

Structural Dynamics Analysis of USP14 Activation by AKT-Mediated Phosphorylation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
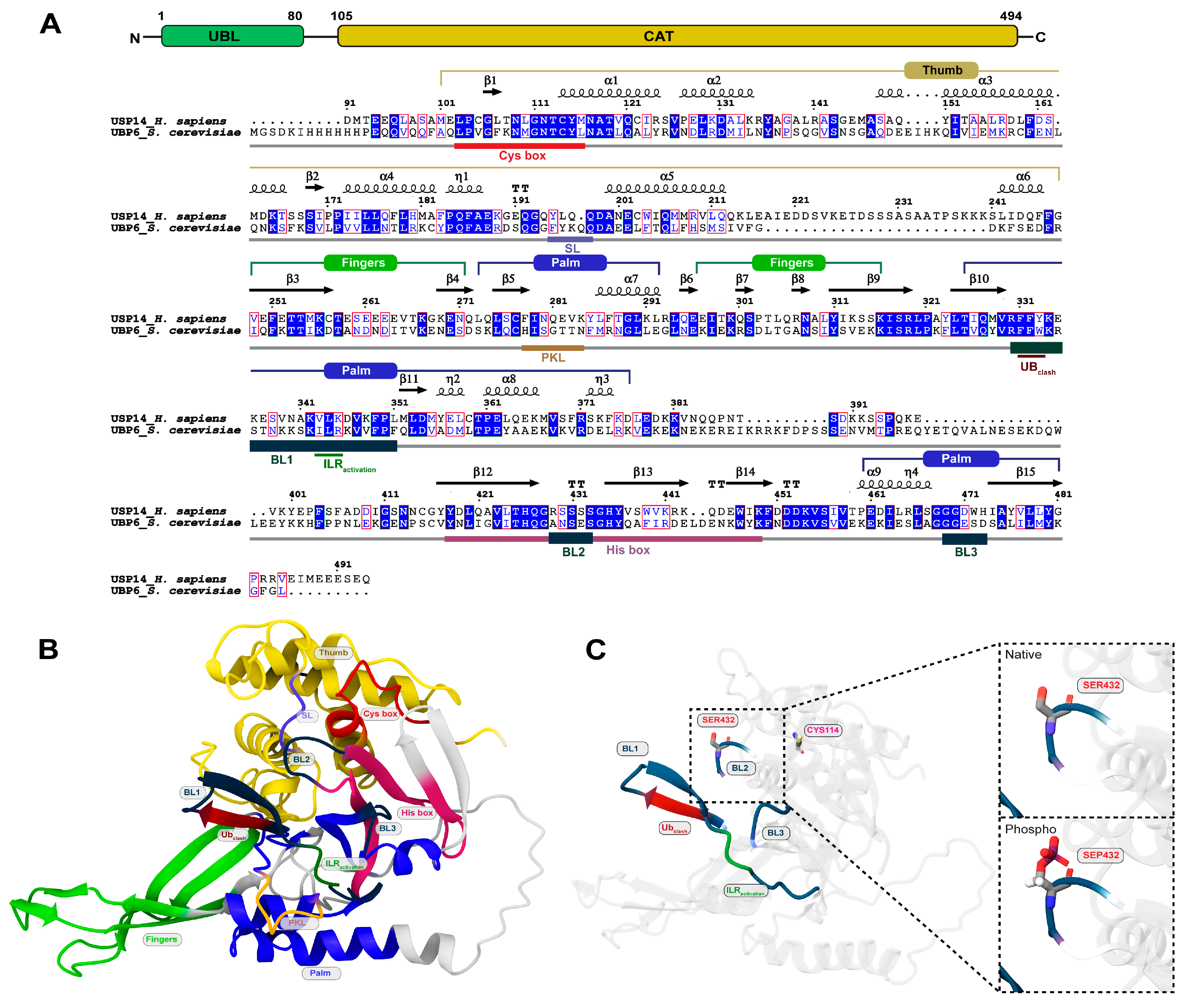
2. Materials and Methods
2.1. Model Building
2.2. Molecular Dynamics (MD) Simulation
2.3. Trajectory Clustering Analysis
2.4. Free Energy Landscape (FEL)
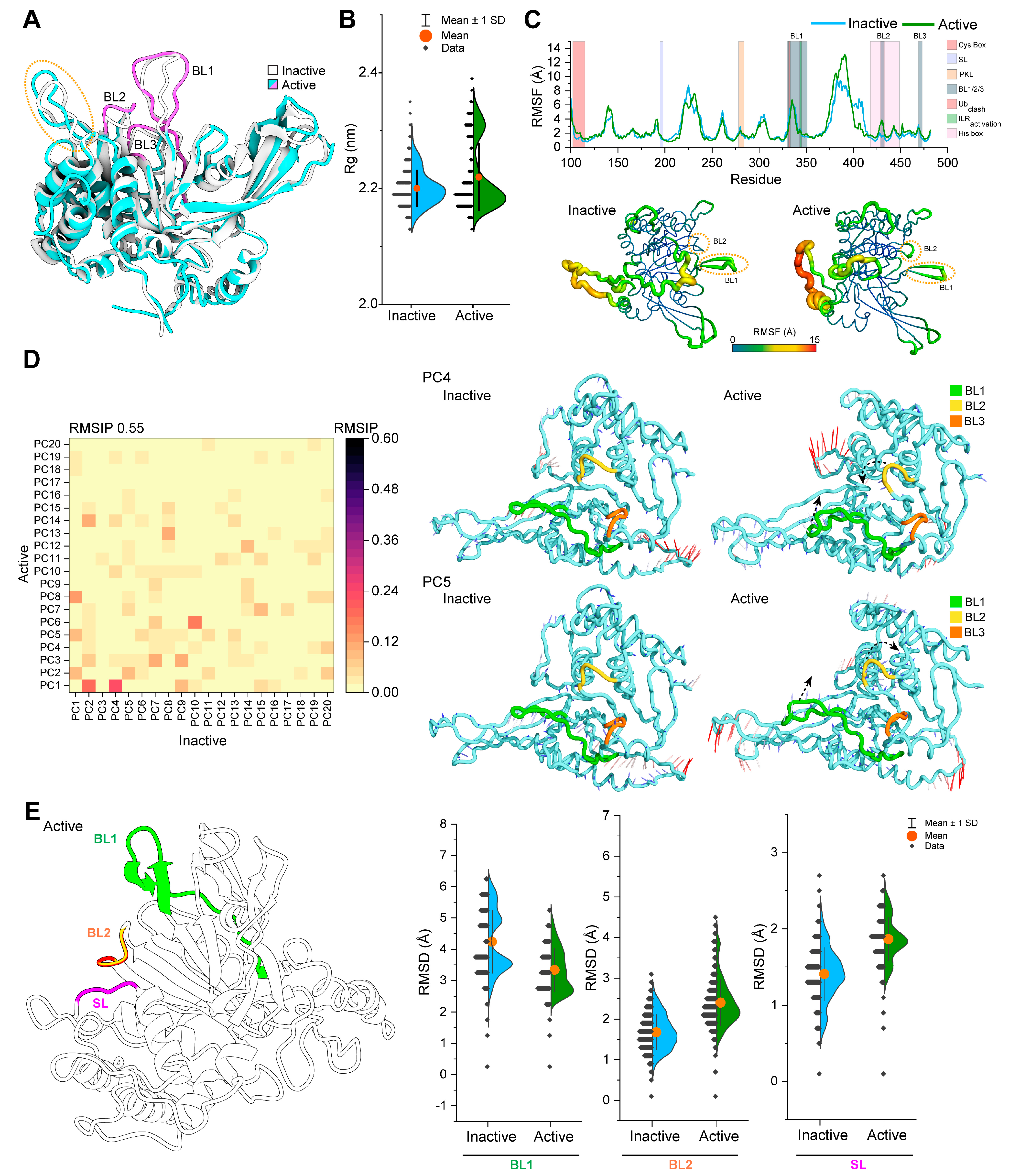
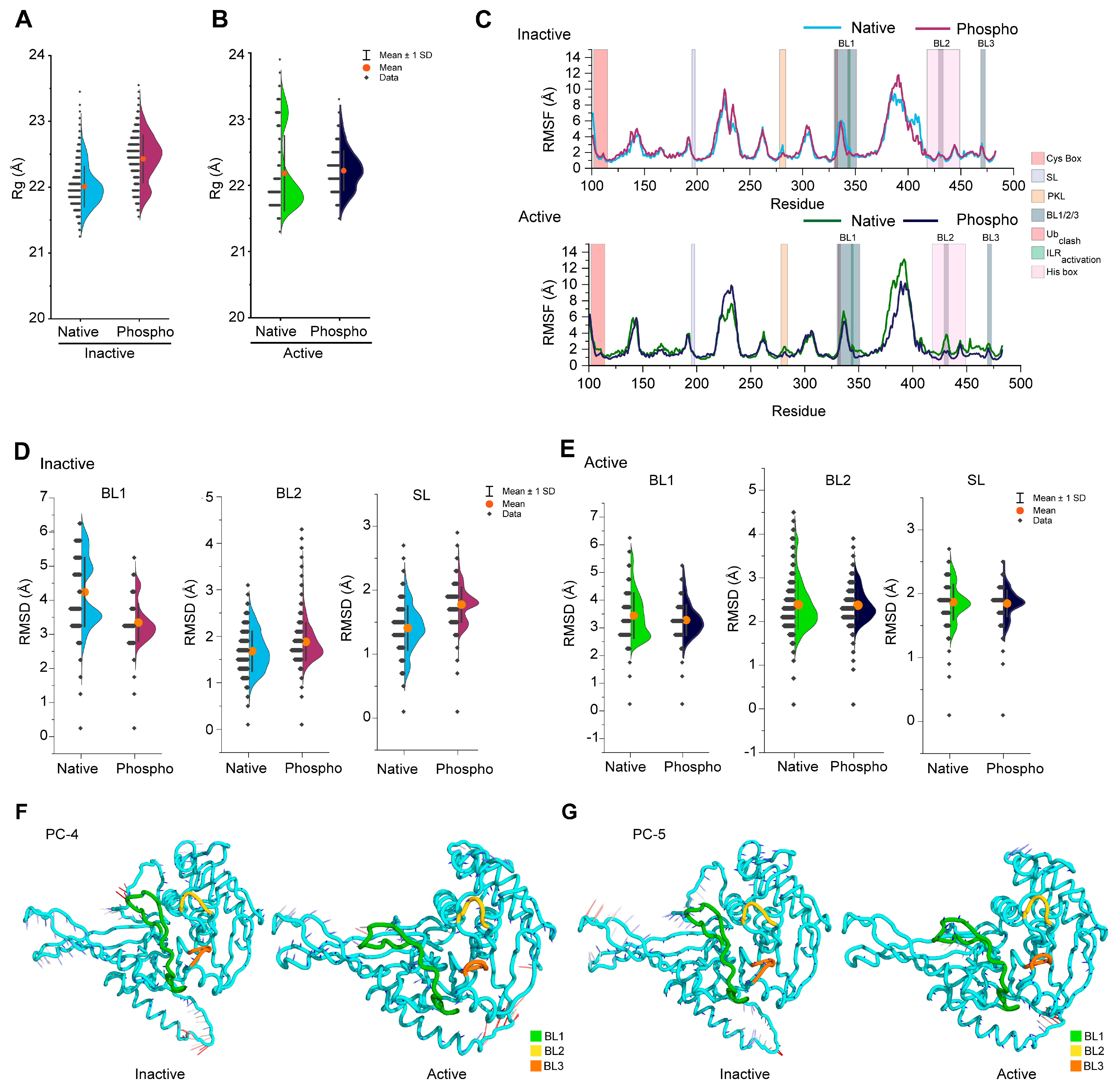
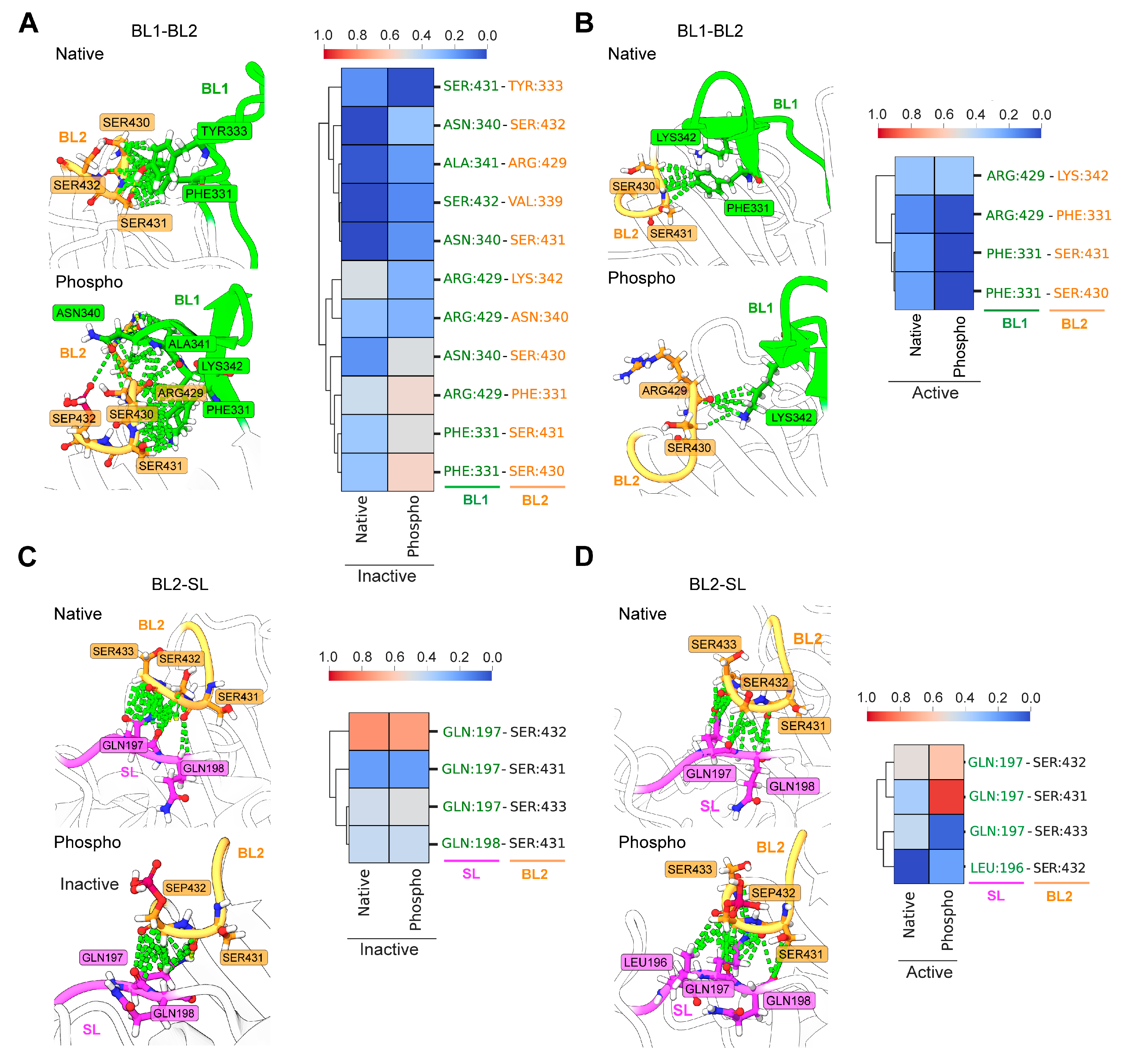
3. Results
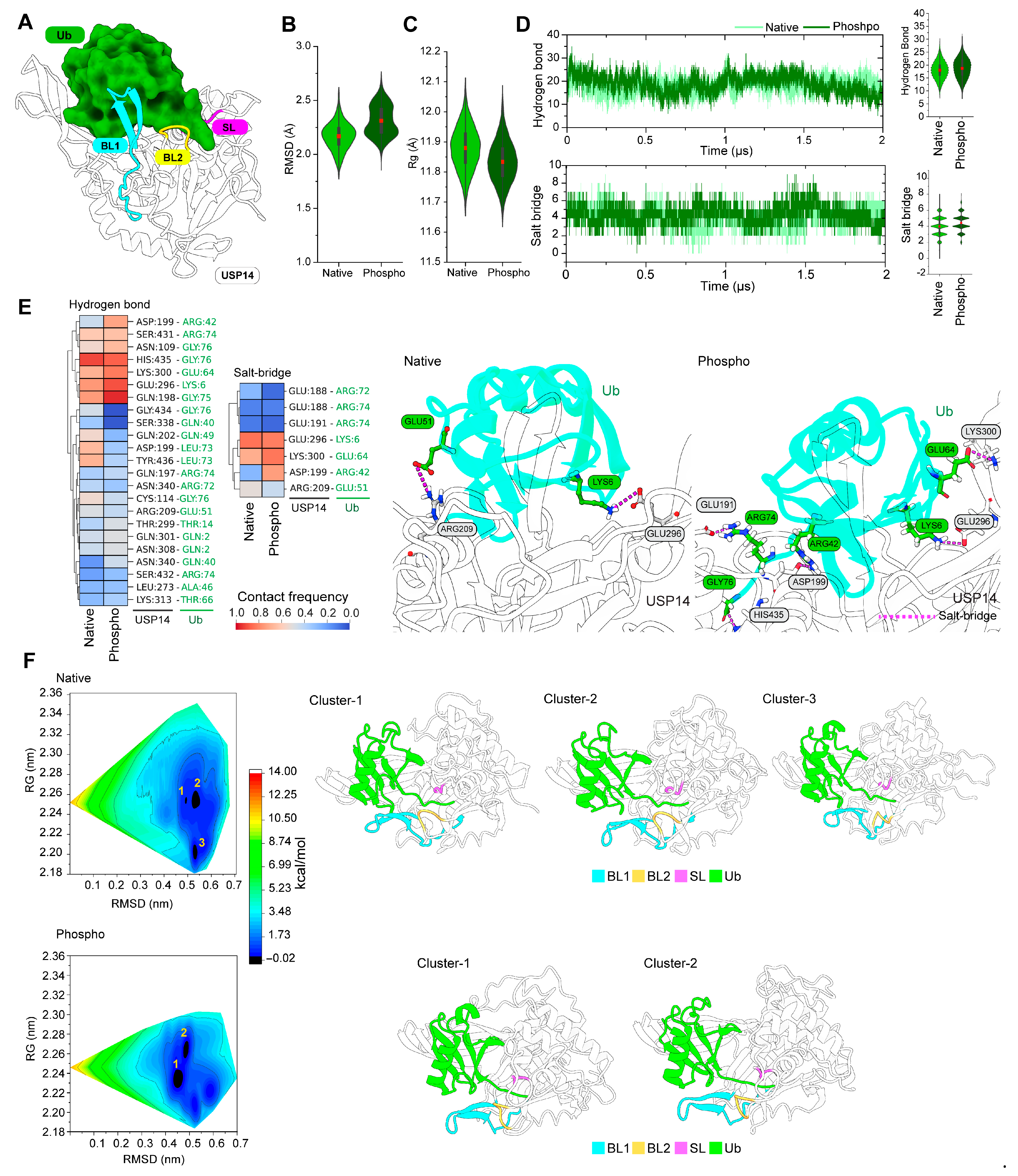
3.1. Active USP14 Displays Different Dynamics from Its Inactive state
3.2. Phosphorylation Changes the Conformational Dynamics of Blocking Loops in the USP14
3.3. Phosphorylation Affects the Dynamic Interactions in the Inhibitory Network of Blocking Loops
3.4. Phosphorylation Induces a β-Sheet Structure in the Inactive USP14 BL1 Loop
3.5. Phosphorylation Increases the Salt Bridge Interactions between USP14 and Ub
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Höhfeld, J.; Benzing, T.; Bloch, W.; Fürst, D.O.; Gehlert, S.; Hesse, M.; Hoffmann, B.; Hoppe, T.; Huesgen, P.F.; Köhn, M.; et al. Maintaining proteostasis under mechanical stress. EMBO Rep. 2021, 22, e52507. [Google Scholar] [CrossRef] [PubMed]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef] [PubMed]
- Muniyappan, S.; Lee, B.H. In vitro analysis of proteasome-associated USP14 activity for substrate degradation and deubiquitylation. Methods Enzym. 2019, 619, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef] [PubMed]
- Finley, D.; Prado, M.A. The Proteasome and Its Network: Engineering for Adaptability. Cold Spring Harb. Perspect. Biol. 2020, 12, a033985. [Google Scholar] [CrossRef] [PubMed]
- Leggett, D.S.; Hanna, J.; Borodovsky, A.; Crosas, B.; Schmidt, M.; Baker, R.T.; Walz, T.; Ploegh, H.; Finley, D. Multiple associated proteins regulate proteasome structure and function. Mol. Cell 2002, 10, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Lee, M.J.; Park, S.; Oh, D.C.; Elsasser, S.; Chen, P.C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Lu, Y.; Prado, M.A.; Shi, Y.; Tian, G.; Sun, S.; Elsasser, S.; Gygi, S.P.; King, R.W.; Finley, D. USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature 2016, 532, 398–401. [Google Scholar] [CrossRef]
- Kim, H.T.; Goldberg, A.L. The deubiquitinating enzyme Usp14 allosterically inhibits multiple proteasomal activities and ubiquitin-independent proteolysis. J. Biol. Chem. 2017, 292, 9830–9839. [Google Scholar] [CrossRef]
- Peth, A.; Kukushkin, N.; Bossé, M.; Goldberg, A.L. Ubiquitinated proteins activate the proteasomal ATPases by binding to Usp14 or Uch37 homologs. J. Biol. Chem. 2013, 288, 7781–7790. [Google Scholar] [CrossRef] [PubMed]
- Peth, A.; Besche, H.C.; Goldberg, A.L. Ubiquitinated proteins activate the proteasome by binding to Usp14/Ubp6, which causes 20S gate opening. Mol. Cell 2009, 36, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zou, S.; Yin, D.; Zhao, L.; Finley, D.; Wu, Z.; Mao, Y. USP14-regulated allostery of the human proteasome by time-resolved cryo-EM. Nature 2022, 605, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Wang, X. From Discovery to Bedside: Targeting the Ubiquitin System. Cell Chem. Biol. 2019, 26, 156–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mazurkiewicz, M.; Hillert, E.K.; Olofsson, M.H.; Pierrou, S.; Hillertz, P.; Gullbo, J.; Selvaraju, K.; Paulus, A.; Akhtar, S.; et al. The proteasome deubiquitinase inhibitor VLX1570 shows selectivity for ubiquitin-specific protease-14 and induces apoptosis of multiple myeloma cells. Sci. Rep. 2016, 6, 26979. [Google Scholar] [CrossRef] [PubMed]
- Boselli, M.; Lee, B.H.; Robert, J.; Prado, M.A.; Min, S.W.; Cheng, C.; Silva, M.C.; Seong, C.; Elsasser, S.; Hatle, K.M.; et al. An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. J. Biol. Chem. 2017, 292, 19209–19225. [Google Scholar] [CrossRef] [PubMed]
- Homma, T.; Ishibashi, D.; Nakagaki, T.; Fuse, T.; Mori, T.; Satoh, K.; Atarashi, R.; Nishida, N. Ubiquitin-specific protease 14 modulates degradation of cellular prion protein. Sci. Rep. 2015, 5, 11028. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Li, P.; Song, L.; Jeffrey, P.D.; Chenova, T.A.; Wilkinson, K.D.; Cohen, R.E.; Shi, Y. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. Embo J. 2005, 24, 3747–3756. [Google Scholar] [CrossRef] [PubMed]
- Hung, K.Y.S.; Klumpe, S.; Eisele, M.R.; Elsasser, S.; Tian, G.; Sun, S.; Moroco, J.A.; Cheng, T.C.; Joshi, T.; Seibel, T.; et al. Allosteric control of Ubp6 and the proteasome via a bidirectional switch. Nat. Commun. 2022, 13, 838. [Google Scholar] [CrossRef]
- Shin, J.Y.; Muniyappan, S.; Tran, N.N.; Park, H.; Lee, S.B.; Lee, B.H. Deubiquitination Reactions on the Proteasome for Proteasome Versatility. Int. J. Mol. Sci. 2020, 21, 5312. [Google Scholar] [CrossRef]
- Huang, X.; Luan, B.; Wu, J.; Shi, Y. An atomic structure of the human 26S proteasome. Nat. Struct. Mol. Biol. 2016, 23, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Shan, B.; Lee, B.H.; Zhu, K.; Zhang, T.; Sun, H.; Liu, M.; Shi, L.; Liang, W.; Qian, L.; et al. Phosphorylation and activation of ubiquitin-specific protease-14 by Akt regulates the ubiquitin-proteasome system. Elife 2015, 4, e10510. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, LLC. Tools: Schrödinger Suite 2017-1; Schrödinger, LLC: New York, NY, USA, 2017. [Google Scholar]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Shaw, D.E. A fast, scalable method for the parallel evaluation of distance-limited pairwise particle interactions. J. Comput. Chem. 2005, 26, 1318–1328. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 84-es. [Google Scholar]
- Shaik, M.S.; Liem, S.Y.; Popelier, P.L.A. Properties of liquid water from a systematic refinement of a high-rank multipolar electrostatic potential. J. Chem. Phys. 2010, 132, 174504. [Google Scholar] [CrossRef]
- Arpit Kumar, P.; Ashwin, J.; Nithishwer, M.A.; Shyamasree, G. Insilico analysis of structural and functional impact of SNPs in Pleckstrin. bioRxiv 2019, 782623. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Toukmaji, A.Y.; Board, J.A. Ewald summation techniques in perspective: A survey. Comput. Phys. Commun. 1996, 95, 73–92. [Google Scholar] [CrossRef]
- Humphreys, D.D.; Friesner, R.A.; Berne, B.J. A multiple-time-step molecular dynamics algorithm for macromolecules. J. Phys. Chem. 1994, 98, 6885–6892. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- Westerlund, A.M.; Harpole, T.J.; Blau, C.; Delemotte, L. Inference of Calmodulin’s Ca(2+)-Dependent Free Energy Landscapes via Gaussian Mixture Model Validation. J. Chem. Theory Comput. 2018, 14, 63–71. [Google Scholar] [CrossRef]
- Westerlund, A.M.; Delemotte, L. InfleCS: Clustering Free Energy Landscapes with Gaussian Mixtures. J. Chem. Theory Comput. 2019, 15, 6752–6759. [Google Scholar] [CrossRef]
- Maisuradze, G.G.; Liwo, A.; Scheraga, H.A. Relation between free energy landscapes of proteins and dynamics. J. Chem. Theory Comput. 2010, 6, 583–595. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dash, R.; Tran, N.-N.; Lee, S.B.; Lee, B.-H. Structural Dynamics Analysis of USP14 Activation by AKT-Mediated Phosphorylation. Cells 2024, 13, 955. https://doi.org/10.3390/cells13110955
Dash R, Tran N-N, Lee SB, Lee B-H. Structural Dynamics Analysis of USP14 Activation by AKT-Mediated Phosphorylation. Cells. 2024; 13(11):955. https://doi.org/10.3390/cells13110955
Chicago/Turabian StyleDash, Raju, Non-Nuoc Tran, Sung Bae Lee, and Byung-Hoon Lee. 2024. "Structural Dynamics Analysis of USP14 Activation by AKT-Mediated Phosphorylation" Cells 13, no. 11: 955. https://doi.org/10.3390/cells13110955
APA StyleDash, R., Tran, N.-N., Lee, S. B., & Lee, B.-H. (2024). Structural Dynamics Analysis of USP14 Activation by AKT-Mediated Phosphorylation. Cells, 13(11), 955. https://doi.org/10.3390/cells13110955