
A Highly Sensitive Molecular Technique for RNA Virus Detection

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Samples
2.2. Recombinase Polymerase Amplification-Loop-Mediated Isothermal Amplification Primer Design
2.3. Recombinase Polymerase Amplification-Loop-Mediated Isothermal Amplification Reaction
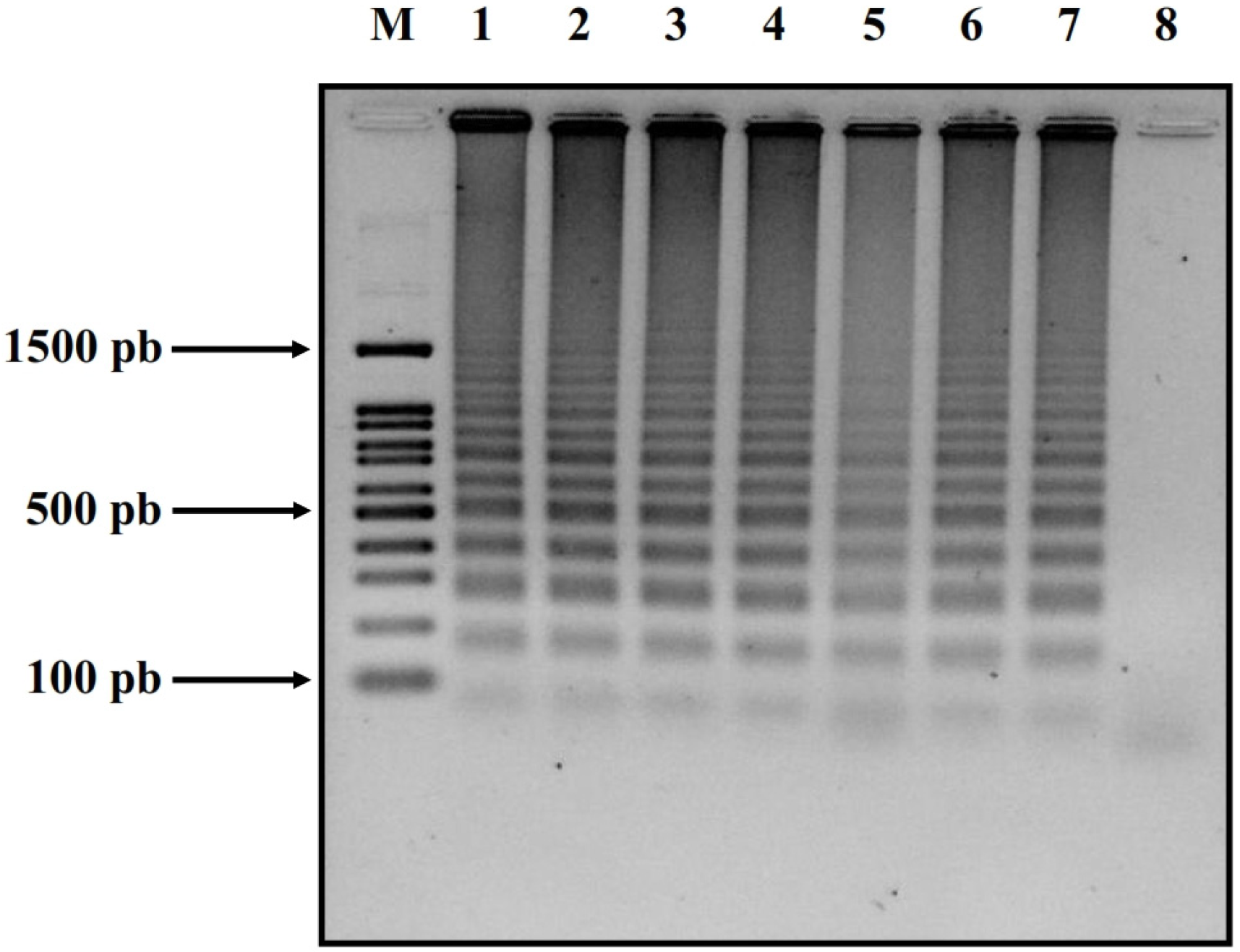
2.4. Confirmation via Gel Electrophoresis
2.5. Statistical Analysis
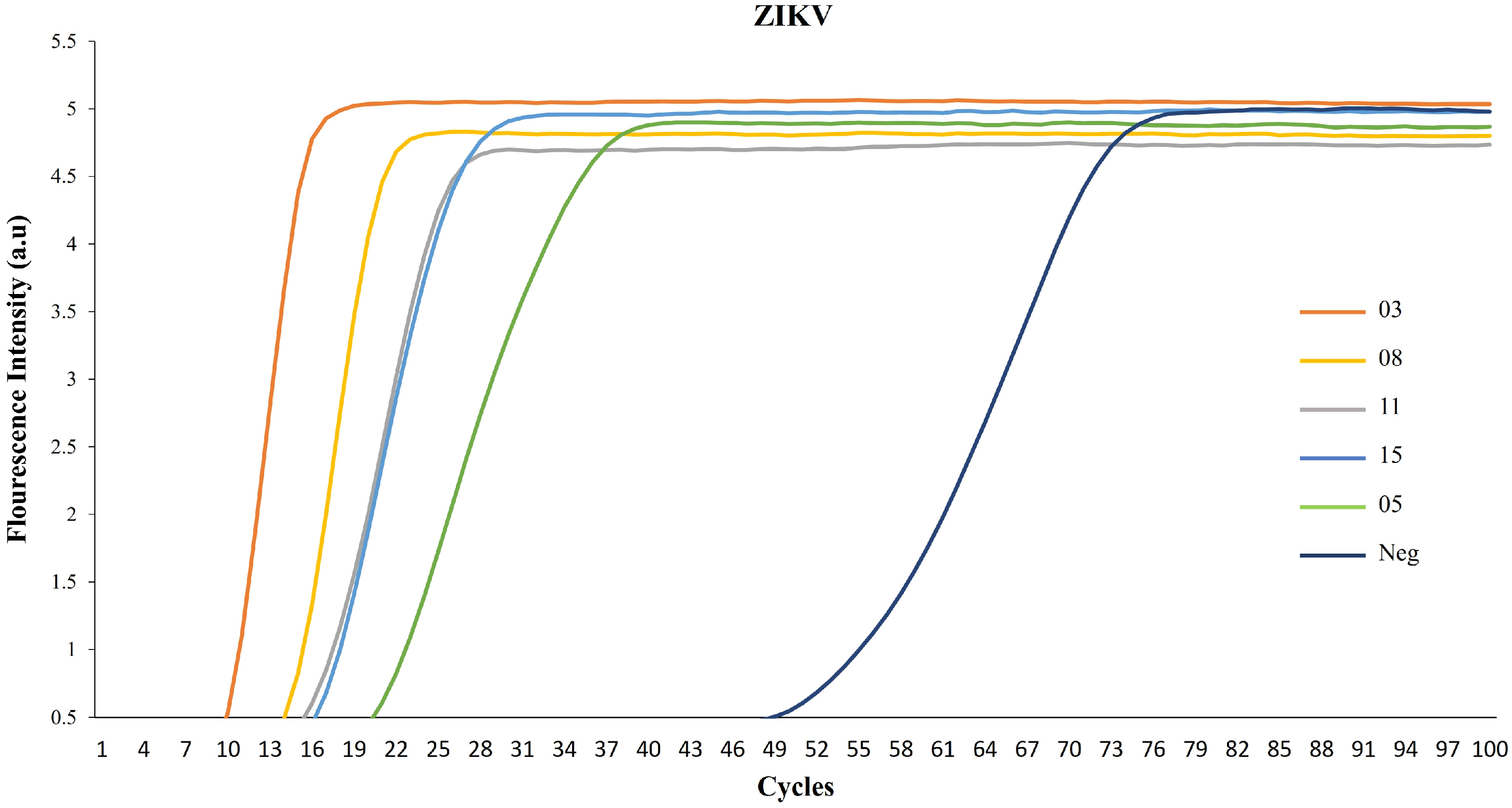
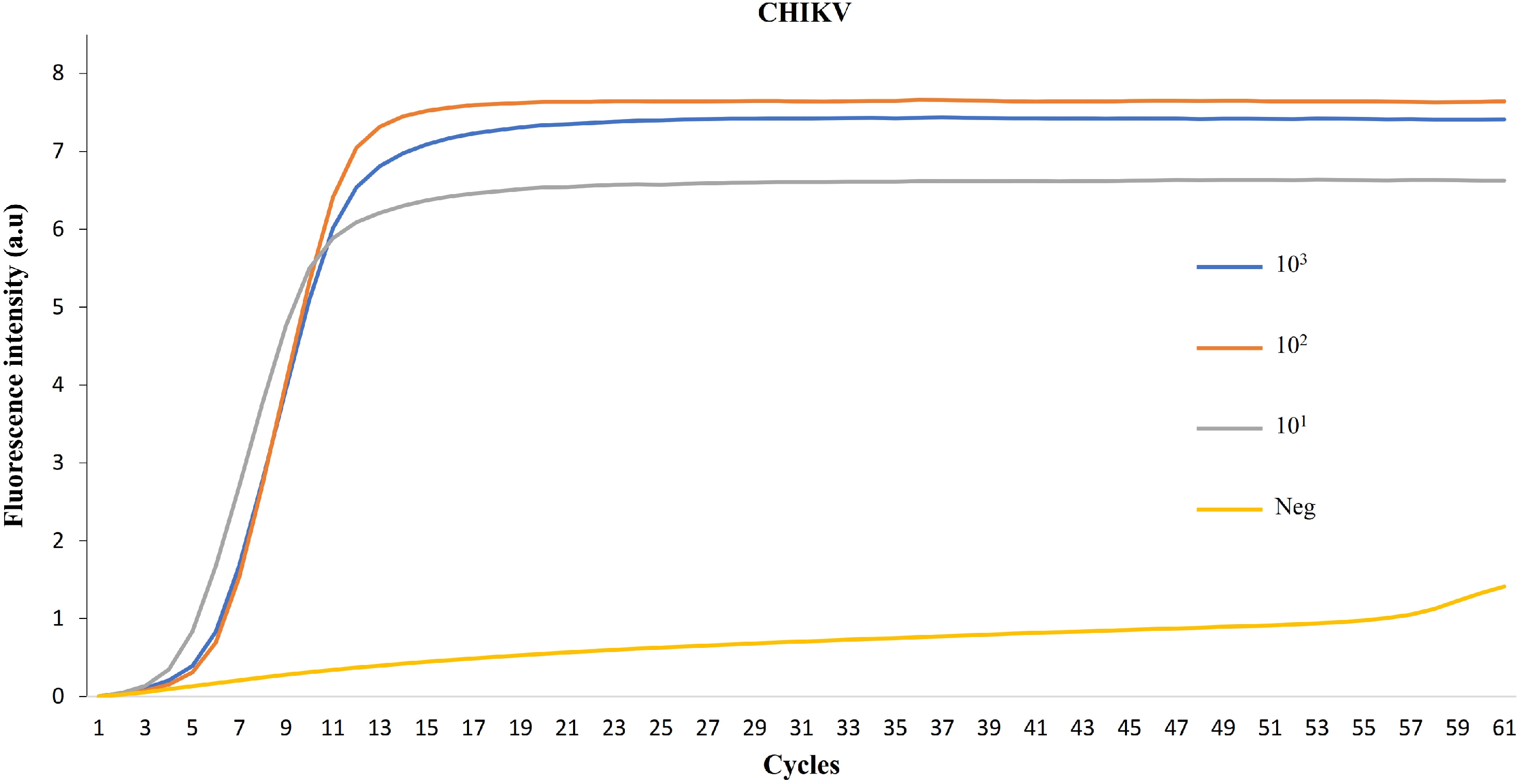
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharma, S.; Pardasani, D.; Dash, P.K.; Parida, M.; Dubey, D.K. Development of magnetic bead based sample extraction coupled polymerase spiral reaction for rapid on-site detection of Chikungunya virus. Sci. Rep. 2020, 10, 11651. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, Y.; Martins, D.B.G.; Kimura, M.; Catena, A.D.S.; Borba, M.A.C.S.M.; Mattos, S.D.S.; Abe, H.; Yoshikawa, R.; de Lima Filho, J.L.; Yasuda, J. Development and evaluation of a rapid molecular diagnostic test for Zika virus infection by reverse transcription loop-mediated isothermal amplification. Sci. Rep. 2017, 7, 13503. [Google Scholar] [CrossRef] [PubMed]
- Shimelis, T.; Mulu, A.; Mengesha, M.; Alemu, A.; Mihret, A.; Tadesse, B.T.; Bartlett, A.W.; Belay, F.W.; Schierhout, G.; Dittrich, S.; et al. Detection of dengue virus infection in children presenting with fever in Hawassa, southern Ethiopia. Sci. Rep. 2023, 13, 7997. [Google Scholar] [CrossRef] [PubMed]
- Amaya-Larios, I.Y.; Martínez-Vega, R.A.; Diaz-Quijano, F.A.; Sarti, E.; Puentes-Rosas, E.; Chihu, L.; Ramos-Castañeda, J. Risk of dengue virus infection according to serostatus in individuals from dengue endemic areas of Mexico. Sci. Rep. 2020, 10, 19017. [Google Scholar] [CrossRef] [PubMed]
- In Vitro Diagnostics. Available online: https://www.fda.gov/medical-devices/in-vitro-diagnostics/nucleic-acid-based-tests (accessed on 26 September 2023).
- Sirohi, D.; Chen, Z.; Sun, L.; Klose, T.; Pierson, T.C.; Rossmann, M.G.; Kuhn, R.J. The 3.8 Å resolution cryo-EM structure of Zika virus. Science 2016, 352, 467–470. [Google Scholar] [CrossRef] [PubMed]
- van Leur, S.W.; Heunis, T.; Munnur, D.; Sanyal, S. Pathogenesis and virulence of flavivirus infections. Virulence 2021, 12, 2814–2838. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, C.J.; Cortese, M.; Acosta, E.G.; Bartenschlager, R. Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Microbiol. 2018, 16, 125–142. [Google Scholar] [CrossRef]
- Brasil, P.; Pereira, J.P., Jr.; Moreira, M.E.; Ribeiro Nogueira, R.M.; Damasceno, L.; Wakimoto, M.; Rabello, R.S.; Valderramos, S.G.; Halai, U.A.; Salles, T.S.; et al. Zika Virus Infection in Pregnant Women in Rio de Janeiro. N. Engl. J. Med. 2016, 375, 2321–2334. [Google Scholar] [CrossRef]
- Horwood, P.F.; Reimer, L.J.; Dagina, R.; Susapu, M.; Bande, G.; Katusele, M.; Koimbu, G.; Jimmy, S.; Ropa, B.; Siba, P.M.; et al. Outbreak of chikungunya virus infection, Vanimo, Papua New Guinea. Emerg. Infect. Dis. 2013, 19, 1535–1538. [Google Scholar] [CrossRef] [PubMed]
- Staples, J.E.; Fischer, M. Chikungunya virus in the Americas—What a vectorborne pathogen can do. N. Engl. J. Med. 2014, 371, 887–889. [Google Scholar] [CrossRef]
- Rasmussen, S.A.; Jamieson, D.J.; Honein, M.A.; Petersen, L.R. Zika Virus and Birth Defects—Reviewing the Evidence for Causality. N. Engl. J. Med. 2016, 374, 1981–1987. [Google Scholar] [CrossRef] [PubMed]
- Cao-Lormeau, V.M.; Blake, A.; Mons, S.; Lastère, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- Richner, J.M.; Himansu, S.; Dowd, K.A.; Butler, S.L.; Salazar, V.; Fox, J.M.; Julander, J.G.; Tang, W.W.; Shresta, S.; Pierson, T.C.; et al. Modified mRNA Vaccines Protect against Zika Virus Infection. Cell 2017, 169, 176. [Google Scholar] [CrossRef] [PubMed]
- Arévalo Romero, H.; Vargas Pavía, T.A.; Velázquez Cervantes, M.A.; Flores Pliego, A.; Helguera Repetto, A.C.; León Juárez, M. The Dual Role of the Immune Response in Reproductive Organs During Zika Virus Infection. Front. Immunol. 2019, 10, 1617. [Google Scholar] [CrossRef]
- Morens, D.M.; Fauci, A.S. Chikungunya at the door—Déjà vu all over again? N. Engl. J. Med. 2014, 371, 885–887. [Google Scholar] [CrossRef]
- Silva, L.A.; Dermody, T.S. Chikungunya virus: Epidemiology, replication, disease mechanisms, and prospective intervention strategies. J. Clin. Investig. 2017, 127, 737–749. [Google Scholar] [CrossRef]
- Solignat, M.; Gay, B.; Higgs, S.; Briant, L.; Devaux, C. Replication cycle of chikungunya: A re-emerging arbovirus. Virology 2009, 393, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Zhao, Z.; Chai, Y.; Jin, X.; Li, C.; Yuan, F.; Liu, S.; Gao, Z.; Wang, H.; Song, J.; et al. Molecular Basis of Arthritogenic Alphavirus Receptor MXRA8 Binding to Chikungunya Virus Envelope Protein. Cell 2019, 177, 1714–1724.e12. [Google Scholar] [CrossRef]
- De Caluwé, L.; Coppens, S.; Vereecken, K.; Daled, S.; Dhaenens, M.; Van Ostade, X.; Deforce, D.; Ariën, K.K.; Bartholomeeusen, K. The CD147 Protein Complex Is Involved in Entry of Chikungunya Virus and Related Alphaviruses in Human Cells. Front. Microbiol. 2021, 12, 615165. [Google Scholar] [CrossRef]
- Schnierle, B.S. Cellular Attachment and Entry Factors for Chikungunya Virus. Viruses 2019, 11, 1078. [Google Scholar] [CrossRef]
- Thomas, S.; Rai, J.; John, L.; Günther, S.; Drosten, C.; Pützer, B.M.; Schaefer, S. Functional dissection of the alphavirus capsid protease: Sequence requirements for activity. Virol. J. 2010, 7, 327. [Google Scholar] [CrossRef] [PubMed]
- Yap, M.L.; Klose, T. Structural studies of Chikungunya virus maturation. Proc. Natl. Acad. Sci. USA 2017, 114, 13703–13707. [Google Scholar] [CrossRef]
- Enserink, M. Infectious diseases. Chikungunya: No longer a third world disease. Science 2007, 318, 1860–1861. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; Chen, R.; Sherman, M.B.; Weaver, S.C. Chikungunya virus: Evolution and genetic determinants of emergence. Curr. Opin. Virol. 2011, 1, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Schilte, C.; Staikowsky, F.; Couderc, T.; Madec, Y.; Carpentier, F.; Kassab, S.; Albert, M.L.; Lecuit, M.; Michault, A. Chikungunya virus-associated long-term arthralgia: A 36-month prospective longitudinal study. PLoS Neglected Trop. Dis. 2013, 7, e2137. [Google Scholar] [CrossRef] [PubMed]
- Burt, F.J.; Rolph, M.S.; Rulli, N.E.; Mahalingam, S.; Heise, M.T. Chikungunya: A re-emerging virus. Lancet 2012, 379, 662–671. [Google Scholar] [CrossRef]
- Sourisseau, M.; Schilte, C.; Casartelli, N.; Trouillet, C.; Guivel-Benhassine, F.; Rudnicka, D.; Sol-Foulon, N.; Le Roux, K.; Prevost, M.C.; Fsihi, H.; et al. Characterization of reemerging chikungunya virus. PLoS Pathog. 2007, 3, e89. [Google Scholar] [CrossRef] [PubMed]
- Hoarau, J.J.; Jaffar Bandjee, M.C.; Krejbich Trotot, P.; Das, T.; Li-Pat-Yuen, G.; Dassa, B.; Denizot, M.; Guichard, E.; Ribera, A.; Henni, T.; et al. Persistent chronic inflammation and infection by Chikungunya arthritogenic alphavirus in spite of a robust host immune response. J. Immunol. 2010, 184, 5914–5927. [Google Scholar] [CrossRef]
- Available online: https://www.cdc.gov/zika/pregnancy/index.html (accessed on 30 June 2023).
- Available online: https://www.who.int/publications/m/item/current-zika-product-pipeline (accessed on 30 June 2023).
- Liu, C.; Geva, E.; Mauk, M.; Qiu, X.; Abrams, W.R.; Malamud, D.; Curtis, K.; Owen, S.M.; Bau, H.H. An isothermal amplification reactor with an integrated isolation membrane for point-of-care detection of infectious diseases. Analyst 2011, 136, 2069–2076. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Mauk, M.G.; Hart, R.; Qiu, X.; Bau, H.H. A self-heating cartridge for molecular diagnostics. Lab Chip 2011, 11, 2686–2692. [Google Scholar] [CrossRef]
- Liao, S.C.; Peng, J.; Mauk, M.G.; Awasthi, S.; Song, J.; Friedman, H.; Bau, H.H.; Liu, C. Smart Cup: A Minimally-Instrumented, Smartphone-Based Point-of-Care Molecular Diagnostic Device. Sens. Actuators B Chem. 2016, 229, 232–238. [Google Scholar] [CrossRef]
- Liu, C.; Liao, S.C.; Song, J.; Mauk, M.G.; Li, X.; Wu, G.; Ge, D.; Greenberg, R.M.; Yang, S.; Bau, H.H. A high-efficiency superhydrophobic plasma separator. Lab Chip 2016, 16, 553–560. [Google Scholar] [CrossRef]
- Song, J.; Liu, C.; Bais, S.; Mauk, M.G.; Bau, H.H.; Greenberg, R.M. Molecular Detection of Schistosome Infections with a Disposable Microfluidic Cassette. PLoS Neglected Trop. Dis. 2015, 9, e0004318. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Mauk, M.G.; Hart, R.; Bonizzoni, M.; Yan, G.; Bau, H.H. A low-cost microfluidic chip for rapid genotyping of malaria-transmitting mosquitoes. PLoS ONE 2012, 7, e42222. [Google Scholar] [CrossRef] [PubMed]
- Enfissi, A.; Codrington, J.; Roosblad, J.; Kazanji, M.; Rousset, D. Zika virus genome from the Americas. Lancet 2016, 387, 227–228. [Google Scholar] [CrossRef]
- Mansuy, J.M.; Dutertre, M.; Mengelle, C.; Fourcade, C.; Marchou, B.; Delobel, P.; Izopet, J.; Martin-Blondel, G. Zika virus: High infectious viral load in semen, a new sexually transmitted pathogen? Lancet Infect. Dis. 2016, 16, 405. [Google Scholar] [CrossRef]
- Gourinat, A.C.; O’Connor, O.; Calvez, E.; Goarant, C.; Dupont-Rouzeyrol, M. Detection of Zika virus in urine. Emerg. Infect. Dis. 2015, 21, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Lustig, Y.; Mendelson, E.; Paran, N.; Melamed, S.; Schwartz, E. Detection of Zika virus RNA in whole blood of imported Zika virus disease cases up to 2 months after symptom onset, Israel, December 2015 to April 2016. Euro Surveill. 2016, 21, 30269. [Google Scholar] [CrossRef]
- Musso, D.; Teissier, A.; Rouault, E.; Teururai, S.; de Pina, J.J.; Nhan, T.X. Detection of chikungunya virus in saliva and urine. Virol. J. 2016, 13, 102. [Google Scholar] [CrossRef] [PubMed]
- Driggers, R.W.; Ho, C.Y.; Korhonen, E.M.; Kuivanen, S.; Jääskeläinen, A.J.; Smura, T.; Rosenberg, A.; Hill, D.A.; DeBiasi, R.L.; Vezina, G.; et al. Zika Virus Infection with Prolonged Maternal Viremia and Fetal Brain Abnormalities. N. Engl. J. Med. 2016, 374, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Pardee, K.; Green, A.A.; Ferrante, T.; Cameron, D.E.; DaleyKeyser, A.; Yin, P.; Collins, J.J. Paper-based synthetic gene networks. Cell 2014, 159, 940–954. [Google Scholar] [CrossRef]
- Pardee, K.; Green, A.A.; Takahashi, M.K.; Braff, D.; Lambert, G.; Lee, J.W.; Ferrante, T.; Ma, D.; Donghia, N.; Fan, M.; et al. Rapid, Low-Cost Detection of Zika Virus Using Programmable Biomolecular Components. Cell 2016, 165, 1255–1266. [Google Scholar] [CrossRef] [PubMed]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, E63. [Google Scholar] [CrossRef]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA detection using recombination proteins. PLoS Biol. 2006, 4, e204. [Google Scholar] [CrossRef] [PubMed]
- Thézé, J.; Li, T.; du Plessis, L.; Bouquet, J.; Kraemer, M.U.G.; Somasekar, S.; Yu, G.; de Cesare, M.; Balmaseda, A.; Kuan, G.; et al. Genomic Epidemiology Reconstructs the Introduction and Spread of Zika Virus in Central America and Mexico. Cell Host Microbe 2018, 23, 855–864.e7. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, B.; Candido, D.d.S.; Bajaj, S.; Maldonado, A.P.R.; Ayala, F.G.; Rodriguez, M.d.l.L.T.; Rodriguez, A.A.; Arámbula, C.W.; González, E.R.; Martínez, I.L.; et al. Convergent trends and spatiotemporal patterns of arboviruses in Mexico and Central America. medRxiv 2023. [Google Scholar] [CrossRef]
- Grajales-Muñiz, C.; Borja-Aburto, V.H.; Cabrera-Gaytán, D.A.; Rojas-Mendoza, T.; Arriaga-Nieto, L.; Vallejos-Parás, A. Zika virus: Epidemiological surveillance of the Mexican Institute of Social Security. PLoS ONE 2019, 14, e0212114. [Google Scholar] [CrossRef] [PubMed]
- Kautz, T.F.; Díaz-González, E.E.; Erasmus, J.H.; Malo-García, I.R.; Langsjoen, R.M.; Patterson, E.I.; Auguste, D.I.; Forrester, N.L.; Sanchez-Casas, R.M.; Hernández-Ávila, M.; et al. Chikungunya Virus as Cause of Febrile Illness Outbreak, Chiapas, Mexico, 2014. Emerg. Infect. Dis. 2015, 21, 2070–2073. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Mauk, M.G.; Hackett, B.A.; Cherry, S.; Bau, H.H.; Liu, C. Instrument-Free Point-of-Care Molecular Detection of Zika Virus. Anal. Chem. 2016, 88, 7289–7294. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Weaver, S.C.; Wong, P.Y.; Lie, S.; Wang, E.; Guerbois, M.; Vayugundla, S.P.; Wong, S. Rapid, Affordable and Portable Medium-Throughput Molecular Device for Zika Virus. Sci. Rep. 2016, 6, 38223. [Google Scholar] [CrossRef] [PubMed]
- Yaren, O.; Alto, B.W.; Gangodkar, P.V.; Ranade, S.R.; Patil, K.N.; Bradley, K.M.; Yang, Z.; Phadke, N.; Benner, S.A. Point of sampling detection of Zika virus within a multiplexed kit capable of detecting dengue and chikungunya. BMC Infect. Dis. 2017, 17, 293. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yin, F.; Bi, Y.; Cheng, G.; Li, J.; Hou, L.; Li, Y.; Yang, B.; Liu, W.; Yang, L. Rapid and sensitive detection of Zika virus by reverse transcription loop-mediated isothermal amplification. J. Virol. Methods 2016, 238, 86–93. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rozmyslowicz, T.; Arévalo-Romero, H.; Conover, D.O.; Fuentes-Pananá, E.M.; León-Juárez, M.; Gaulton, G.N. A Highly Sensitive Molecular Technique for RNA Virus Detection. Cells 2024, 13, 804. https://doi.org/10.3390/cells13100804
Rozmyslowicz T, Arévalo-Romero H, Conover DO, Fuentes-Pananá EM, León-Juárez M, Gaulton GN. A Highly Sensitive Molecular Technique for RNA Virus Detection. Cells. 2024; 13(10):804. https://doi.org/10.3390/cells13100804
Chicago/Turabian StyleRozmyslowicz, Tomasz, Haruki Arévalo-Romero, Dareus O. Conover, Ezequiel M. Fuentes-Pananá, Moisés León-Juárez, and Glen N. Gaulton. 2024. "A Highly Sensitive Molecular Technique for RNA Virus Detection" Cells 13, no. 10: 804. https://doi.org/10.3390/cells13100804
APA StyleRozmyslowicz, T., Arévalo-Romero, H., Conover, D. O., Fuentes-Pananá, E. M., León-Juárez, M., & Gaulton, G. N. (2024). A Highly Sensitive Molecular Technique for RNA Virus Detection. Cells, 13(10), 804. https://doi.org/10.3390/cells13100804