
Chronic Astrocytic TNFα Production in the Preoptic-Basal Forebrain Causes Aging-like Sleep–Wake Disturbances in Young Mice

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Experimental Subjects
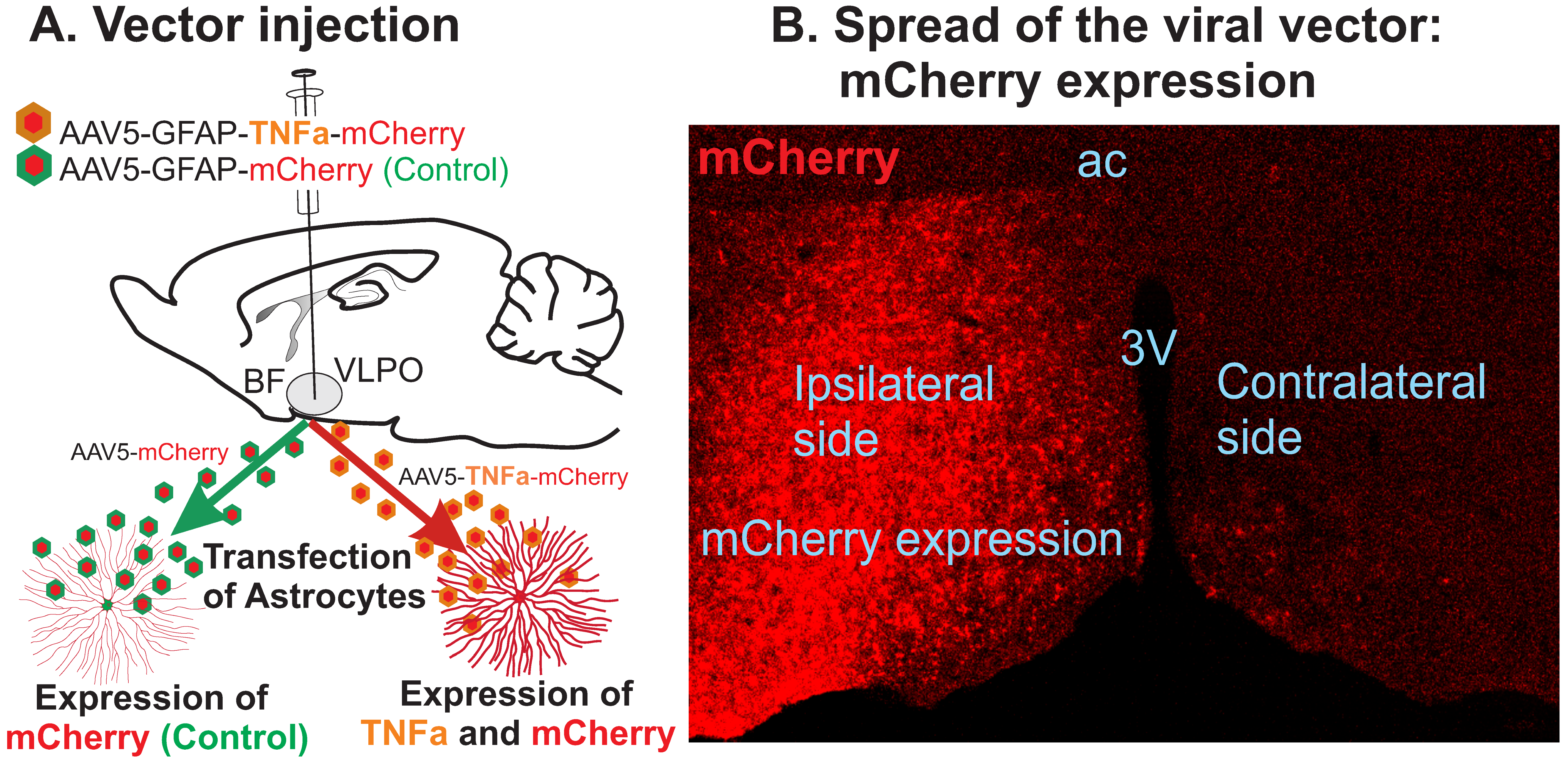
2.2. Viral Vector
2.3. Surgical Procedures
2.4. Recovery and Adaptation
2.5. Data Acquisition
2.6. Perfusion and Histological Processing
2.7. Data Analyses
3. Results
3.1. TNF-α Expression in the POA-BF
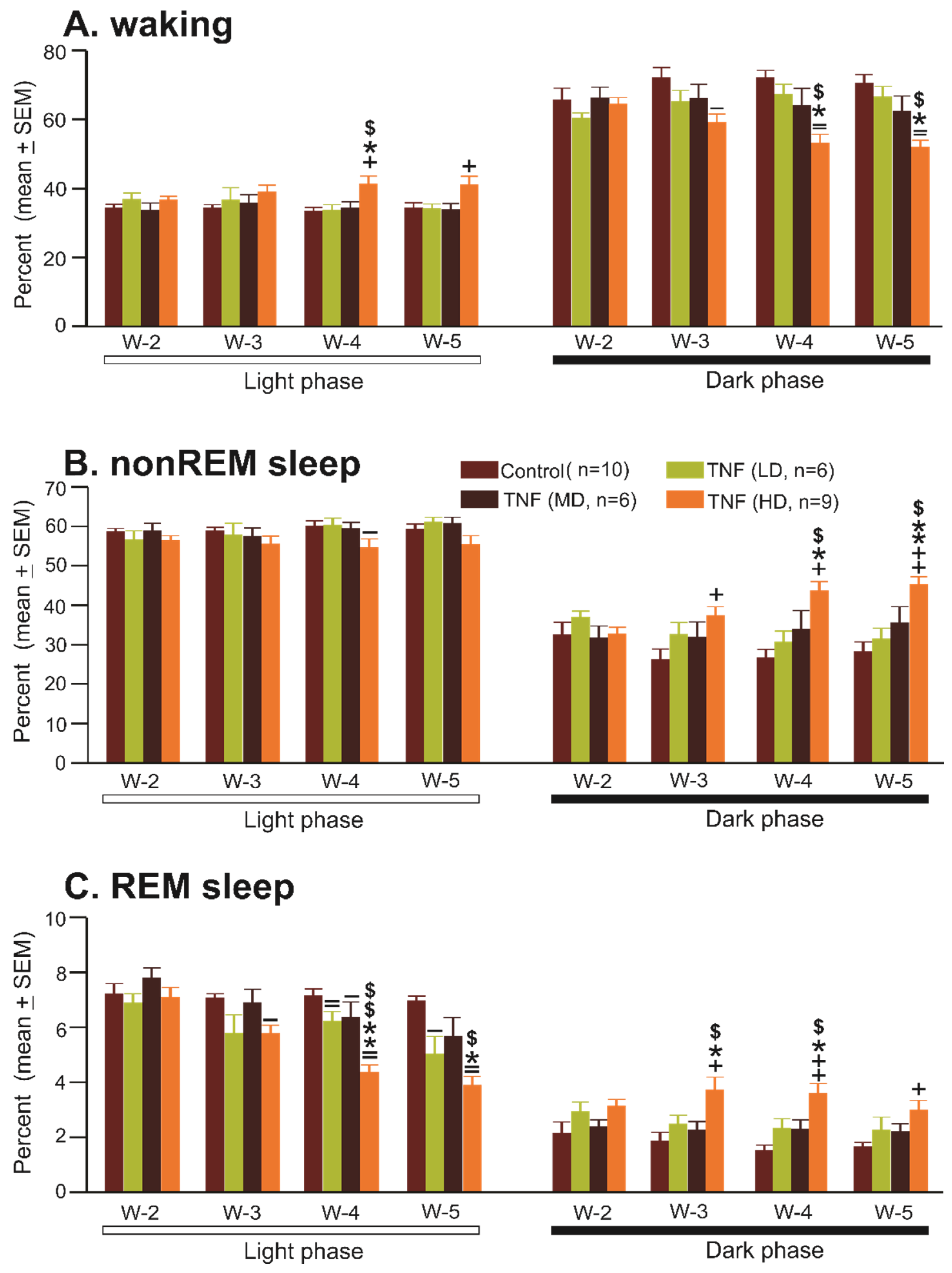
3.2. Effects of Chronic TNFα Expression/Production in the POA-BF on Sleep–Wake Architecture
3.2.1. Effects of AAV5-GFAP-mCherry Injections (Control Group) in the POA-BF on Sleep–Waking
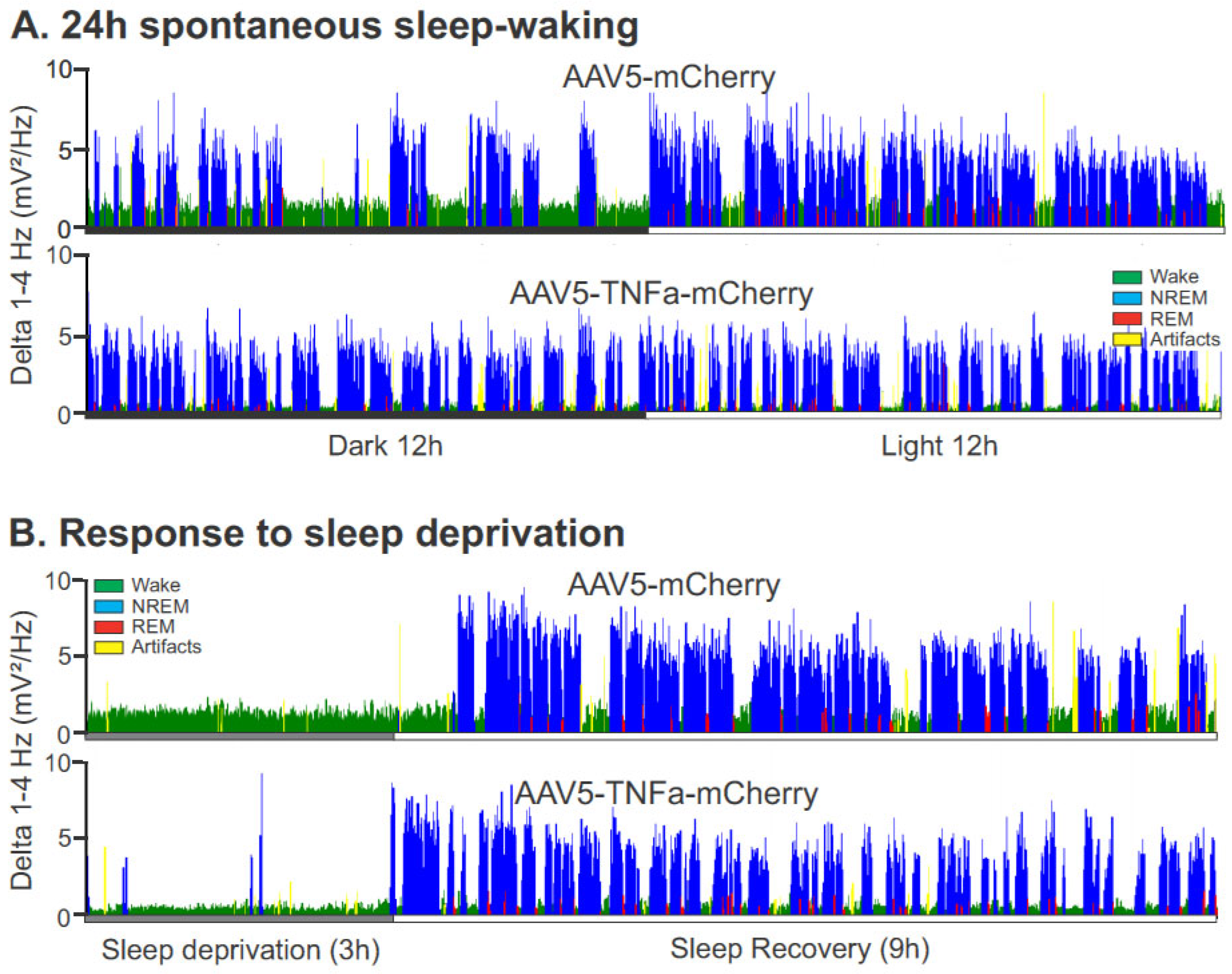
3.2.2. Effects of AAV5-GFAP-TNFα-mCherry Injections (TNFα Group) in the POA-BF on Sleep–Waking
- Sleep–wake amounts
- 2.
- Sleep–wake stability
- 3.
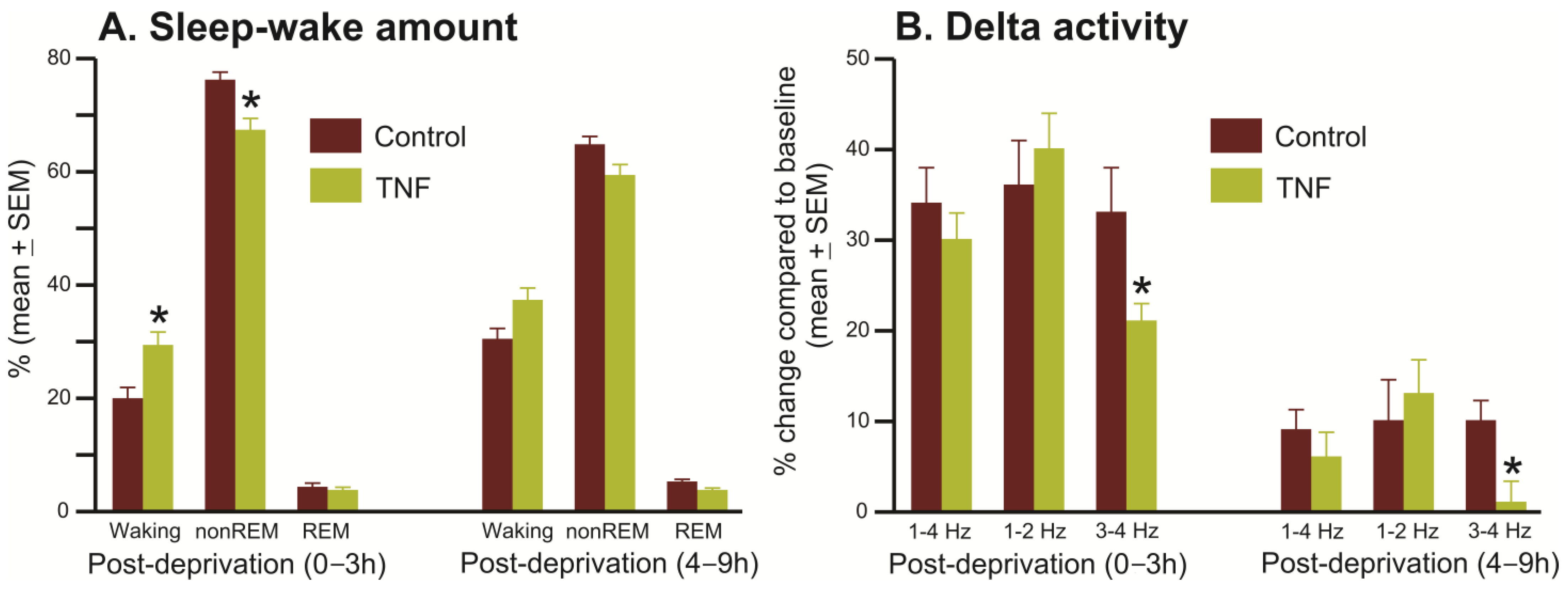
- Homeostatic sleep response
- 4.
- Response to Pharmacologically induced sleep
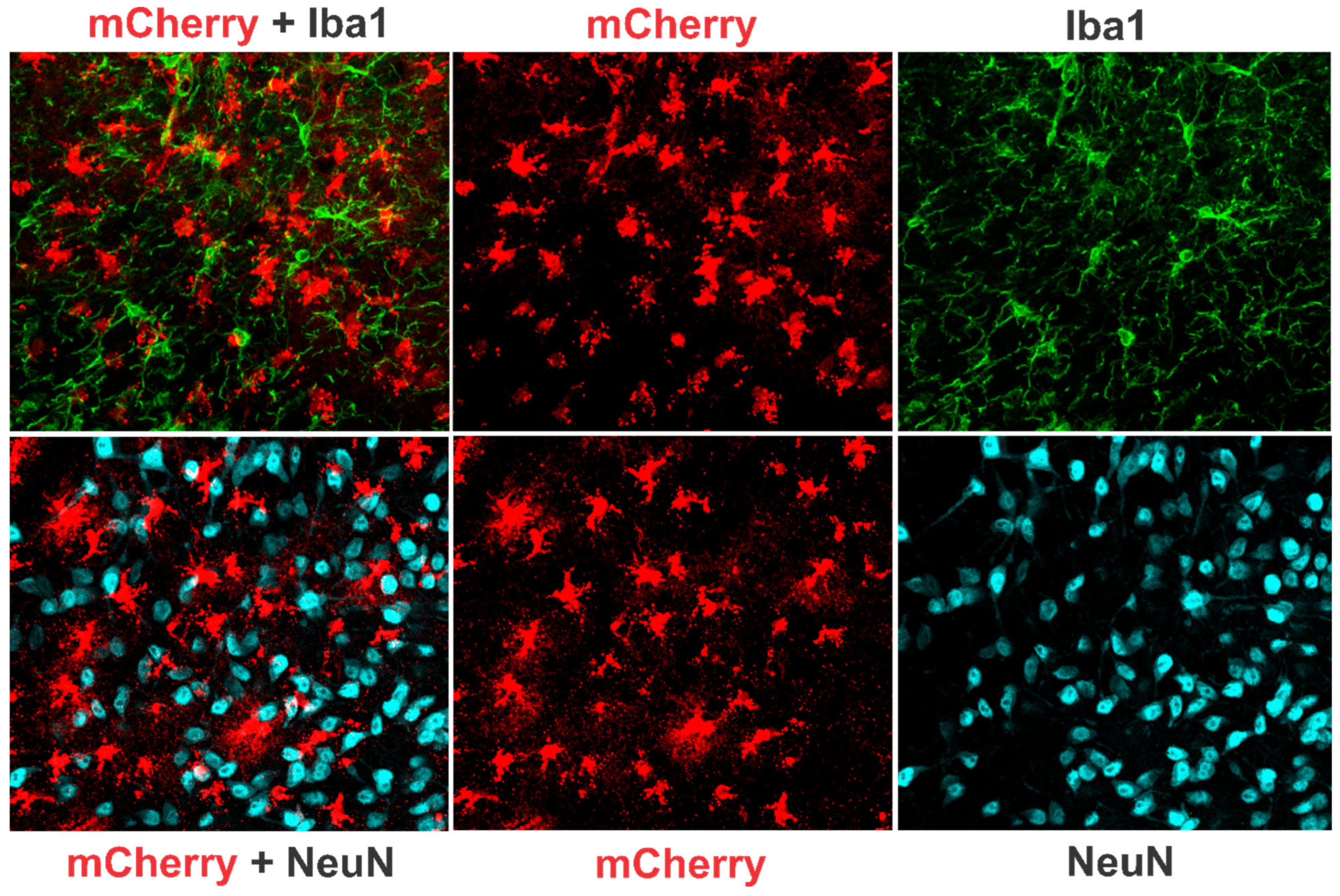
3.2.3. Effects of Chronic TNFα Expression/Production in the POA-BF on Microglia Activation
3.2.4. Effects of Chronic TNFα Expression/Production in the POA-BF on Locomotor Activity and Motor Functions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, J.; Vitiello, M.V.; Gooneratne, N.S. Sleep in Normal Aging. Sleep. Med. Clin. 2022, 17, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Taillard, J.; Gronfier, C.; Bioulac, S.; Philip, P.; Sagaspe, P. Sleep in Normal Aging, Homeostatic and Circadian Regulation and Vulnerability to Sleep Deprivation. Brain Sci. 2021, 11, 1003. [Google Scholar] [CrossRef]
- Mander, B.A.; Winer, J.R.; Walker, M.P. Sleep and Human Aging. Neuron 2017, 94, 19–36. [Google Scholar] [CrossRef]
- Hood, S.; Amir, S. The aging clock: Circadian rhythms and later life. J. Clin. Investig. 2017, 127, 437–446. [Google Scholar] [CrossRef]
- A Stowe, T.; A McClung, C. How Does Chronobiology Contribute to the Development of Diseases in Later Life. Clin. Interv. Aging 2023, 18, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Kostin, A.; Alam, M.A.; McGinty, D.; Szymusiak, R.; Alam, M.N. Chronic Suppression of Hypothalamic Cell Proliferation and Neurogenesis Induces Aging-Like Changes in Sleep–Wake Organization in Young Mice. Neuroscience 2019, 404, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Kostin, A.; Alam, M.A.; Siegel, J.M.; McGinty, D.; Alam, M.N. Sex- and Age-dependent Differences in Sleep-wake Characteristics of Fisher-344 Rats. Neuroscience 2020, 427, 29–42. [Google Scholar] [CrossRef]
- Kostin, A.; Alam, M.A.; McGinty, D.; Alam, M.N. Adult hypothalamic neurogenesis and sleep-wake dysfunction in aging. Sleep 2021, 44, zsaa173. [Google Scholar] [CrossRef]
- Shiromani, P.J.; Lu, J.; Wagner, D.; Thakkar, J.; Greco, M.A.; Basheer, R.; Thakkar, M. Compensatory sleep response to 12 h wakefulness in young and old rats. Am. J. Physiol. Integr. Comp. Physiol. 2000, 278, R125–R133. [Google Scholar] [CrossRef]
- Perry, V.H. Contribution of systemic inflammation to chronic neurodegeneration. Acta Neuropathol. 2010, 120, 277–286. [Google Scholar] [CrossRef]
- Rosano, C.; Marsland, A.L.; Gianaros, P.J. Maintaining brain health by monitoring inflammatory processes: A mechanism to promote successful aging. Aging Dis. 2012, 3, 16–33. [Google Scholar] [PubMed]
- Li, X.; Li, C.; Zhang, W.; Wang, Y.; Qian, P.; Huang, H. Inflammation and aging: Signaling pathways and intervention therapies. Signal Transduct. Targeted Ther. 2023, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lv, H.; Yin, D.; Zhou, X.; Zhu, H.; Guo, L.; Wang, J. Systemic Immune-Inflammation Index Predicts Long-Term Outcomes in Patients with Three-Vessel Coronary Disease After Revascularization: Results from a Large Cohort of 3561 Patients. J. Inflamm. Res. 2022, 15, 5283–5292. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Shin, J.; Lee, K.W. Effects of Inflammation and Depression on Telomere Length in Young Adults in the United States. J. Clin. Med. 2019, 8, 711. [Google Scholar] [CrossRef] [PubMed]
- Kordinas, V.; Ioannidis, A.; Chatzipanagiotou, S. The Telomere/Telomerase System in Chronic Inflammatory Diseases. Cause or Effect? Genes 2016, 7, 60. [Google Scholar] [CrossRef] [PubMed]
- Ekdahl, C.T.; Claasen, J.H.; Bonde, S.; Kokaia, Z.; Lindvall, O. Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13632–13637. [Google Scholar] [CrossRef] [PubMed]
- Zonis, S.; Pechnick, R.N.; A Ljubimov, V.; Mahgerefteh, M.; Wawrowsky, K.; Michelsen, K.S.; Chesnokova, V. Chronic intestinal inflammation alters hippocampal neurogenesis. J. Neuroinflamm. 2015, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.E.; Lucey, B.P.; Holtzman, D.M. Sleep and Alzheimer disease pathology—A bidirectional relationship. Nat. Rev. Neurol. 2014, 10, 115–119. [Google Scholar] [CrossRef]
- Musiek, E.S.; Xiong, D.D.; Holtzman, D.M. Sleep, circadian rhythms, and the pathogenesis of Alzheimer disease. Exp. Mol. Med. 2015, 47, e148. [Google Scholar] [CrossRef]
- Sixel-Doring, F.; Trautmann, E.; Mollenhauer, B.; Trenkwalder, C. Associated factors for REM sleep behavior disorder in Parkinson disease. Neurology 2011, 77, 1048–1054. [Google Scholar] [CrossRef]
- Alam, M.N. NREM Sleep: Anatomy and Physiology. Encyclopaedia of Sleep and Circadian Rhythms, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2023; pp. 99–108. [Google Scholar]
- Saper, C.B.; Fuller, P.M.; Pedersen, N.P.; Lu, J.; Scammell, T.E. Sleep state switching. Neuron 2010, 68, 1023–1042. [Google Scholar] [CrossRef] [PubMed]
- Rothhaas, R.; Chung, S. Role of the Preoptic Area in Sleep and Thermoregulation. Front. Neurosci. 2021, 15, 664781. [Google Scholar] [CrossRef]
- Lu, J.; Greco, M.A.; Shiromani, P.; Saper, C.B. Effect of lesions of the ventrolateral preoptic nucleus on NREM and REM sleep. J. Neurosci. 2000, 20, 3830–3842. [Google Scholar] [CrossRef]
- Kostin, A.; Alam, M.A.; Saevskiy, A.; McGinty, D.; Alam, M.N. Activation of the Ventrolateral Preoptic Neurons Projecting to the Perifornical-Hypothalamic Area Promotes Sleep: DREADD Activation in Wild-Type Rats. Cells 2022, 11, 2140. [Google Scholar] [CrossRef] [PubMed]
- Gvilia, I.; Xu, F.; McGinty, D.; Szymusiak, R. Homeostatic regulation of sleep: A role for preoptic area neurons. J. Neurosci. 2006, 26, 9426–9433. [Google Scholar] [CrossRef] [PubMed]
- Sherin, J.E.; Shiromani, P.J.; McCarley, R.W.; Saper, C.B. Activation of ventrolateral preoptic neurons during sleep. Science 1996, 271, 216–219. [Google Scholar] [CrossRef]
- Kostin, A.; Alam, M.A.; Saevskiy, A.; Yang, C.; Golshani, P.; Alam, M.N. Calcium Dynamics of the Ventrolateral Preoptic GABAergic Neurons during Spontaneous Sleep-Waking and in Response to Homeostatic Sleep Demands. Int. J. Mol. Sci. 2023, 24, 8311. [Google Scholar] [CrossRef]
- Szymusiak, R.; Alam, N.; Steininger, T.L.; McGinty, D. Sleep-waking discharge patterns of ventrolateral preoptic/anterior hypothalamic neurons in rats. Brain Res. 1998, 803, 178–188. [Google Scholar] [CrossRef]
- Alam, M.A.; Kumar, S.; McGinty, D.; Alam, M.N.; Szymusiak, R. Neuronal activity in the preoptic hypothalamus during sleep deprivation and recovery sleep. J. Neurophysiol. 2014, 111, 287–299. [Google Scholar] [CrossRef]
- Kroeger, D.; Absi, G.; Gagliardi, C.; Bandaru, S.S.; Madara, J.C.; Ferrari, L.L.; Arrigoni, E.; Munzberg, H.; Scammell, T.E.; Saper, C.B.; et al. Galanin neurons in the ventrolateral preoptic area promote sleep and heat loss in mice. Nat. Commun. 2018, 9, 4129. [Google Scholar] [CrossRef] [PubMed]
- Vanini, G.; Bassana, M.; Mast, M.; Mondino, A.; Cerda, I.; Phyle, M.; Chen, V.; Colmenero, A.; Hambrecht-Wiedbusch, V.S.; Mashour, G.A. Activation of Preoptic GABAergic or Glutamatergic Neurons Modulates Sleep-Wake Architecture, but Not Anesthetic State Transitions. Curr. Biol. 2020, 30, 779–787.e4. [Google Scholar] [CrossRef]
- Takahashi, K.; Lin, J.S.; Sakai, K. Characterization and mapping of sleep-waking specific neurons in the basal forebrain and preoptic hypothalamus in mice. Neuroscience 2009, 161, 269–292. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Brown, R.E.; Hussain Shuler, M.G.; Petersen, C.C.; Kepecs, A. Optogenetic Dissection of the Basal Forebrain Neuromodulatory Control of Cortical Activation, Plasticity, and Cognition. J. Neurosci. 2015, 35, 13896–13903. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Chung, S.; Zhang, S.; Zhong, P.; Ma, C.; Chang, W.-C.; Weissbourd, B.; Sakai, N.; Luo, L.; Nishino, S.; et al. Basal forebrain circuit for sleep-wake control. Nat. Neurosci. 2015, 18, 1641–1647. [Google Scholar] [CrossRef] [PubMed]
- Fournier, G.N.; Materi, L.M.; Semba, K.; Rasmusson, D.D. Cortical acetylcholine release and electroencephalogram activation evoked by ionotropic glutamate receptor agonists in the rat basal forebrain. Neuroscience 2004, 123, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, M.M.; Winston, S.; McCarley, R.W. A1 receptor and adenosinergic homeostatic regulation of sleep-wakefulness: Effects of antisense to the A1 receptor in the cholinergic basal forebrain. J. Neurosci. 2003, 23, 4278–4287. [Google Scholar] [CrossRef] [PubMed]
- Gritti, I.; Manns, I.D.; Mainville, L.; Jones, B.E. Parvalbumin, calbindin, or calretinin in cortically projecting and GABAergic, cholinergic, or glutamatergic basal forebrain neurons of the rat. J. Comp. Neurol. 2003, 458, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Hassani, O.K.; Lee, M.G.; Henny, P.; Jones, B.E. Discharge profiles of identified GABAergic in comparison to cholinergic and putative glutamatergic basal forebrain neurons across the sleep-wake cycle. J. Neurosci. 2009, 29, 11828–11840. [Google Scholar] [CrossRef]
- Chu, W.M. Tumor necrosis factor. Cancer Lett. 2013, 328, 222–225. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Gupta, S.C.; Kim, J.H. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 2012, 119, 651–665. [Google Scholar] [CrossRef] [PubMed]
- van Loo, G.; Bertrand, M.J.M. Death by TNF: A road to inflammation. Nat. Rev. Immunol. 2023, 23, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, A.P.; Pitha, P.M.; Shin, H.S.; Shin, M.L. Production of tumor necrosis factor and other cytokines by astrocytes stimulated with lipopolysaccharide or a neurotropic virus. Proc. Natl. Acad. Sci. USA 1989, 86, 6348–6352. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.T.; Yu, A.C. Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. J. Neurotrauma 2001, 18, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Heir, R.; Abbasi, Z.; Komal, P.; Altimimi, H.F.; Franquin, M.; Moschou, D.; Chambon, J.; Stellwagen, D. Astrocytes are the source of TNF mediating homeostatic synaptic plasticity. J. Neurosci. 2024, 44, e2278222024. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.M.; Fackelmeier, B.; Fong, D.M.; Mouravlev, A.; Young, D.; O’Carroll, S.J. Astrocyte-selective AAV gene therapy through the endogenous GFAP promoter results in robust transduction in the rat spinal cord following injury. Gene Ther. 2019, 26, 198–210. [Google Scholar] [CrossRef] [PubMed]
- O’Carroll, S.J.; Cook, W.H.; Young, D. AAV Targeting of Glial Cell Types in the Central and Peripheral Nervous System and Relevance to Human Gene Therapy. Front. Mol. Neurosci. 2020, 13, 618020. [Google Scholar] [CrossRef] [PubMed]
- Kostin, A.; McGinty, D.; Szymusiak, R.; Alam, M.N. Sleep-wake and diurnal modulation of nitric oxide in the perifornical-lateral hypothalamic area: Real-time detection in freely behaving rats. Neuroscience 2013, 254, 275–284. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K.J. The Mouse Brain in Stereotaxic Coordinates, 2nd ed.; Academic Press: San Diego, CA, USA, 2001. [Google Scholar]
- Seibenhener, M.L.; Wooten, M.C. Use of the Open Field Maze to measure locomotor and anxiety-like behavior in mice. J. Vis. Exp. 2015, 96, e52434. [Google Scholar]
- Luo, L.; Sun, T.; Yang, L.; Liu, A.; Liu, Q.Q.; Tian, Q.Q.; Wang, Y.; Zhao, M.G.; Yang, Q. Scopoletin ameliorates anxiety-like behaviors in complete Freund’s adjuvant-induced mouse model. Mol. Brain 2020, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Wang, T.; Chen, X.; Zhao, Z.; Chen, Z. Gut microbiota relieves inflammation in the substantia nigra of chronic Parkinson’s disease by protecting the function of dopamine neurons. Exp. Ther. Med. 2022, 23, 52. [Google Scholar] [CrossRef] [PubMed]
- Shiotsuki, H.; Yoshimi, K.; Shimo, Y.; Funayama, M.; Takamatsu, Y.; Ikeda, K.; Takahashi, R.; Kitazawa, S.; Hattori, N. A rotarod test for evaluation of motor skill learning. J. Neurosci. Methods 2010, 189, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Deacon, R.M. Measuring motor coordination in mice. J. Vis. Exp. 2013, 75, e2609. [Google Scholar]
- Klein, S.M.; Vykoukal, J.; Lechler, P.; Zeitler, K.; Gehmert, S.; Schreml, S.; Alt, E.; Bogdahn, U.; Prantl, L. Noninvasive in vivo assessment of muscle impairment in the mdx mouse model—A comparison of two common wire hanging methods with two different results. J. Neurosci. Methods 2012, 203, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Bonetto, A.; Andersson, D.C.; Waning, D.L. Assessment of muscle mass and strength in mice. Bonekey Rep. 2015, 4, 732. [Google Scholar] [CrossRef] [PubMed]
- Winrow, C.J.; Gotter, A.L.; Cox, C.D.; Doran, S.M.; Tannenbaum, P.L.; Breslin, M.J.; Garson, S.L.; Fox, S.V.; Harrell, C.M.; Stevens, J.; et al. Promotion of sleep by suvorexant-a novel dual orexin receptor antagonist. J. Neurogenet. 2011, 25, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.V.; Aspesi, A.V.; Evoy, K.E. Suvorexant: A dual orexin receptor antagonist for the treatment of sleep onset and sleep maintenance insomnia. Ann. Pharmacother. 2015, 49, 477–483. [Google Scholar] [CrossRef]
- Etori, K.; Saito, Y.C.; Tsujino, N.; Sakurai, T. Effects of a newly developed potent orexin-2 receptor-selective antagonist, compound 1 m, on sleep/wakefulness states in mice. Front. Neurosci. 2014, 8, 8. [Google Scholar] [CrossRef]
- Caplan, H.W.; Cardenas, F.; Gudenkauf, F.; Zelnick, P.; Xue, H.; Cox, C.S.; Bedi, S.S. Spatiotemporal Distribution of Microglia After Traumatic Brain Injury in Male Mice. ASN Neuro 2020, 12, 1759091420911770. [Google Scholar] [CrossRef]
- Woodburn, S.C.; Bollinger, J.L.; Wohleb, E.S. The semantics of microglia activation: Neuroinflammation, homeostasis, and stress. J. Neuroinflamm. 2021, 18, 258. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.A.; Vissel, B. Inflammation-sleep interface in brain disease: TNF, insulin, orexin. J. Neuroinflamm. 2014, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Heir, R.; Stellwagen, D. TNF-Mediated Homeostatic Synaptic Plasticity: From in vitro to in vivo Models. Front. Cell. Neurosci. 2020, 14, 565841. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Tansey, M.G. TNF signaling inhibition in the CNS: Implications for normal brain function and neurodegenerative disease. J. Neuroinflamm. 2008, 5, 45. [Google Scholar] [CrossRef] [PubMed]
- Neniskyte, U.; Vilalta, A.; Brown, G.C. Tumour necrosis factor alpha-induced neuronal loss is mediated by microglial phagocytosis. FEBS Lett. 2014, 588, 2952–2956. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.A.; Alleva, L.M.; Vissel, B. The roles of TNF in brain dysfunction and disease. Pharmacol. Ther. 2010, 128, 519–548. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Caldito, N. Role of tumor necrosis factor-alpha in the central nervous system: A focus on autoimmune disorders. Front. Immunol. 2023, 14, 1213448. [Google Scholar] [CrossRef] [PubMed]
- Coborn, J.E.; DePorter, D.P.; Mavanji, V.; Sinton, C.M.; Kotz, C.M.; Billington, C.J.; Teske, J.A. Role of orexin-A in the ventrolateral preoptic area on components of total energy expenditure. Int. J. Obes. 2017, 41, 1256–1262. [Google Scholar] [CrossRef]
- Mavanji, V.; Perez-Leighton, C.E.; Kotz, C.M.; Billington, C.J.; Parthasarathy, S.; Sinton, C.M.; Teske, J.A. Promotion of Wakefulness and Energy Expenditure by Orexin-A in the Ventrolateral Preoptic Area. Sleep 2015, 38, 1361–1370. [Google Scholar] [CrossRef]
- Alam, M.N.; McGinty, D.; Bashir, T.; Kumar, S.; Imeri, L.; Opp, M.R.; Szymusiak, R. Interleukin-1beta modulates state-dependent discharge activity of preoptic area and basal forebrain neurons: Role in sleep regulation. Eur. J. Neurosci. 2004, 20, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Baker, F.C.; Shah, S.; Stewart, D.; Angara, C.; Gong, H.; Szymusiak, R.; Opp, M.R.; McGinty, D. Interleukin 1β enhances non-rapid eye movement sleep and increases c-Fos protein expression in the median preoptic nucleus of the hypothalamus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R998–R1005. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Chen, J.; Ji, W.; Xu, L.; Xie, Y.; He, S.; Lai, C.; Hou, K.; Li, Z.; Chen, G.; et al. High-titer AAV disrupts cerebrovascular integrity and induces lymphocyte infiltration in adult mouse brain. Mol. Ther. Methods Clin. Dev. 2023, 31, 101102. [Google Scholar] [CrossRef] [PubMed]
- Kohler, S.; Winkler, U.; Hirrlinger, J. Heterogeneity of Astrocytes in Grey and White Matter. Neurochem. Res. 2021, 46, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.A.; Ao, Y.; Sofroniew, M.V. Heterogeneity of reactive astrocytes. Neurosci. Lett. 2014, 565, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Holt, M.G. Astrocyte heterogeneity and interactions with local neural circuits. Essays Biochem. 2023, 67, 93–106. [Google Scholar] [PubMed]
- Biesmans, S.; Meert, T.F.; Bouwknecht, J.A.; Acton, P.D.; Davoodi, N.; De Haes, P.; Kuijlaars, J.; Langlois, X.; Matthews, L.J.; Ver Donck, L.; et al. Systemic immune activation leads to neuroinflammation and sickness behavior in mice. Mediat. Inflamm. 2013, 2013, 271359. [Google Scholar] [CrossRef] [PubMed]
- Kondziella, W. A New Method for the Measurement of Muscle Relaxation in White Mice. Arch. Int. Pharmacodyn. Ther. 1964, 152, 277–284. [Google Scholar] [PubMed]
- Norman, K.; Stobaus, N.; Kulka, K.; Schulzke, J. Effect of inflammation on handgrip strength in the non-critically ill is independent from age, gender and body composition. Eur. J. Clin. Nutr. 2014, 68, 155–158. [Google Scholar] [CrossRef]
- Wu, D.; Gao, X.; Shi, Y.; Wang, H.; Wang, W.; Li, Y.; Zheng, Z. Association between Handgrip Strength and the Systemic Immune-Inflammation Index: A Nationwide Study, NHANES 2011–2014. Int. J. Environ. Res. Public Health 2022, 19, 13616. [Google Scholar] [CrossRef]
- Reid, W.D.; Rurak, J.; Harris, R.L. Skeletal muscle response to inflammation—Lessons for chronic obstructive pulmonary disease. Crit. Care Med. 2009, 37, S372–S383. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, C.S.L.; Thang, L.A.N.; Maier, A.B. Markers of inflammation and their association with muscle strength and mass: A systematic review and meta-analysis. Ageing Res. Rev. 2020, 64, 101185. [Google Scholar] [CrossRef] [PubMed]
- Klaus, F.; Paterna, J.C.; Marzorati, E.; Sigrist, H.; Gotze, L.; Schwendener, S.; Bergamini, G.; Jehli, E.; Azzinnari, D.; Fuertig, R.; et al. Differential effects of peripheral and brain tumor necrosis factor on inflammation, sickness, emotional behavior and memory in mice. Brain Behav. Immun. 2016, 58, 310–326. [Google Scholar] [CrossRef] [PubMed]
- Chennaoui, M.; Gomez-Merino, D.; Drogou, C.; Geoffroy, H.; Dispersyn, G.; Langrume, C.; Ciret, S.; Gallopin, T.; Sauvet, F. Effects of exercise on brain and peripheral inflammatory biomarkers induced by total sleep deprivation in rats. J. Inflamm. 2015, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Zhai, S.; Tao, S.; Wu, X.; Zou, L.; Yang, Y.; Xie, Y.; Li, T.; Zhang, D.; Qu, Y.; Tao, F. Associations of Sleep Insufficiency and Chronotype with Inflammatory Cytokines in College Students. Nat. Sci. Sleep 2021, 13, 1675–1685. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.T.; Kim, Y.J.; Nguyen, P.T.; Koh, Y.H.; Nguyen, T.T.; Ma, H.I.; Kim, Y.E. Temporal Evolution of Inflammation and Neurodegeneration with Alpha-Synuclein Propagation in Parkinson’s Disease Mouse Model. Front. Integr. Neurosci. 2021, 15, 715190. [Google Scholar] [CrossRef]
- Zhang, Z.; Ma, Z.; Zou, W.; Guo, H.; Liu, M.; Ma, Y.; Zhang, L. The Appropriate Marker for Astrocytes: Comparing the Distribution and Expression of Three Astrocytic Markers in Different Mouse Cerebral Regions. Biomed. Res. Int. 2019, 2019, 9605265. [Google Scholar] [CrossRef] [PubMed]
- Bushong, E.A.; Martone, M.E.; Jones, Y.Z.; Ellisman, M.H. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 2002, 22, 183–192. [Google Scholar] [CrossRef]
- Walz, W.; Lang, M.K. Immunocytochemical evidence for a distinct GFAP-negative subpopulation of astrocytes in the adult rat hippocampus. Neurosci. Lett. 1998, 257, 127–130. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Light Phase | Week 2 | Week 3 | Week 4 | Week 5 | F Value | p Value |
| Control vector | ||||||
| Waking | 34.2 ± 1.1 | 34.1 ± 1.0 | 33.2 ± 1.3 | 36.3 ± 2.1 | F9,3 = 1.07 | 0.38 |
| nonREM sleep | 58.6 ± 0.9 | 58.8 ± 1.0 | 59.7 ± 1.3 | 59.1 ± 1.5 | 0.67 | 0.58 |
| REM sleep | 7.2 ± 0.4 | 7.0 ± 0.2 | 7.2 ± 0.2 | 6.9 ± 0.2 | 0 | 0.76 |
| TNF vector-Low | ||||||
| Waking | 36.7 ± 2.4 | 36.6 ± 3.6 | 33.6 ± 1.7 | 34.0 ± 1.5 | F5,3 = 0.69 | 0.57 |
| nonREM sleep | 61.0 ± 1.3 | 60.2 ± 1.9 | 57.65 ± 3.2 | 56.5 ± 2.4 | 1.26 | 0.32 |
| REM sleep | 6.9 ± 0.4 | 5.8 ± 0.7 | 6.2 ± 0.4 | 5.0 ± 0.7 *,$ | 5.85 | <0.01 |
| TNF-vector Medium | ||||||
| Waking | 33.5 ± 2.3 | 35.7 ± 2.4 | 34.2 ± 1.9 | 33.7 ± 1.9 | F5,3 = 0.66 | 0.58 |
| nonREM sleep | 58.8 ± 2.0 | 57.4 ± 2.1 | 59.4 ± 1.6 | 60.7 ± 1.6 | 2.68 | 0.08 |
| REM sleep | 7.7 ± 0.4 | 6.9 ± 0.5 | 6.4 ± 0.6 | 5.7 ± 0.7 * | 3.76 | <0.05 |
| TNF-vector High | ||||||
| Waking | 36.5 ± 1.2 | 38.8 ± 2.1 | 41.1 ± 2.4 * | 40.9 ± 2.6 * | F8,3 = 4.86 | 0.009 |
| nonREM sleep | 56.4 ± 1.3 | 55.4 ± 2.1 | 54.5 ± 2.3 | 55.3 ± 2.4 | 0.77 | 0.51 |
| REM sleep | 7.1 ± 0.4 | 5.8 ± 0.3 ** | 4.3 ± 0.3 **,$$ | 3.9 ± 0.3 **,$$ | 34.95 | <0.001 |
| Dark Phase | Week 2 | Week 3 | Week 4 | Week 5 | F Value | p Value |
| Control vector | ||||||
| Waking | 65.4 ± 3.6 | 72.0 ± 3.0 | 71.9 ± 2.3 | 71.2 ± 2.3 | 3.12 | 0.04 |
| nonREM sleep | 32.4 ± 3.3 | 26.1 ± 2.8 | 26.6 ± 2.2 | 27.0 ± 2.3 | 3.18 | 0.04 |
| REM sleep | 2.1 ± 0.4 | 1.9 ± 0.3 | 1.5 ± 0.2 | 1.7 ± 0.2 | 1.62 | 0.2 |
| TNF vector-Low | ||||||
| Waking | 60.2 ± 1.9 | 65.0 ± 3.4 | 67.1 ± 3.0 | 66.4 ± 3.2 | 2.41 | 0.11 |
| nonREM sleep | 36.8 ± 1.7 | 32.5 ± 3.1 | 30.6 ± 2.8 | 31.3 ± 2.8 | 2.35 | 0.11 |
| REM sleep | 3.0 ± 0.4 | 2.5 ± 0.3 | 2.3 ± 0.4 | 2.3 ± 0.5 | 1.85 | 0.18 |
| TNF-vector Medium | ||||||
| Waking | 66.0 ± 2.3 | 65.9 ± 4.2 | 63.9 ± 5.1 | 62.3 ± 4.4 | 2.37 | 0.11 |
| nonREM sleep | 31.6 ± 3.2 | 31.9 ± 3.9 | 33.9 ± 4.8 | 35.5 ± 4.1 | 2.67 | 0.09 |
| REM sleep | 2.4 ± 0.3 | 2.2 ± 0.3 | 2.3 ± 0.3 | 2.2 ± 0.3 | 0.22 | 0.87 |
| TNF-vector High | ||||||
| Waking | 64.3 ± 2.0 | 58.9 ± 2.5 * | 52.9 ± 2.7 *,$ | 51.8 ± 2.14 *,$ | 28.97 | <0.001 |
| nonREM sleep | 32.6 ± 1.8 | 37.3 ± 2.2 ** | 43.5 ± 2.5 **,$$ | 45.2 ± 2.0 **,$$ | 39.3 | <0.001 |
| REM sleep | 3.1 ± 0.3 | 3.7 ± 0.5 ** | 3.6 ± 0.4 | 2.98 ± 0.4 | 3.76 | <0.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostin, A.; Alam, M.A.; Saevskiy, A.; Alam, M.N. Chronic Astrocytic TNFα Production in the Preoptic-Basal Forebrain Causes Aging-like Sleep–Wake Disturbances in Young Mice. Cells 2024, 13, 894. https://doi.org/10.3390/cells13110894
Kostin A, Alam MA, Saevskiy A, Alam MN. Chronic Astrocytic TNFα Production in the Preoptic-Basal Forebrain Causes Aging-like Sleep–Wake Disturbances in Young Mice. Cells. 2024; 13(11):894. https://doi.org/10.3390/cells13110894
Chicago/Turabian StyleKostin, Andrey, Md. Aftab Alam, Anton Saevskiy, and Md. Noor Alam. 2024. "Chronic Astrocytic TNFα Production in the Preoptic-Basal Forebrain Causes Aging-like Sleep–Wake Disturbances in Young Mice" Cells 13, no. 11: 894. https://doi.org/10.3390/cells13110894