol. 2019, 139, 1516–1525. [Google Scholar] [CrossRef]
- Hazzan, T.; Eberle, J.; Worm, M.; Babina, M. Apoptotic resistance of human skin mast cells is mediated by Mcl-1. Cell Death Discov. 2017, 3, 17048. [Google Scholar] [CrossRef] [PubMed]
- Babina, M.; Wang, Z.; Artuc, M.; Guhl, S.; Zuberbier, T. MRGPRX2 is negatively targeted by SCF and IL-4 to diminish pseudo-allergic stimulation of skin mast cells in culture. Exp. Dermatol. 2018, 27, 1298–1303. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, S.; Willmes, D.M.; Nassiri, M.; Babina, M.; Worm, M. PGE(2) deficiency predisposes to anaphylaxis by causing mast cell hyperresponsiveness. J. Allergy Clin. Immunol. 2020, 146, 1387–1396.e1313. [Google Scholar] [CrossRef]
- Babina, M.; Artuc, M.; Guhl, S.; Zuberbier, T. Retinoic Acid Negatively Impacts Proliferation and MC(TC) Specific Attributes of Human Skin Derived Mast Cells, but Reinforces Allergic Stimulability. Int. J. Mol. Sci. 2017, 18, 525. [Google Scholar] [CrossRef]
- Guhl, S.; Neou, A.; Artuc, M.; Zuberbier, T.; Babina, M. Skin mast cells develop non-synchronized changes in typical lineage characteristics upon culture. Exp. Dermatol. 2014, 23, 933–935. [Google Scholar] [CrossRef]
- Guhl, S.; Artuc, M.; Neou, A.; Babina, M.; Zuberbier, T. Long-term cultured human skin mast cells are suitable for pharmacological studies of anti-allergic drugs due to high responsiveness to FcepsilonRI cross-linking. Biosci. Biotechnol. Biochem. 2011, 75, 382–384. [Google Scholar] [CrossRef]
- Xie, F.; Li, B.X.; Kassenbrock, A.; Xue, C.; Wang, X.; Qian, D.Z.; Sears, R.C.; Xiao, X. Identification of a Potent Inhibitor of CREB-Mediated Gene Transcription with Efficacious in Vivo Anticancer Activity. J. Med. Chem. 2015, 58, 5075–5087. [Google Scholar] [CrossRef] [PubMed]
- Li, B.X.; Gardner, R.; Xue, C.; Qian, D.Z.; Xie, F.; Thomas, G.; Kazmierczak, S.C.; Habecker, B.A.; Xiao, X. Systemic Inhibition of CREB is Well-tolerated in vivo. Sci. Rep. 2016, 6, 34513. [Google Scholar] [CrossRef]
- Probst, S.; Scharner, B.; McErlean, R.; Lee, W.K.; Thevenod, F. Inverse Regulation of Lipocalin-2/24p3 Receptor/SLC22A17 and Lipocalin-2 Expression by Tonicity, NFAT5/TonEBP and Arginine Vasopressin in Mouse Cortical Collecting Duct Cells mCCD(cl.1): Implications for Osmotolerance. Int. J. Mol. Sci. 2019, 20, 5398. [Google Scholar] [CrossRef] [PubMed]
- Hasel, K.W.; Glass, J.R.; Godbout, M.; Sutcliffe, J.G. An endoplasmic reticulum-specific cyclophilin. Mol. Cell Biol. 1991, 11, 3484–3491. [Google Scholar] [CrossRef] [PubMed]
- Hazzan, T.; Eberle, J.; Worm, M.; Babina, M. Thymic Stromal Lymphopoietin Interferes with the Apoptosis of Human Skin Mast Cells by a Dual Strategy Involving STAT5/Mcl-1 and JNK/Bcl-x(L). Cells 2019, 8, 829. [Google Scholar] [CrossRef] [PubMed]
- Hazzan, T.; Guhl, S.; Artuc, M.; Franke, K.; Worm, M.; Zuberbier, T.; Babina, M. An efficient method for gene knock-down by RNA interference in human skin mast cells. Exp. Dermatol. 2017, 26, 1136–1139. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Z.; Bal, G.; Franke, K.; Zuberbier, T.; Babina, M. beta-arrestin-1 and beta-arrestin-2 Restrain MRGPRX2-Triggered Degranulation and ERK1/2 Activation in Human Skin Mast Cells. Front. Allergy 2022, 3, 930233. [Google Scholar] [CrossRef]
- Babina, M.; Wang, Z.; Roy, S.; Guhl, S.; Franke, K.; Artuc, M.; Ali, H.; Zuberbier, T. MRGPRX2 Is the Codeine Receptor of Human Skin Mast Cells: Desensitization through beta-Arrestin and Lack of Correlation with the FcepsilonRI Pathway. J. Investig. Dermatol. 2021, 141, 1286–1296.e1284. [Google Scholar] [CrossRef]
- Babina, M.; Wang, Z.; Franke, K.; Zuberbier, T. Thymic Stromal Lymphopoietin Promotes MRGPRX2-Triggered Degranulation of Skin Mast Cells in a STAT5-Dependent Manner with Further Support from JNK. Cells 2021, 10, 102. [Google Scholar] [CrossRef]
- Guo, Y.; Olle, L.; Proano-Perez, E.; Aparicio, C.; Guerrero, M.; Munoz-Cano, R.; Martin, M. MRGPRX2 signaling involves the Lysyl-tRNA synthetase and MITF pathway. Front. Immunol. 2023, 14, 1154108. [Google Scholar] [CrossRef]
- Babina, M.; Guhl, S.; Artuc, M.; Trivedi, N.N.; Zuberbier, T. Phenotypic variability in human skin mast cells. Exp. Dermatol. 2016, 25, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Motakis, E.; Guhl, S.; Ishizu, Y.; Itoh, M.; Kawaji, H.; de Hoon, M.; Lassmann, T.; Carninci, P.; Hayashizaki, Y.; Zuberbier, T.; et al. Redefinition of the human mast cell transcriptome by deep-CAGE sequencing. Blood 2014, 123, e58–e67. [Google Scholar] [CrossRef] [PubMed]
- Babina, M.; Guhl, S.; Starke, A.; Kirchhof, L.; Zuberbier, T.; Henz, B.M. Comparative cytokine profile of human skin mast cells from two compartments--strong resemblance with monocytes at baseline but induction of IL-5 by IL-4 priming. J. Leukoc. Biol. 2004, 75, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, D.; Maurer, M.; Gedrich, R.; Seibel, S.B.; Murphy, M.B.; Crew, L.; Goldstein, J.; Crocker, A.; Vitale, L.A.; Morani, P.A.; et al. Anti-KIT monoclonal antibody CDX-0159 induces profound and durable mast cell suppression in a healthy volunteer study. Allergy 2022, 77, 2393–2403. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Morii, E.; Ogihara, H.; Lee, Y.M.; Jippo, T.; Adachi, S.; Maeyama, K.; Kim, H.M.; Kitamura, Y. Different effect of various mutant MITF encoded by mi, Mior, or Miwh allele on phenotype of murine mast cells. Blood 1999, 93, 4179–4186. [Google Scholar] [CrossRef] [PubMed]
- Shahlaee, A.H.; Brandal, S.; Lee, Y.N.; Jie, C.; Takemoto, C.M. Distinct and shared transcriptomes are regulated by microphthalmia-associated transcription factor isoforms in mast cells. J. Immunol. 2007, 178, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Candelas, E.; Ainsua-Enrich, E.; Navines-Ferrer, A.; Rodrigues, P.; Garcia-Valverde, A.; Bazzocco, S.; Macaya, I.; Arribas, J.; Serrano, C.; Sayos, J.; et al. Silencing of adaptor protein SH3BP2 reduces KIT/PDGFRA receptors expression and impairs gastrointestinal stromal tumors growth. Mol. Oncol. 2018, 12, 1383–1397. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.M.; Akin, C.; Gilfillan, A.M. Pharmacological targeting of the KIT growth factor receptor: A therapeutic consideration for mast cell disorders. Br. J. Pharmacol. 2008, 154, 1572–1582. [Google Scholar] [CrossRef]
- The FANTOM Consortium and the RIKEN PMI and CLST (DGT). A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef]
- Noguchi, S.; Arakawa, T.; Fukuda, S.; Furuno, M.; Hasegawa, A.; Hori, F.; Ishikawa-Kato, S.; Kaida, K.; Kaiho, A.; Kanamori-Katayama, M.; et al. FANTOM5 CAGE profiles of human and mouse samples. Sci. Data 2017, 4, 170112. [Google Scholar] [CrossRef]
- Grabbe, J.; Welker, P.; Rosenbach, T.; Nurnberg, W.; Kruger-Krasagakes, S.; Artuc, M.; Fiebiger, E.; Henz, B.M. Release of stem cell factor from a human keratinocyte line, HaCaT, is increased in differentiating versus proliferating cells. J. Investig. Dermatol. 1996, 107, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Mascarenhas, N.; Eckmann, L.; Miyamoto, Y.; Sun, X.; Kawakami, T.; Di Nardo, A. Skin microbiome promotes mast cell maturation by triggering stem cell factor production in keratinocytes. J. Allergy Clin. Immunol. 2017, 139, 1205–1216.e1206. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Shibata, T.; Kwon, S.; Park, T.J.; Kang, H.Y. Ultraviolet-irradiated endothelial cells secrete stem cell factor and induce epidermal pigmentation. Sci. Rep. 2018, 8, 4235. [Google Scholar] [CrossRef] [PubMed]
- Meininger, C.J.; Brightman, S.E.; Kelly, K.A.; Zetter, B.R. Increased stem cell factor release by hemangioma-derived endothelial cells. Lab. Investig. 1995, 72, 166–173. [Google Scholar]
- Yamamoto, T.; Katayama, I.; Nishioka, K. Impaired expression of stem cell factor in dermatofibroma fibroblasts. Acta Derm. Venereol. 1996, 76, 257–259. [Google Scholar] [CrossRef]
- Shin, J.; Kim, J.H.; Kim, E.K. Repeated exposure of human fibroblasts to UVR induces secretion of stem cell factor and senescence. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1577–1580. [Google Scholar] [CrossRef]
- Hibberts, N.A.; Messenger, A.G.; Randall, V.A. Dermal papilla cells derived from beard hair follicles secrete more stem cell factor (SCF) in culture than scalp cells or dermal fibroblasts. Biochem. Biophys. Res. Commun. 1996, 222, 401–405. [Google Scholar] [CrossRef]
- Okayama, Y.; Petit-Frere, C.; Kassel, O.; Semper, A.; Quint, D.; Tunon-de-Lara, M.J.; Bradding, P.; Holgate, S.T.; Church, M.K. IgE-dependent expression of mRNA for IL-4 and IL-5 in human lung mast cells. J. Immunol. 1995, 155, 1796–1808. [Google Scholar] [CrossRef]
- Ravindran, A.; Ronnberg, E.; Dahlin, J.S.; Mazzurana, L.; Safholm, J.; Orre, A.C.; Al-Ameri, M.; Peachell, P.; Adner, M.; Dahlen, S.E.; et al. An Optimized Protocol for the Isolation and Functional Analysis of Human Lung Mast Cells. Front. Immunol. 2018, 9, 2193. [Google Scholar] [CrossRef]
- Ronnberg, E.; Boey, D.Z.H.; Ravindran, A.; Safholm, J.; Orre, A.C.; Al-Ameri, M.; Adner, M.; Dahlen, S.E.; Dahlin, J.S.; Nilsson, G. Immunoprofiling Reveals Novel Mast Cell Receptors and the Continuous Nature of Human Lung Mast Cell Heterogeneity. Front. Immunol. 2021, 12, 804812. [Google Scholar] [CrossRef]
- Bischoff, S.C.; Schwengberg, S.; Raab, R.; Manns, M.P. Functional properties of human intestinal mast cells cultured in a new culture system: Enhancement of IgE receptor-dependent mediator release and response to stem cell factor. J. Immunol. 1997, 159, 5560–5567. [Google Scholar] [CrossRef]
- Bischoff, S.C.; Sellge, G.; Lorentz, A.; Sebald, W.; Raab, R.; Manns, M.P. IL-4 enhances proliferation and mediator release in mature human mast cells. Proc. Natl. Acad. Sci. USA 1999, 96, 8080–8085. [Google Scholar] [CrossRef] [PubMed]
- Babina, M.; Guhl, S.; Artuc, M.; Zuberbier, T. IL-4 and human skin mast cells revisited: Reinforcement of a pro-allergic phenotype upon prolonged exposure. Arch. Dermatol. Res. 2016, 308, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Oskeritzian, C.A.; Zhao, W.; Pozez, A.L.; Cohen, N.M.; Grimes, M.; Schwartz, L.B. Neutralizing endogenous IL-6 renders mast cells of the MCT type from lung, but not the MCTC type from skin and lung, susceptible to human recombinant IL-4-induced apoptosis. J. Immunol. 2004, 172, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Bawazir, M.; Amponnawarat, A.; Hui, Y.; Oskeritzian, C.A.; Ali, H. Inhibition of MRGPRX2 but not FcepsilonRI or MrgprB2-mediated mast cell degranulation by a small molecule inverse receptor agonist. Front. Immunol. 2022, 13, 1033794. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Li, Y.; Guan, X.; Mohammed, Z.; Gomez, G.; Hui, Y.; Zhao, D.; Oskeritzian, C.A.; Huang, H. IL-33 priming and antigenic stimulation synergistically promote the transcription of proinflammatory cytokine and chemokine genes in human skin mast cells. BMC Genom. 2023, 24, 592. [Google Scholar] [CrossRef] [PubMed]
- McHale, C.; Mohammed, Z.; Gomez, G. Human Skin-Derived Mast Cells Spontaneously Secrete Several Angiogenesis-Related Factors. Front. Immunol. 2019, 10, 1445. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Giuditta, A. Role of a transcription factor (CREB) in memory processes. Riv. Biol. 1997, 90, 371–384. [Google Scholar]
- Chowdhury, M.A.R.; An, J.; Jeong, S. The Pleiotropic Face of CREB Family Transcription Factors. Mol. Cells 2023, 46, 399–413. [Google Scholar] [CrossRef]
- Kinjo, K.; Sandoval, S.; Sakamoto, K.M.; Shankar, D.B. The role of CREB as a proto-oncogene in hematopoiesis. Cell Cycle 2005, 4, 1134–1135. [Google Scholar] [CrossRef]
- Sapio, L.; Salzillo, A.; Ragone, A.; Illiano, M.; Spina, A.; Naviglio, S. Targeting CREB in Cancer Therapy: A Key Candidate or One of Many? An Update. Cancers 2020, 12, 3166. [Google Scholar] [CrossRef]
- Babina, M.; Guhl, S.; Motakis, E.; Artuc, M.; Hazzan, T.; Worm, M.; Forrest, A.R.; Zuberbier, T. Retinoic acid potentiates inflammatory cytokines in human mast cells: Identification of mast cells as prominent constituents of the skin retinoid network. Mol. Cell Endocrinol. 2015, 406, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Mery, A.G.; Beller, E.M.; Favot, C.; Boyce, J.A. Adenine nucleotides inhibit cytokine generation by human mast cells through a Gs-coupled receptor. J. Immunol. 2004, 173, 7539–7547. [Google Scholar] [CrossRef] [PubMed]
- Mortaz, E.; Redegeld, F.A.; Sarir, H.; Karimi, K.; Raats, D.; Nijkamp, F.P.; Folkerts, G. Cigarette smoke stimulates the production of chemokines in mast cells. J. Leukoc. Biol. 2008, 83, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.H.; Min, D.; Kim, H.P.; Song, K.J.; Kim, K.A.; Lee, Y.A.; Kim, S.H.; Shin, M.H. Leukotriene B4 receptor BLT-mediated phosphorylation of NF-kappaB and CREB is involved in IL-8 production in human mast cells induced by Trichomonas vaginalis-derived secretory products. Microbes Infect. 2011, 13, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, H.; Tao, X.; Luo, Y.; Wang, H.; He, J.; Fang, Q.; Guo, S.; Song, C. SCF promotes the production of IL-13 via the MEK-ERK-CREB signaling pathway in mast cells. Exp. Ther. Med. 2019, 18, 2491–2496. [Google Scholar] [CrossRef] [PubMed]
- Laresgoiti, U.; Apraiz, A.; Olea, M.; Mitxelena, J.; Osinalde, N.; Rodriguez, J.A.; Fullaondo, A.; Zubiaga, A.M. E2F2 and CREB cooperatively regulate transcriptional activity of cell cycle genes. Nucleic Acids Res. 2013, 41, 10185–10198. [Google Scholar] [CrossRef]
- Hamers, A.A.; Hanna, R.N.; Nowyhed, H.; Hedrick, C.C.; de Vries, C.J. NR4A nuclear receptors in immunity and atherosclerosis. Curr. Opin. Lipidol. 2013, 24, 381–385. [Google Scholar] [CrossRef]
- Nishiyama, C.; Ito, T.; Nishiyama, M.; Masaki, S.; Maeda, K.; Nakano, N.; Ng, W.; Fukuyama, K.; Yamamoto, M.; Okumura, K.; et al. GATA-1 is required for expression of FcepsilonRI on mast cells: Analysis of mast cells derived from GATA-1 knockdown mouse bone marrow. Int. Immunol. 2005, 17, 847–856. [Google Scholar] [CrossRef]
- Sasaki, H.; Kurotaki, D.; Tamura, T. Regulation of basophil and mast cell development by transcription factors. Allergol. Int. 2016, 65, 127–134. [Google Scholar] [CrossRef]
- Nechushtan, H.; Razin, E. The function of MITF and associated proteins in mast cells. Mol. Immunol. 2002, 38, 1177–1180. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.N.; Nechushtan, H.; Figov, N.; Razin, E. The function of lysyl-tRNA synthetase and Ap4A as signaling regulators of MITF activity in FcepsilonRI-activated mast cells. Immunity 2004, 20, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Nechushtan, H.; Kim, S.; Kay, G.; Razin, E. Chapter 1: The physiological role of lysyl tRNA synthetase in the immune system. Adv. Immunol. 2009, 103, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Babina, M.; Schulke, Y.; Kirchhof, L.; Guhl, S.; Franke, R.; Bohm, S.; Zuberbier, T.; Henz, B.M.; Gombart, A.F. The transcription factor profile of human mast cells in comparison with monocytes and granulocytes. Cell Mol. Life Sci. 2005, 62, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Proano-Perez, E.; Olle, L.; Guo, Y.; Aparicio, C.; Guerrero, M.; Munoz-Cano, R.; Martin, M. MITF Downregulation Induces Death in Human Mast Cell Leukemia Cells and Impairs IgE-Dependent Degranulation. Int. J. Mol. Sci. 2023, 24, 3515. [Google Scholar] [CrossRef]
- Prince, L.R.; Prosseda, S.D.; Higgins, K.; Carlring, J.; Prestwich, E.C.; Ogryzko, N.V.; Rahman, A.; Basran, A.; Falciani, F.; Taylor, P.; et al. NR4A orphan nuclear receptor family members, NR4A2 and NR4A3, regulate neutrophil number and survival. Blood 2017, 130, 1014–1025. [Google Scholar] [CrossRef]
- Murphy, M.P.; Caraher, E. Mcl-1 is vital for neutrophil survival. Immunol. Res. 2015, 62, 225–233. [Google Scholar] [CrossRef]
- Luo, Y.; Fernandez Vallone, V.; He, J.; Frischbutter, S.; Kolkhir, P.; Monino-Romero, S.; Stachelscheid, H.; Streu-Haddad, V.; Maurer, M.; Siebenhaar, F.; et al. A novel approach for studying mast cell-driven disorders: Mast cells derived from induced pluripotent stem cells. J. Allergy Clin. Immunol. 2022, 149, 1060–1068.e1064. [Google Scholar] [CrossRef]
- de Toledo, M.A.S.; Fu, X.; Maie, T.; Buhl, E.M.; Gotz, K.; Schmitz, S.; Kaiser, A.; Boor, P.; Braunschweig, T.; Chatain, N.; et al. KIT D816V Mast Cells Derived from Induced Pluripotent Stem Cells Recapitulate Systemic Mastocytosis Transcriptional Profile. Int. J. Mol. Sci. 2023, 24, 5275. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward 5′-3′ | Reverse 5′-3′ |
|---|---|---|
| CREB1 | GAGAAGCGGAGTGTTGGTGA | TCCGTCACTGCTTTCGTTCA |
| MITF | CCCTTATTCCATCCACGGGTCTC | ATACTGCTCCTCCGGCTGCTTGT |
| HPRT | GCCTCCCATCTCCTTCATCA | CCTGGCGTCGTGATTAGTGA |
| PPIB * | AAGATGTCCCTGTGCCCTAC | ATGGCAAGCATGTGGTGTTT |
| GAPDH | ATCTCGCTCCTGGAAGATGG | AGGTCGGAGTCAACGGATTT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bal, G.; Schneikert, J.; Li, Z.; Franke, K.; Tripathi, S.R.; Zuberbier, T.; Babina, M. CREB Is Indispensable to KIT Function in Human Skin Mast Cells—A Positive Feedback Loop between CREB and KIT Orchestrates Skin Mast Cell Fate. Cells 2024, 13, 42. https://doi.org/10.3390/cells13010042
Bal G, Schneikert J, Li Z, Franke K, Tripathi SR, Zuberbier T, Babina M. CREB Is Indispensable to KIT Function in Human Skin Mast Cells—A Positive Feedback Loop between CREB and KIT Orchestrates Skin Mast Cell Fate. Cells. 2024; 13(1):42. https://doi.org/10.3390/cells13010042
Chicago/Turabian StyleBal, Gürkan, Jean Schneikert, Zhuoran Li, Kristin Franke, Shiva Raj Tripathi, Torsten Zuberbier, and Magda Babina. 2024. "CREB Is Indispensable to KIT Function in Human Skin Mast Cells—A Positive Feedback Loop between CREB and KIT Orchestrates Skin Mast Cell Fate" Cells 13, no. 1: 42. https://doi.org/10.3390/cells13010042