

A Microfluidics Approach for Ovarian Cancer Immune Monitoring in an Outpatient Setting

 ,
,  , , , and
, , , and
Abstract
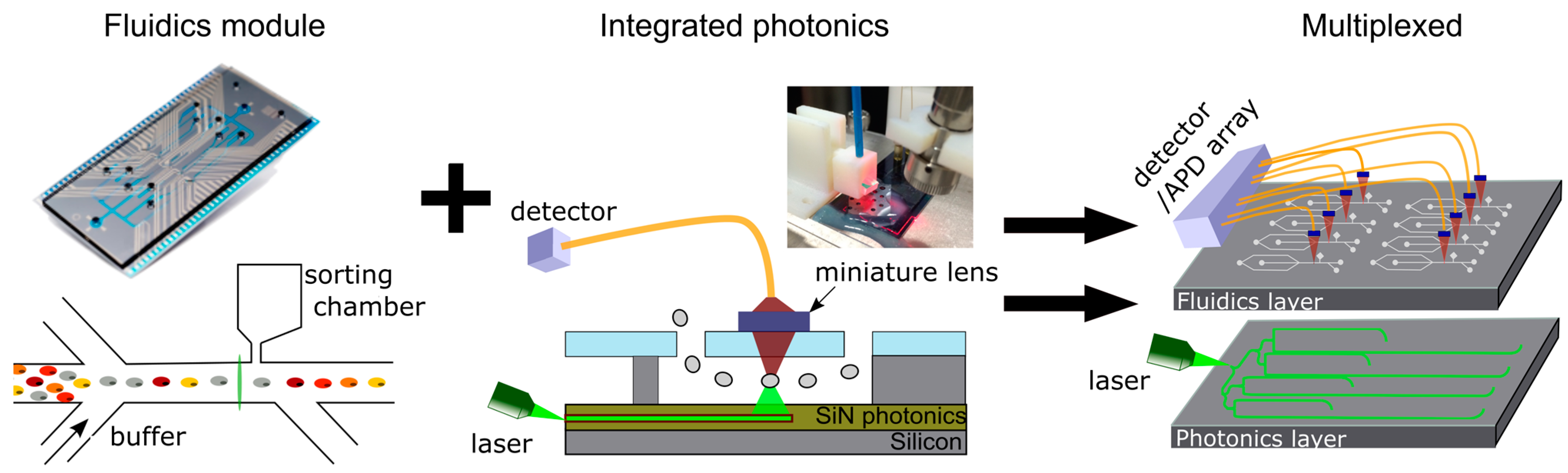
:1. Introduction
2. Materials and Methods
2.1. Study Design
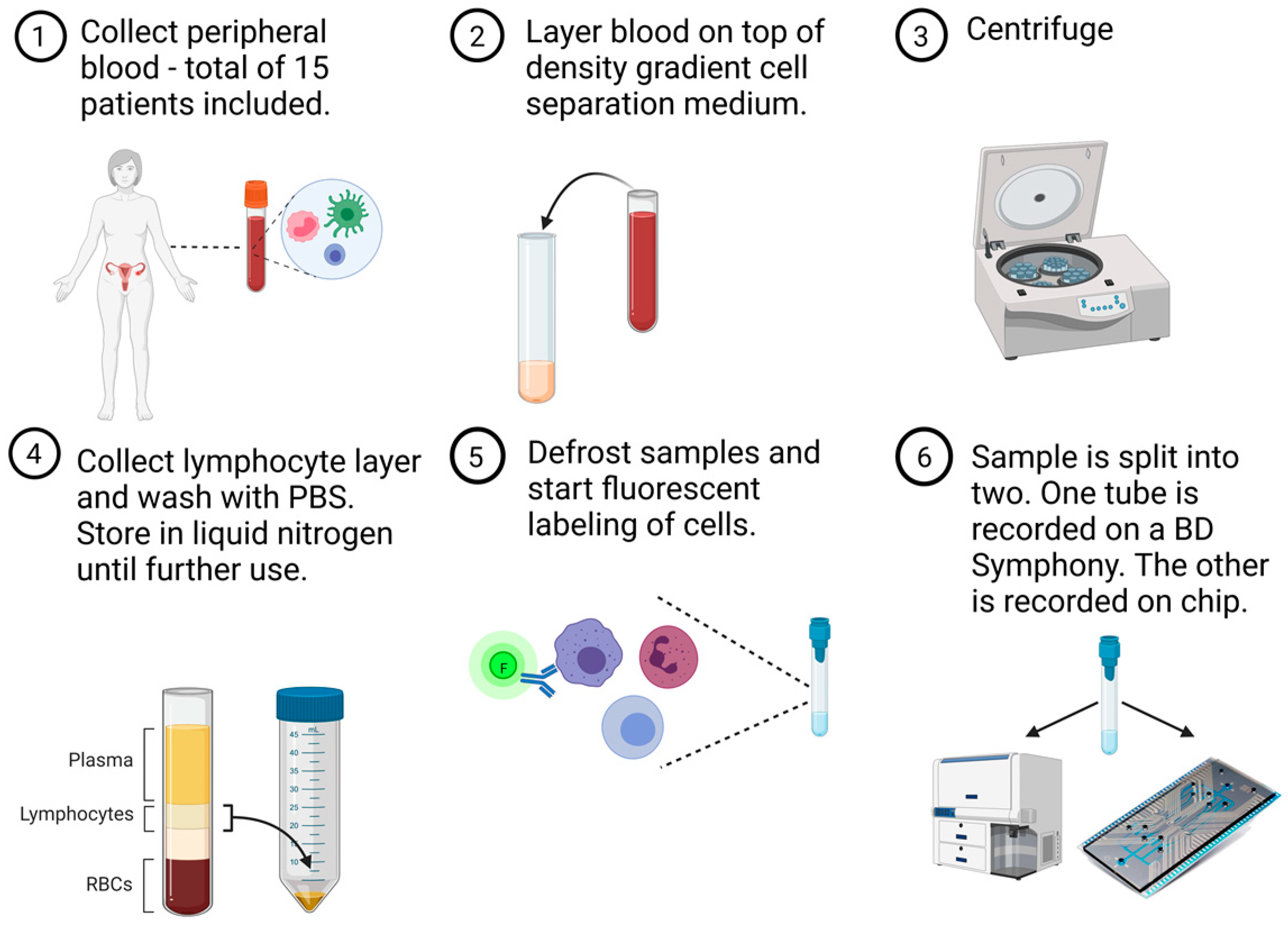
2.2. Sample Preparation
2.3. Conventional Flow Cytometry Measurement
2.4. Microfluidic Chip and Cytometry Measurement
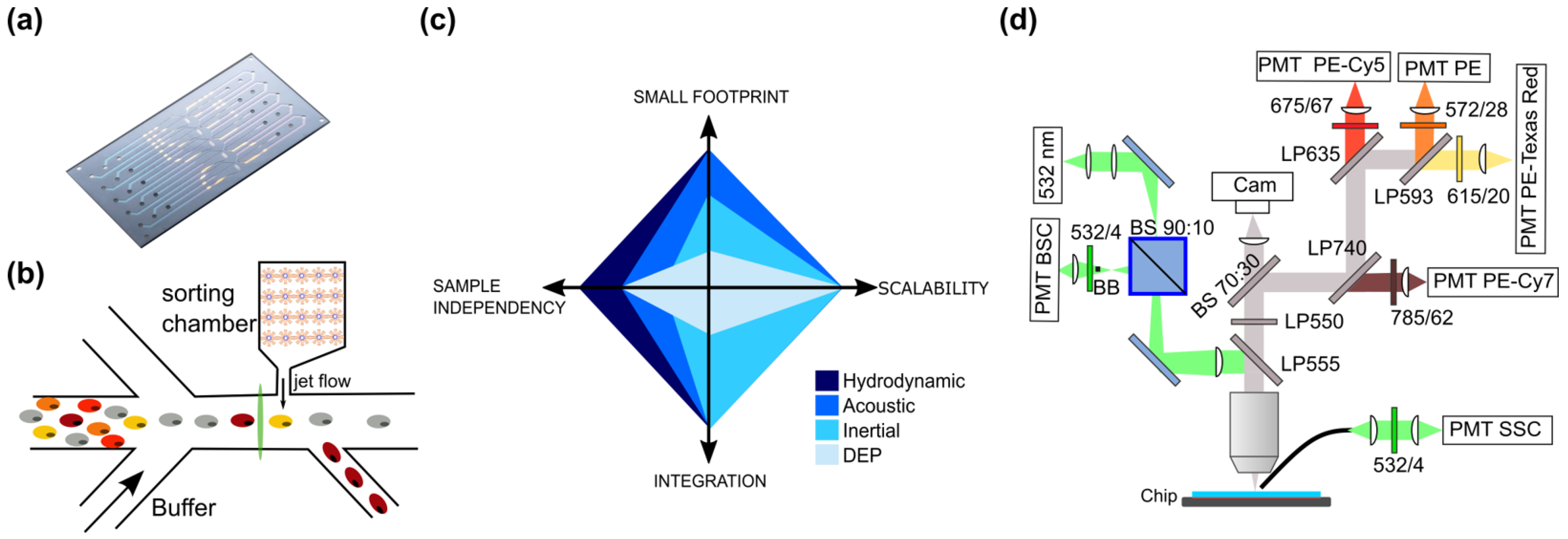
2.5. Data Analysis and Gating Principle
2.6. Statistics
3. Results
3.1. Patient Demographics
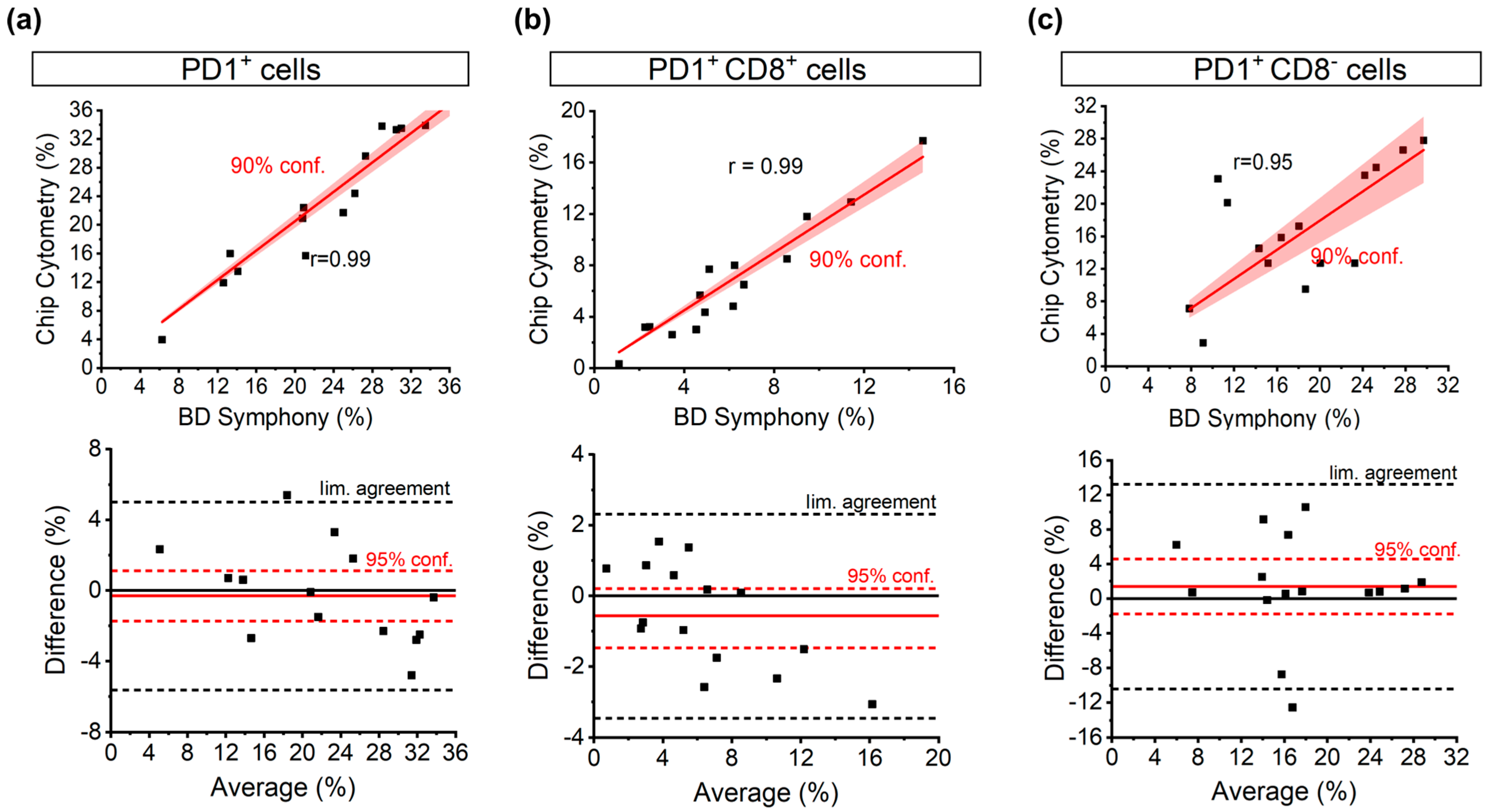
3.2. Correlation of PD1 Populations between FACSymphony and the Chip
3.3. Clinically Relevant Patterns Found on Conventional Cytometry Are Replicated through Chip Cytometry
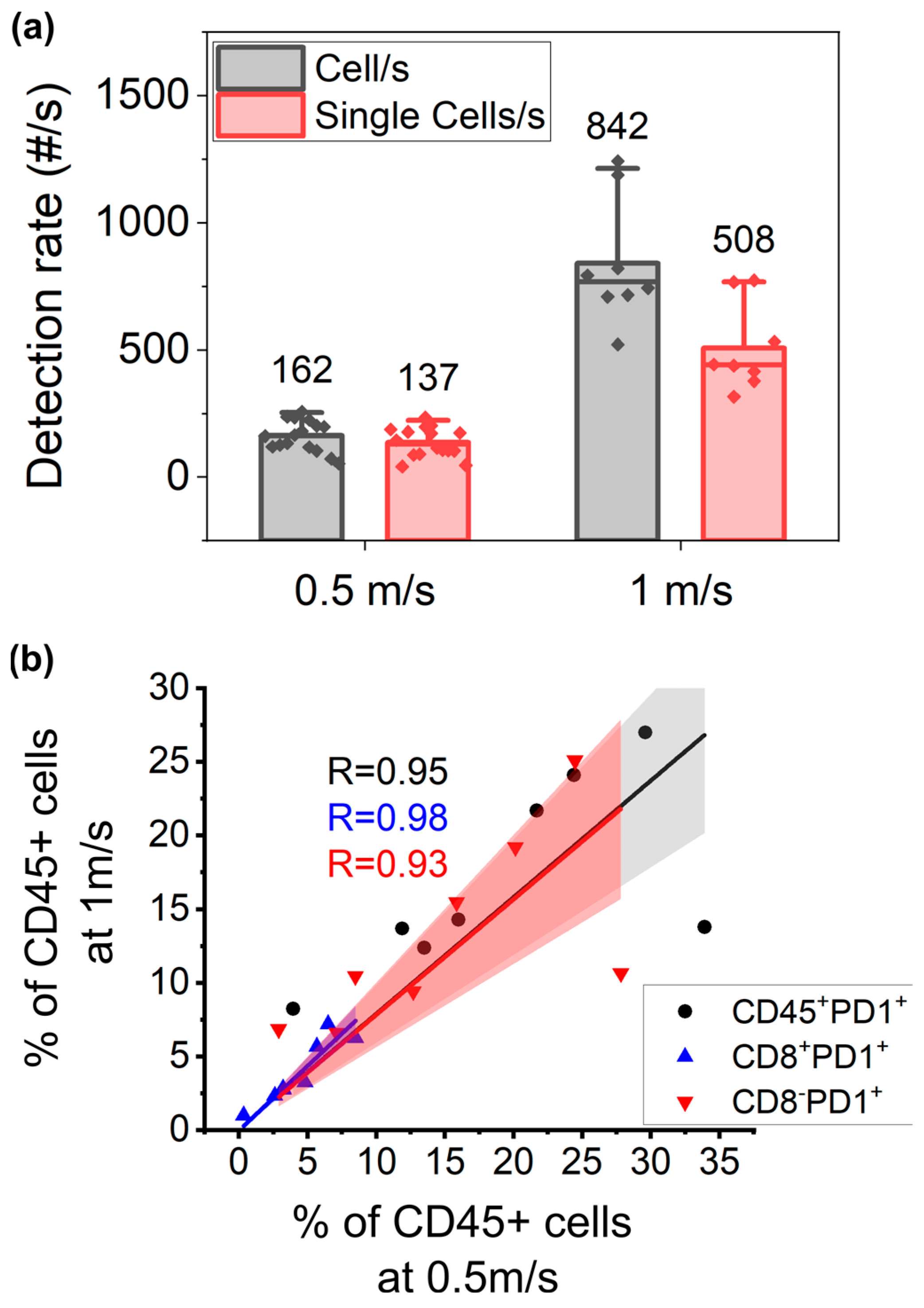
3.4. Higher Throughput Acquisition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Globocan. Available online: https://gco.iarc.fr/survival/survmark/visualizations/viz1/?groupby=%22cancer_site%22&period=%225%22&cancer_site=%22Colon%22&country=%22Australia%22&year=%222014%22&gender=%22Females%22&sorting=%222%22 (accessed on 26 January 2023).
- Peres, L.C.; Cushing-Haugen, K.L.; Köbel, M.; Harris, H.R.; Berchuck, A.; Rossing, M.A.; Schildkraut, J.M.; Doherty, J.A. Invasive Epithelial Ovarian Cancer Survival by Histotype and Disease Stage. J. Natl. Cancer Inst. 2019, 111, 60–68. [Google Scholar] [CrossRef] [PubMed]
- SEER Cancer Statistics. Available online: https://seer.cancer.gov/statfacts/html/ovary.html (accessed on 26 January 2023).
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data from Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Taylor, M.H.; Kelly, K.; Beck, J.T.; Gordon, M.; Moore, K.M.; Patel, M.R.; Chaves, J.; Park, H.; Mita, A.C.; et al. Efficacy and Safety of Avelumab for Patients with Recurrent or Refractory Ovarian Cancer: Phase 1b Results from the JAVELIN Solid Tumor Trial. JAMA Oncol. 2019, 5, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Heindl, A.; Lan, C.; Rodrigues, D.N.; Koelble, K.; Yuan, Y. Similarity and Diversity of the Tumor Microenvironment in Multiple Metastases: Critical Implications for Overall and Progression-Free Survival of High-Grade Serous Ovarian Cancer. Oncotarget 2016, 7, 71123–71135. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Sánchez, A.; Memon, D.; Pourpe, S.; Veeraraghavan, H.; Li, Y.; Vargas, H.A.; Gill, M.B.; Park, K.J.; Zivanovic, O.; Konner, J.; et al. Heterogeneous Tumor-Immune Microenvironments among Differentially Growing Metastases in an Ovarian Cancer Patient. Cell 2017, 170, 927–938.e20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.W.; McPherson, A.; Milne, K.; Kroeger, D.R.; Hamilton, P.T.; Miranda, A.; Funnell, T.; Little, N.; de Souza, C.P.E.; Laan, S.; et al. Interfaces of Malignant and Immunologic Clonal Dynamics in Ovarian Cancer. Cell 2018, 173, 1755–1769.e22. [Google Scholar] [CrossRef] [PubMed]
- de Bruyn, C.; Ceusters, J.; Landolfo, C.; Baert, T.; Thirion, G.; Claes, S.; Vankerckhoven, A.; Wouters, R.; Schols, D.; Timmerman, D.; et al. Neo-Adjuvant Chemotherapy Reduces, and Surgery Increases Immunosuppression in First-Line Treatment for Ovarian Cancer. Cancers 2021, 13, 5899. [Google Scholar] [CrossRef]
- Park, Y.H.; Lal, S.; Lee, J.E.; Choi, Y.-L.; Wen, J.; Ram, S.; Ding, Y.; Lee, S.H.; Powell, E.; Lee, S.K.; et al. Chemotherapy Induces Dynamic Immune Responses in Breast Cancers That Impact Treatment Outcome. Nat. Commun. 2020, 11, 6175. [Google Scholar] [CrossRef]
- Chen, W.; Huang, N.T.; Li, X.; Yu, Z.T.F.; Kurabayashi, K.; Fu, J. Emerging Microfluidic Tools for Functional Cellular Immunophenotyping: A New Potential Paradigm for Immune Status Characterization. Front. Oncol. 2013, 3, 98. [Google Scholar] [CrossRef]
- Yang, R.J.; Fu, L.M.; Hou, H.H. Review and Perspectives on Microfluidic Flow Cytometers. Sens. Actuators B Chem. 2018, 266, 26–45. [Google Scholar] [CrossRef]
- Verellen, N.; Vercruysse, D.; Rochus, V.; Du Bois, B.; Dusa, A.; Kerman, S.; Mahmud-Ul-Hasan, M.; Van Dorpe, P.; Rottenberg, X.; Lagae, L. Integrated Photonics for Miniature Flow Cytometry. In Proceedings of the 49th International Conferece on Solid State Devices and Materials—SSDM, Tsukuba, Japan, 19–22 September 2017. [Google Scholar]
- Zhang, Y.; Watts, B.R.; Guo, T.; Zhang, Z.; Xu, C.; Fang, Q. Optofluidic Device Based Microflow Cytometers for Particle/Cell Detection: A Review. Micromachines 2016, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Friis, P.; Hoppe, K.; Leistiko, O.; Mogensen, K.B.; Rg Hü Bner, J.; Rg, J.; Kutter, P. Monolithic Integration of Microfluidic Channels and Optical Waveguides in Silica on Silicon. Appl. Opt. 2001, 40, 6246–6251. [Google Scholar] [CrossRef] [PubMed]
- Kerman, S.; Vercruysse, D.; Claes, T.; Stassen, A.; Mahmud Ul Hasan, M.; Neutens, P.; Mukund, V.; Verellen, N.; Rottenberg, X.; Lagae, L.; et al. Integrated Nanophotonic Excitation and Detection of Fluorescent Microparticles. ACS Photon. 2017, 4, 1937–1944. [Google Scholar] [CrossRef]
- Rosenauer, M.; Vellekoop, M.J. Characterization of a Microflow Cytometer with an Integrated Three-Dimensional Optofluidic Lens System. Biomicrofluidics 2010, 4, 043005. [Google Scholar] [CrossRef] [PubMed]
- Porcel, M.A.G.; Hinojosa, A.; Jans, H.; Stassen, A.; Goyvaerts, J.; Geuzebroek, D.; Geiselmann, M.; Dominguez, C.; Artundo, I. Silicon Nitride Photonic Integration for Visible Light Applications. Opt. Laser Technol. 2019, 112, 299–306. [Google Scholar] [CrossRef]
- Ozcelik, D.; Jain, A.; Stambaugh, A.; Stott, M.A.; Parks, J.W.; Hawkins, A.; Schmidt, H. Scalable Spatial-Spectral Multiplexing of Single-Virus Detection Using Multimode Interference Waveguides. Sci. Rep. 2017, 7, 12199. [Google Scholar] [CrossRef]
- Dumitru, A.; Dobrica, E.C.; Croitoru, A.; Cretoiu, S.M.; Gaspar, B.S. Focus on PD-1/PD-L1 as a Therapeutic Target in Ovarian Cancer. Int. J. Mol. Sci. 2022, 23, 12067. [Google Scholar] [CrossRef]
- De Wijs, K.; Liu, C.; Dusa, A.; Vercruysse, D.; Majeed, B.; Tezcan, D.S.; Blaszkiewicz, K.; Loo, J.; Lagae, L. Micro Vapor Bubble Jet Flow for Safe and High-Rate Fluorescence-Activated Cell Sorting. Lab Chip 2017, 17, 1287–1296. [Google Scholar] [CrossRef]
- Kung, Y.C.; Huang, K.W.; Chong, W.; Chiou, P.Y. Microfluidics: Tunnel Dielectrophoresis for Tunable, Single-Stream Cell Focusing in Physiological Buffers in High-Speed Microfluidic Flows (Small 32/2016). Small 2016, 12, 4302. [Google Scholar] [CrossRef]
- Yu, C.; Vykoukal, J.; Vykoukal, D.M.; Schwartz, J.A.; Shi, L.; Gascoyne, P.R.C. A Three-Dimensional Dielectrophoretic Particle Focusing Channel for Microcytometry Applications. J. Microelectromech. Syst. 2005, 14, 480–487. [Google Scholar] [CrossRef]
- Yan, S.; Zhang, J.; Li, M.; Alici, G.; Du, H.; Sluyter, R.; Li, W. On-Chip High-Throughput Manipulation of Particles in a Dielectrophoresis- Active Hydrophoretic Focuser. Sci. Rep. 2014, 4, 5060. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wu, M.; Lin, Y.; Xu, J. Acoustic Microfluidic Separation Techniques and Bioapplications: A Review. Micromachines 2020, 11, 921. [Google Scholar] [CrossRef] [PubMed]
- Zalis, M.C.; Reyes, J.F.; Augustsson, P.; Holmqvist, S.; Roybon, L.; Laurell, T.; Deierborg, T. Label-Free Concentration of Viable Neurons, HESCs and Cancer Cells by Means of Acoustophoresis. Integr. Biol. 2016, 8, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Dykes, J.; Lenshof, A.; Åstrand-Grundström, I.B.; Laurell, T.; Scheding, S. Efficient Removal of Platelets from Peripheral Blood Progenitor Cell Products Using a Novel Micro-Chip Based Acoustophoretic Platform. PLoS ONE 2011, 6, e23074. [Google Scholar] [CrossRef] [PubMed]
- Dao, M.; Suresh, S.; Huang, T.J.; Li, P.; Mao, Z.; Peng, Z.; Zhou, L.; Chen, Y.; Huang, P.H.; Truica, C.I.; et al. Acoustic Separation of Circulating Tumor Cells. Proc. Natl. Acad. Sci. USA 2015, 112, 4970–4975. [Google Scholar] [CrossRef]
- Zmijan, R.; Jonnalagadda, U.S.; Carugo, D.; Kochi, Y.; Lemm, E.; Packham, G.; Hill, M.; Glynne-Jones, P. High Throughput Imaging Cytometer with Acoustic Focussing. RSC Adv. 2015, 5, 83206–83216. [Google Scholar] [CrossRef] [PubMed]
- Piyasena, M.E.; Suthanthiraraj, P.P.A.; Applegate, R.W.; Goumas, A.M.; Woods, T.A.; López, G.P.; Graves, S.W. Multinode Acoustic Focusing for Parallel Flow Cytometry. Anal. Chem. 2012, 84, 1831–1839. [Google Scholar] [CrossRef]
- Pritchard, R.H.; Zhukov, A.A.; Fullerton, J.N.; Want, A.J.; Hussain, F.; la Cour, M.F.; Bashtanov, M.E.; Gold, R.D.; Hailes, A.; Banham-Hall, E.; et al. Cell Sorting Actuated by a Microfluidic Inertial Vortex. Lab Chip 2019, 19, 2456–2465. [Google Scholar] [CrossRef]
- Hulspas, R.; Villa-Komaroff, L.; Koksal, E.; Etienne, K.; Rogers, P.; Tuttle, M.; Korsgren, O.; Sharpe, J.C.; Berglund, D. Purification of Regulatory T Cells with the Use of a Fully Enclosed High-Speed Microfluidic System. Cytotherapy 2014, 16, 1384–1389. [Google Scholar] [CrossRef]
- Zhukov, A.A.; Pritchard, R.H.; Withers, M.J.; Hailes, T.; Gold, R.D.; Hayes, C.; la Cour, M.F.; Hussein, F.; Rogers, S.S. Extremely High-Throughput Parallel Microfluidic Vortex-Actuated Cell Sorting. Micromachines 2021, 12, 389. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ma, Y.; Chen, Z.; Wu, Y.; Song, F.; Qiu, J.; Shi, M.; Wu, X. Sheatless Microflow Cytometer Utilizing Two Bulk Standing Acoustic Waves. Cytometry 2021, 99, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Pawłowska, A.; Suszczyk, D.; Tarkowski, R.; Paduch, R.; Kotarski, J.; Wertel, I. Programmed Death-1 Receptor (PD-1) as a Potential Prognosis Biomarker for Ovarian Cancer Patients. Cancer Manag. Res. 2020, 12, 9691–9709. [Google Scholar] [CrossRef] [PubMed]
- Piruska, A.; Nikcevic, I.; Lee, S.H.; Ahn, C.; Heineman, W.R.; Limbach, P.A.; Seliskar, C.J. The Autofluorescence of Plastic Materials and Chips Measured under Laser Irradiation. Lab Chip 2005, 5, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Coosemans, A.; Baert, T.; Ceusters, J.; Busschaert, P.; Landolfo, C.; Verschuere, T.; van Rompuy, A.S.; Vanderstichele, A.; Froyman, W.; Neven, P.; et al. Myeloid-Derived Suppressor Cells at Diagnosis May Discriminate between Benign and Malignant Ovarian Tumors. Int. J. Gynecol. Cancer 2019, 29, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.Z.; Neutens, P.; Dhakal, A.; Jansen, R.; Claes, T.; Rottenberg, X.; Peyskens, F.; Selvaraja, S.; Helin, P.; Dubois, B.; et al. Low-Loss Singlemode PECVD Silicon Nitride Photonic Wire Waveguides for 532-900 Nm Wavelength Window Fabricated within a CMOS Pilot Line. IEEE Photon. J. 2013, 5. [Google Scholar] [CrossRef]
- Butement, J.T.; Horak, P.; Wilkinson, J.S.; Butement, J.T.; Holloway, P.M.; Welsh, J.A.; Holloway, J.A.; Englyst, N.A.; West, J.; Holloway, P.M.; et al. Monolithically-Integrated Cytometer for Measuring Particle Diameter in the Extracellular Vesicle Size Range Using Multi-Angle Scattering. Lab Chip 2020, 20, 1267–1280. [Google Scholar] [CrossRef] [PubMed]
- Mangal, N.; Snyder, B.; Van Campenhout, J.; Van Steenberge, G.; Missinne, J. Monolithic Integration of Microlenses on the Backside of a Silicon Photonics Chip for Expanded Beam Coupling. Opt. Express 2021, 29, 7601. [Google Scholar] [CrossRef]
- Picot, J.; Guerin, C.L.; Le Van Kim, C.; Boulanger, C.M. Flow Cytometry: Retrospective, Fundamentals and Recent Instrumentation. Cytotechnology 2012, 64, 109–130. [Google Scholar] [CrossRef]
- Glier, H.; Heijnen, I.; Hauwel, M.; Dirks, J.; Quarroz, S.; Lehmann, T.; Rovo, A.; Arn, K.; Matthes, T.; Hogan, C.; et al. Standardization of 8-Color Flow Cytometry across Different Flow Cytometer Instruments: A Feasibility Study in Clinical Laboratories in Switzerland. J. Immunol. Methods 2019, 475, 112348. [Google Scholar] [CrossRef]
- Ivison, S.; Malek, M.; Garcia, R.V.; Broady, R.; Halpin, A.; Richaud, M.; Brant, R.F.; Wang, S.I.; Goupil, M.; Guan, Q.; et al. A Standardized Immune Phenotyping and Automated Data Analysis Platform for Multicenter Biomarker Studies. JCI Insight 2018, 3, e121867. [Google Scholar] [CrossRef]
- Patibandla, P.K.; Estrada, R.; Kannan, M.; Sethu, P. A Microfluidics-Based Technique for Automated and Rapid Labeling of Cells for Flow Cytometry. J. Micromech. Microeng. 2014, 24, 034002. [Google Scholar] [CrossRef]
- Kim, B.; Kim, K.H.; Chang, Y.; Shin, S.; Shin, E.C.; Choi, S. One-Step Microfluidic Purification of White Blood Cells from Whole Blood for Immunophenotyping. Anal. Chem. 2019, 91, 13230–13236. [Google Scholar] [CrossRef]
- Morotti, M.; Albukhari, A.; Alsaadi, A.; Artibani, M.; Brenton, J.D.; Curbishley, S.M.; Dong, T.; Dustin, M.L.; Hu, Z.; McGranahan, N.; et al. Promises and Challenges of Adoptive T-Cell Therapies for Solid Tumours. Br. J. Cancer 2021, 124, 1759–1776. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Result n (%) | |
|---|---|---|
| Age | Median (years), range | 68 (49–76) |
| FIGO 1 stage at diagnosis | III | 7 (47) |
| IV | 8 (53) | |
| Timing of blood sampling | At diagnosis | 4 (27) |
| During primary treatment | 8 (53) 2 * | |
| After recurrence | 3 (20) 2 # | |
| (Peg)filgrastim | Yes | 5 (33) |
| No | 6 (40) | |
| Not applicable | 4 (27) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Libbrecht, S.; Vankerckhoven, A.; de Wijs, K.; Baert, T.; Thirion, G.; Vandenbrande, K.; Van Gorp, T.; Timmerman, D.; Coosemans, A.; Lagae, L. A Microfluidics Approach for Ovarian Cancer Immune Monitoring in an Outpatient Setting. Cells 2024, 13, 7. https://doi.org/10.3390/cells13010007
Libbrecht S, Vankerckhoven A, de Wijs K, Baert T, Thirion G, Vandenbrande K, Van Gorp T, Timmerman D, Coosemans A, Lagae L. A Microfluidics Approach for Ovarian Cancer Immune Monitoring in an Outpatient Setting. Cells. 2024; 13(1):7. https://doi.org/10.3390/cells13010007
Chicago/Turabian StyleLibbrecht, Sarah, Ann Vankerckhoven, Koen de Wijs, Thaïs Baert, Gitte Thirion, Katja Vandenbrande, Toon Van Gorp, Dirk Timmerman, An Coosemans, and Liesbet Lagae. 2024. "A Microfluidics Approach for Ovarian Cancer Immune Monitoring in an Outpatient Setting" Cells 13, no. 1: 7. https://doi.org/10.3390/cells13010007
APA StyleLibbrecht, S., Vankerckhoven, A., de Wijs, K., Baert, T., Thirion, G., Vandenbrande, K., Van Gorp, T., Timmerman, D., Coosemans, A., & Lagae, L. (2024). A Microfluidics Approach for Ovarian Cancer Immune Monitoring in an Outpatient Setting. Cells, 13(1), 7. https://doi.org/10.3390/cells13010007