
Multiple Endocrine Neoplasia Type 1 Regulates TGFβ-Mediated Suppression of Tumor Formation and Metastasis in Melanoma

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Antibodies
2.3. Cell Lines
2.4. TGFβ Treatment
2.5. Quantitative Real-Time PCR
2.6. Clonogenic Assay
2.7. Flow Cytometry
2.8. Immunoblotting
2.9. Lentiviral Generation and Infection
2.10. Generation of MEN1 CRISPR Knockout Cells
2.11. Luciferase Assay
2.12. Subcutaneous Tumor Xenografts
2.13. Statistics
3. Results
3.1. TGFβ Induces MEN1 Gene Expression in Melanoma Cells through Smad3
3.2. MEN1 Is Essential for Inhibiting Melanoma Cell Growth and Tumorigenesis
3.3. The TGFβ/Smad3/MEN1 Axis Is Essential for Inducing Cell-Cycle Arrest and Apoptosis in Human Melanoma Cells
3.4. Identification of MEN1 Mutations in Melanoma Patients and Loss of TGFβ Responses
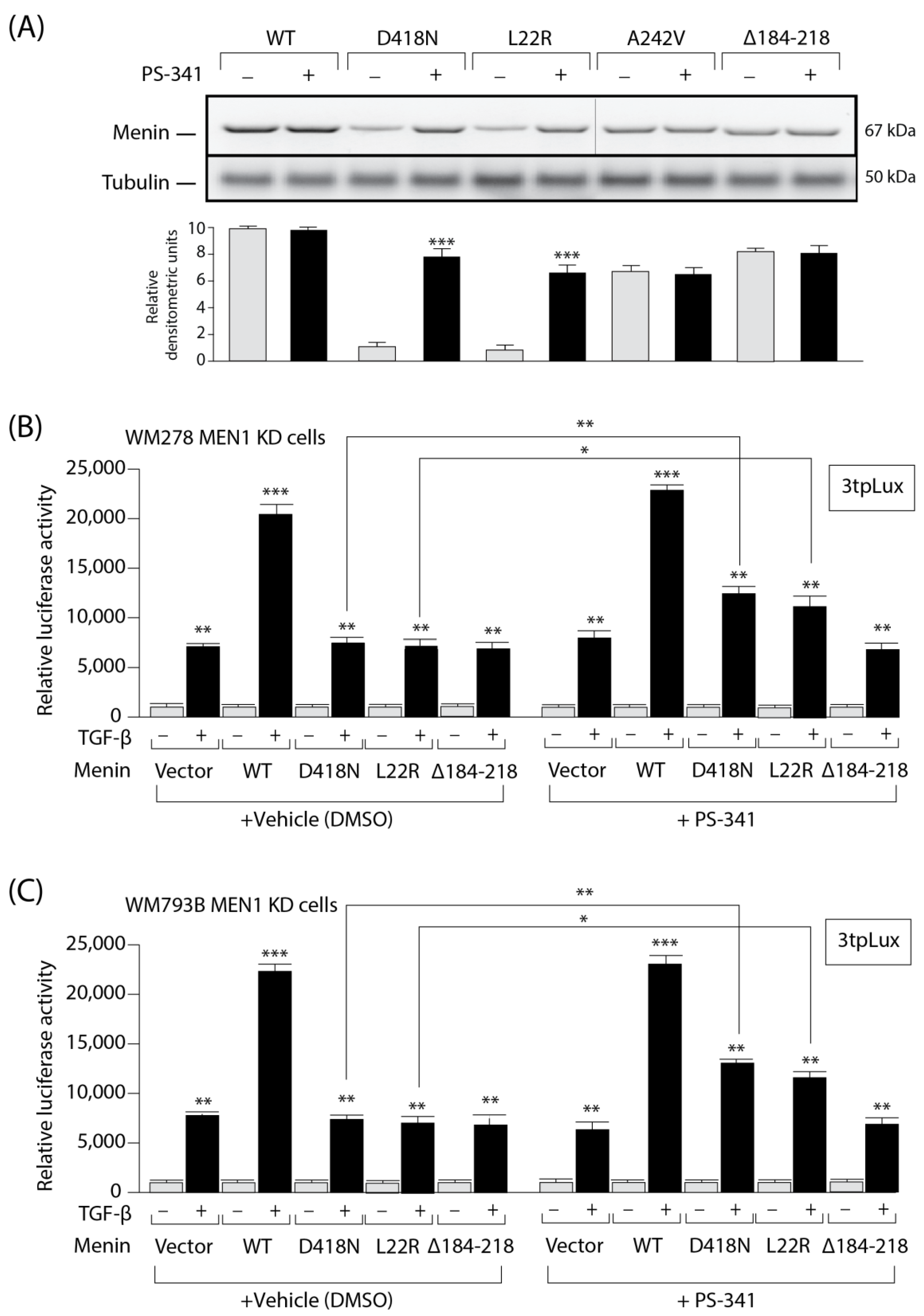
3.5. The Expression and Activity of MEN1 Missense Mutants Can Be Partially Rescued by a Proteasome Inhibitor
3.6. The Expression and Activity of MEN1 Missense Mutants Can Be Rescued by Inhibition of the Ubiquitin Ligase CHIP
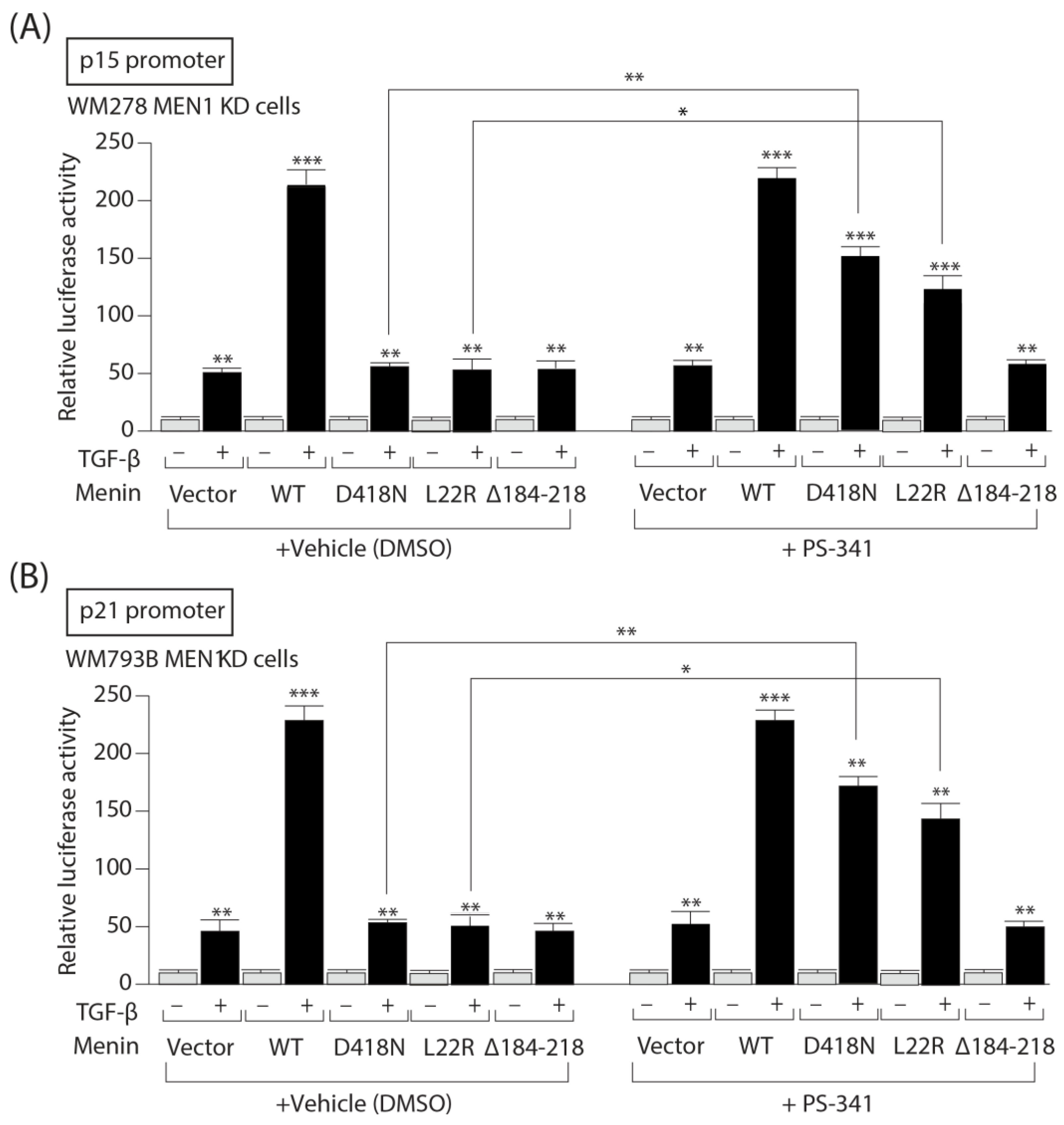
3.7. PS-341 Restores the Ability of Menin Missense Mutants to Mediate TGF-b Upregulation of the CDKI p15 and p21 Gene Promoters
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balch, C.M.; Mihm, M.C. Reply to the article “The AJCC staging proposal for cutaneous melanoma: Comments by the EORTC Melanoma Group”, by D. J. Ruiter et al. (Ann Oncol 2001; 12: 9–11). Ann. Oncol. 2002, 13, 175–176. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Ross, M.I.; Gershenwald, J.E. Evidence-based treatment of early-stage melanoma. J. Surg. Oncol. 2011, 104, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Houghton, A.N.; Polsky, D. Focus on melanoma. Cancer Cell 2002, 2, 275–278. [Google Scholar] [CrossRef]
- Enninga, E.A.L.; Moser, J.C.; Weaver, A.L.; Markovic, S.N.; Brewer, J.D.; Leontovich, A.A.; Hieken, T.J.; Shuster, L.; Kottschade, L.A.; Olariu, A.; et al. Survival of cutaneous melanoma based on sex, age, and stage in the United States, 1992–2011. Cancer Med. 2017, 6, 2203–2212. [Google Scholar] [CrossRef]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; Mccubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef]
- Lopez-Bergami, P.; Fitchman, B.; Ronai, Z. Understanding signaling cascades in melanoma. Photochem. Photobiol. 2008, 84, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Humbert, L.; Ghozlan, M.; Canaff, L.; Tian, J.; Lebrun, J.J. The leukemia inhibitory factor (LIF) and p21 mediate the TGFβ tumor suppressive effects in human cutaneous melanoma. BMC Cancer 2015, 15, 200. [Google Scholar] [CrossRef] [PubMed]
- Humbert, L.; Lebrun, J.J. TGF-beta inhibits human cutaneous melanoma cell migration and invasion through regulation of the plasminogen activator system. Cell. Signal. 2013, 25, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Ramont, L.; Pasco, S.; Hornebeck, W.; Maquart, F.X.; Monboisse, J.C. Transforming growth factor-β1 inhibits tumor growth in a mouse melanoma model by down-regulating the plasminogen activation system. Exp. Cell Res. 2003, 291, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Boudreault, J.; Wang, N.; Ghozlan, M.; Lebrun, J.-J. Transforming Growth Factor-β/Smad Signaling Inhibits Melanoma Cancer Stem Cell Self-Renewal, Tumor Formation and Metastasis. Cancers 2024, 16, 224. [Google Scholar] [CrossRef] [PubMed]
- Balogh, K.; Rácz, K.; Patócs, A.; Hunyady, L. Menin and its interacting proteins: Elucidation of menin function. Trends Endocrinol. Metab. 2006, 17, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Milne, T.A.; Hughes, C.M.; Lloyd, R.; Yang, Z.; Rozenblatt-Rosen, O.; Dou, Y.; Schnepp, R.W.; Krankel, C.; LiVolsi, V.A.; Gibbs, D.; et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Karnik, S.K.; Hughes, C.M.; Gu, X.; Rozenblatt-Rosen, O.; McLean, G.W.; Xiong, Y.; Meyerson, M.; Kim, S.K. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc. Natl. Acad. Sci. USA 2005, 102, 14659–14664. [Google Scholar] [CrossRef] [PubMed]
- Kaji, H.; Canaff, L.; Lebrun, J.J.; Goltzman, D.; Hendy, G.N. Inactivation of menin, a Smad3-interacting protein, blocks transforming growth factor type β signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 3837–3842. [Google Scholar] [CrossRef]
- Sowa, H.; Kaji, H.; Kitazawa, R.; Kitazawa, S.; Tsukamoto, T.; Yano, S.; Tsukada, T.; Canaff, L.; Hendy, G.N.; Sugimoto, T.; et al. Menin Inactivation Leads to Loss of Transforming Growth Factor β Inhibition of Parathyroid Cell Proliferation and Parathyroid Hormone Secretion. Cancer Res. 2004, 64, 2222–2228. [Google Scholar] [CrossRef]
- Thakker, R.V. Multiple endocrine neoplasia type 1. Endocrinol. Metab. Clin. N. Am. 2000, 29, 541–567. [Google Scholar] [CrossRef]
- Baldauf, C.; Vortmeyer, A.O.; Koch, C.A.; Sticherling, M. Combination of multiple skin malignancies with multiple endocrine neoplasia type 1: Coincidental or pathogenetically related? Dermatology 2009, 219, 365–367. [Google Scholar] [CrossRef]
- Nord, B.; Platz, A.; Smoczynski, K.; Kytölä, S.; Robertson, G.; Calender, A.; Murat, A.; Weintraub, D.; Burgess, J.; Edwards, M.; et al. Malignant melanoma in patients with multiple endocrine neoplasia type 1 and involvement of the MEN1 gene in sporadic melanoma. Int. J. Cancer 2000, 87, 463–467. [Google Scholar] [CrossRef]
- Gray-Schopfer, V.; Wellbrock, C.; Marais, R. Melanoma biology and new targeted therapy. Nature 2007, 445, 851–857. [Google Scholar] [CrossRef]
- Gao, S.B.; Feng, Z.J.; Xu, B.; Chen, Y.; Zheng, H.H.; Yin, P.; Hua, X.; Jin, G.H. Menin represses malignant phenotypes of melanoma through regulating multiple pathways. J. Cell. Mol. Med. 2011, 15, 2353–2363. [Google Scholar] [CrossRef] [PubMed]
- Massey, S.; Khan, M.A.; Rab, S.O.; Mustafa, S.; Khan, A.; Malik, Z.; Shaik, R.; Verma, M.K.; Deo, S.; Husain, S.A. Evaluating the role of MEN1 gene expression and its clinical significance in breast cancer patients. PLoS ONE 2023, 18, e0288482. [Google Scholar] [CrossRef] [PubMed]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Imroved vectors and genome-widees for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed]
- Poncin, J.; Abs, R.; Velkeniers, B.; Bonduelle, M.; Abramowicz, M.; Legros, J.J.; Verloes, A.; Meurisse, M.; Van Gaal, L.; Verellen, C.; et al. Mutation analysis of the MEN1 gene in Belgian patients with multiple endocrine neoplasia type 1 and related diseases. Hum. Mutat. 1999, 13, 54–60. [Google Scholar] [CrossRef]
- Ito, T.; Igarashi, H.; Uehara, H.; Berna, M.J.; Jensen, R.T. Causes of death and prognostic factors in multiple endocrine neoplasia type 1: A prospective study:Comparison of 106 men1/zollinger-ellison syndrome patients with 1613 literature men1 patients with or without pancreatic endocrine tumors. Medicine 2013, 92, 135–181. [Google Scholar] [CrossRef]
- Neel, J.-C.; Humbert, L.; Lebrun, J.-J. The Dual Role of TGFβ in Human Cancer: From Tumor Suppression to Cancer Metastasis. ISRN Mol. Biol. 2012, 2012, 381428. [Google Scholar] [CrossRef]
- Datto, M.B.; Li, Y.; Panus, J.F.; Howe, D.J.; Xiong, Y.; Wang, X.F. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 5545–5549. [Google Scholar] [CrossRef] [PubMed]
- Warner, B.J.; Blain, S.W.; Seoane, J.; Massagué, J. Myc Downregulation by Transforming Growth Factor β Required for Activation of the p15 Ink4b G 1 Arrest Pathway. Mol. Cell. Biol. 1999, 19, 5913–5922. [Google Scholar] [CrossRef] [PubMed]
- Schuster, N.; Krieglstein, K. Mechanisms of TGF-β-mediated apoptosis. Cell Tissue Res. 2002, 307, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Canaff, L.; Vanbellinghen, J.F.; Kaji, H.; Goltzman, D.; Hendy, G.N. Impaired transforming growth factor-β (TGF-β) transcriptional activity and cell proliferation control of a menin in-frame deletion mutant associated with Multiple Endocrine Neoplasia type 1 (MEN1). J. Biol. Chem. 2012, 287, 8584–8597. [Google Scholar] [CrossRef] [PubMed]
- Canaff, L.; Vanbellinghen, J.F.; Kanazawa, I.; Kwak, H.; Garfield, N.; Vautour, L.; Hendy, G.N. Menin missense mutants encoded by the MEN1 gene that are targeted to the proteasome: Restoration of expression and activity by CHIP siRNA. J. Clin. Endocrinol. Metab. 2012, 97, E282–E291. [Google Scholar] [CrossRef] [PubMed]
- Yaguchi, H.; Ohkura, N.; Takahashi, M.; Nagamura, Y.; Kitabayashi, I.; Tsukada, T. Menin Missense Mutants Associated with Multiple Endocrine Neoplasia Type 1 Are Rapidly Degraded via the Ubiquitin-Proteasome Pathway. Mol. Cell. Biol. 2004, 24, 6569–6580. [Google Scholar] [CrossRef] [PubMed]
- Guru, S.C.; Goldsmith, P.K.; Lee Burns, A.; Marx, S.J.; Spiegel, A.M.; Collins, F.S.; Chandrasekharappa, S.C. Menin, the product of the MEN1 gene, is a nuclear protein. Proc. Natl. Acad. Sci. USA 1998, 95, 1630–1634. [Google Scholar] [CrossRef] [PubMed]
- Buac, D.; Shen, M.; Schmitt, S.; Rani Kona, F.; Deshmukh, R.; Zhang, Z.; Neslund-Dudas, C.; Mitra, B.; Dou, Q.P. From Bortezomib to other Inhibitors of the Proteasome and Beyond. Curr. Pharm. Des. 2013, 19, 4025–4038. [Google Scholar] [CrossRef] [PubMed]
- McDonough, H.; Patterson, C. CHIP: A link between the chaperone and proteasome systems. Cell Stress Chaperones 2003, 8, 303–308. [Google Scholar] [CrossRef]
- Lebrun, J.J. Activin, TGF-β and menin in pituitary tumorigenesis. Adv. Exp. Med. Biol. 2009, 668, 69–78. [Google Scholar] [CrossRef]
- Hendy, G.N.; Kaji, H.; Sowa, H.; Lebrun, J.-J.; Canaff, L. Menin and TGF-β Superfamily Member Signaling via the Smad Pathway in Pituitary, Parathyroid and Osteoblast. Horm. Metab. Res. 2005, 37, 375–379. [Google Scholar] [CrossRef]
- Lacerte, A.; Lee, E.H.; Reynaud, R.; Canaff, L.; De Guise, C.; Devost, D.; Ali, S.; Hendy, G.N.; Lebrun, J.J. Activin inhibits pituitary prolactin expression and cell growth through Smads, Pit-1 and menin. Mol. Endocrinol. 2004, 18, 1558–1569. [Google Scholar] [CrossRef]
- Antsiferova, M.; Piwko-Czuchra, A.; Cangkrama, M.; Wietecha, M.; Sahin, D.; Birkner, K.; Amann, V.C.; Levesque, M.; Hohl, D.; Dummer, R.; et al. Activin promotes skin carcinogenesis by attraction and reprogramming of macrophages. EMBO Mol. Med. 2017, 9, 27–45. [Google Scholar] [CrossRef]
- Gutiérrez-Seijo, A.; García-Martínez, E.; Barrio-Alonso, C.; Parra-Blanco, V.; Avilés-Izquierdo, J.A.; Sánchez-Mateos, P.; Samaniego, R. Activin A Sustains the Metastatic Phenotype of Tumor-Associated Macrophages and Is a Prognostic Marker in Human Cutaneous Melanoma. J. Investig. Dermatol. 2022, 142, 653–661.e2. [Google Scholar] [CrossRef]
- Dai, M.; Al-Odaini, A.A.; Arakelian, A.; Rabbani, S.A.; Ali, S.; Lebrun, J.J. A novel function for p21Cip1 and acetyltransferase p/CAF as critical transcriptional regulators of TGFβ-mediated breast cancer cell migration and invasion. Breast Cancer Res. 2012, 14, R127. [Google Scholar] [CrossRef]
- Derynck, R.; Akhurst, R.J. Differentiation plasticity regulated by TGF-β family proteins in development and disease. Nat. Cell Biol. 2007, 9, 1000–1004. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 2003, 15, 740–746. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007, 98, 1512–1520. [Google Scholar] [CrossRef]
- Woodward, J.K.L.; Elshaw, S.R.; Murray, A.K.; Nichols, C.E.; Cross, N.; Laws, D.; Rennie, I.G.; Sisley, K. Stimulation and inhibition of uveal melanoma invasion by HGF, GRO, IL-1α and TGF-β. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3144–3152. [Google Scholar]
- Romo, P.; Madigan, M.C.; Provis, J.M.; Cullen, K.M. Differential effects of TGF-β and FGF-2 on in vitro proliferation and migration of primate retinal endothelial and Müller cells. Acta Ophthalmol. 2011, 89, e263–e268. [Google Scholar] [CrossRef]
- Agarwal, S.K. The future: Genetics advances in MEN1 therapeutic approaches and management strategies. Endocr. Relat. Cancer 2017, 24, T119–T134. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharappa, S.C.; Guru, S.C.; Manickam, P.; Olufemi, S.E.; Collins, F.S.; Emmert-Buck, M.R.; Debelenko, L.V.; Zhuang, Z.; Lubensky, I.A.; Liotta, L.A.; et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997, 276, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Nelakurti, D.D.; Pappula, A.L.; Rajasekaran, S.; Miles, W.O.; Petreaca, R.C. Comprehensive Analysis of MEN1 Mutations and Their Role in Cancer. Cancers 2020, 12, 2616. [Google Scholar] [CrossRef]
- Mohan, M.; Matin, A.; Davies, F.E. Update on the optimal use of Bortezomib in the treatment of multiple myeloma. Cancer Manag. Res. 2017, 9, 51–63. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Cell Type | Origin | Sex of Human Donor | Age | Mutations and Characteristics |
|---|---|---|---|---|---|
| WM278 | Melanoma | Primary | Female | 62 | BRAF (V600E); PTEN (Deletion) |
| WM793B | Melanoma | Primary | Male | 37 | BRAF (V600E); CDK4 (K22Q); PTEN (Deletion) |
| BLM | Melanoma | Derived from lung metastases in nude mice injected with BRO parent cell line | Male | 34 | Nras (Q61R) |
| WM1232 | Melanoma | Metastatic | Female | N/A | BRAF (V600E); PTEN (Deletion) |
| DAUV | Melanoma | Primary | N/A | N/A | BRAF (V600E) |
| SKMEL-28 | Melanoma | Primary | Male | 53 | BRAF (V600E); CDK4 (R24C); EGFR (P753S); PTEN (T167A); TP53 (L145R); Tert (Promoter) |
| a375m | Melanoma | Isolated from a tumor from a nude mice injected with the parent a375 cell line | Female | 54 | BRAF(V600E); CDKN2A (Deletion); Tert (Promoter) |
| HEK293 | Kidney | Embryo | Female | Fetus |
| Gene | Sequence | |
|---|---|---|
| MEN1 | Forward | 5′-GGAAGACGACGAGGAGATCTACA-3′ |
| MEN1 | Reverse | 5′-CAGTAGTTCAGAGGCCTTTGCGCT-3′ |
| GAPDH | Forward | 5′-GCCTCAAGATCATCAGCAATGCCT-3′ |
| GAPDH | Reverse | 5′-TGTGGTCATGAGTCCTTCCACGAT-3′ |
| Gene | Sequence | |
|---|---|---|
| MENsg1 | Forward | 5′-CACCGCACCTGCTGCGATTCTACGA-3′ |
| MENsg1 | Reverse | 5′-AAACTCGTAGAATCGCAGCAGGTGC-3′ |
| MEN2sg2 | Forward | 5′-CACCGACGTCGTCGATGGAGCGCAG-3′ |
| MEN2sg2 | Reverse | 5′-AAACCTGCGCTCCATCGACGACGTC-3′ |
| SCRsg1 | Forward | 5′-CACCGACGGAGGCTAAGCGTCGCAA-3′ |
| SCRsg2 | Reverse | 5′-AAACTTGCGACGCTTAGCCTCCGTC-3′ |
| SMAD2sg1 | Forward | 5′-CACCGTCCCACTGATCTATCGTATT-3′ |
| SMAD2sg1 | Reverse | 5′-AAACAATACGATAGATCAGTGGGAC-3′ |
| SMAD2sg2 | Forward | 5′-CACCGTGGCGGCGTGAATGGCAAGA-3′ |
| SMAD2sg2 | Reverse | 5‘-AAACTCTTGCCATTCACGCCGCCAC-3′ |
| SMAD3sg1 | Forward | 5′-CACCGCCCGATCGTGAAGCGCCTGC-3′ |
| SMAD3sg1 | Reverse | 5′-AAACGCAGGCGCTTCACGATCGGGC-3′ |
| SMAD3sg2 | Forward | 5′-CACCGTTCACGATCGGGGGAGTGAA-3′ |
| SMAD3sg2 | Reverse | 5′-AAACTTCACTCCCCCGATCGTGAAC-3′ |
| SMAD4sg1 | Forward | 5′-CACCGAACTCTGTACAAAGACCGCG-3′ |
| SMAD4sg1 | Reverse | 5′-AAACCGCGGTCTTTGTACAGAGTTC-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boudreault, J.; Canaff, L.; Ghozlan, M.; Wang, N.; Guarnieri, V.; Salcuni, A.S.; Scillitani, A.; Goltzman, D.; Ali, S.; Lebrun, J.-J. Multiple Endocrine Neoplasia Type 1 Regulates TGFβ-Mediated Suppression of Tumor Formation and Metastasis in Melanoma. Cells 2024, 13, 973. https://doi.org/10.3390/cells13110973
Boudreault J, Canaff L, Ghozlan M, Wang N, Guarnieri V, Salcuni AS, Scillitani A, Goltzman D, Ali S, Lebrun J-J. Multiple Endocrine Neoplasia Type 1 Regulates TGFβ-Mediated Suppression of Tumor Formation and Metastasis in Melanoma. Cells. 2024; 13(11):973. https://doi.org/10.3390/cells13110973
Chicago/Turabian StyleBoudreault, Julien, Lucie Canaff, Mostafa Ghozlan, Ni Wang, Vito Guarnieri, Antonio Stefano Salcuni, Alfredo Scillitani, David Goltzman, Suhad Ali, and Jean-Jacques Lebrun. 2024. "Multiple Endocrine Neoplasia Type 1 Regulates TGFβ-Mediated Suppression of Tumor Formation and Metastasis in Melanoma" Cells 13, no. 11: 973. https://doi.org/10.3390/cells13110973