
Mitochondrial Role on Cellular Apoptosis, Autophagy, and Senescence during Osteoarthritis Pathogenesis

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Transmitochondrial Cybrid Preparation
2.3. Detection of Apoptotic Cells
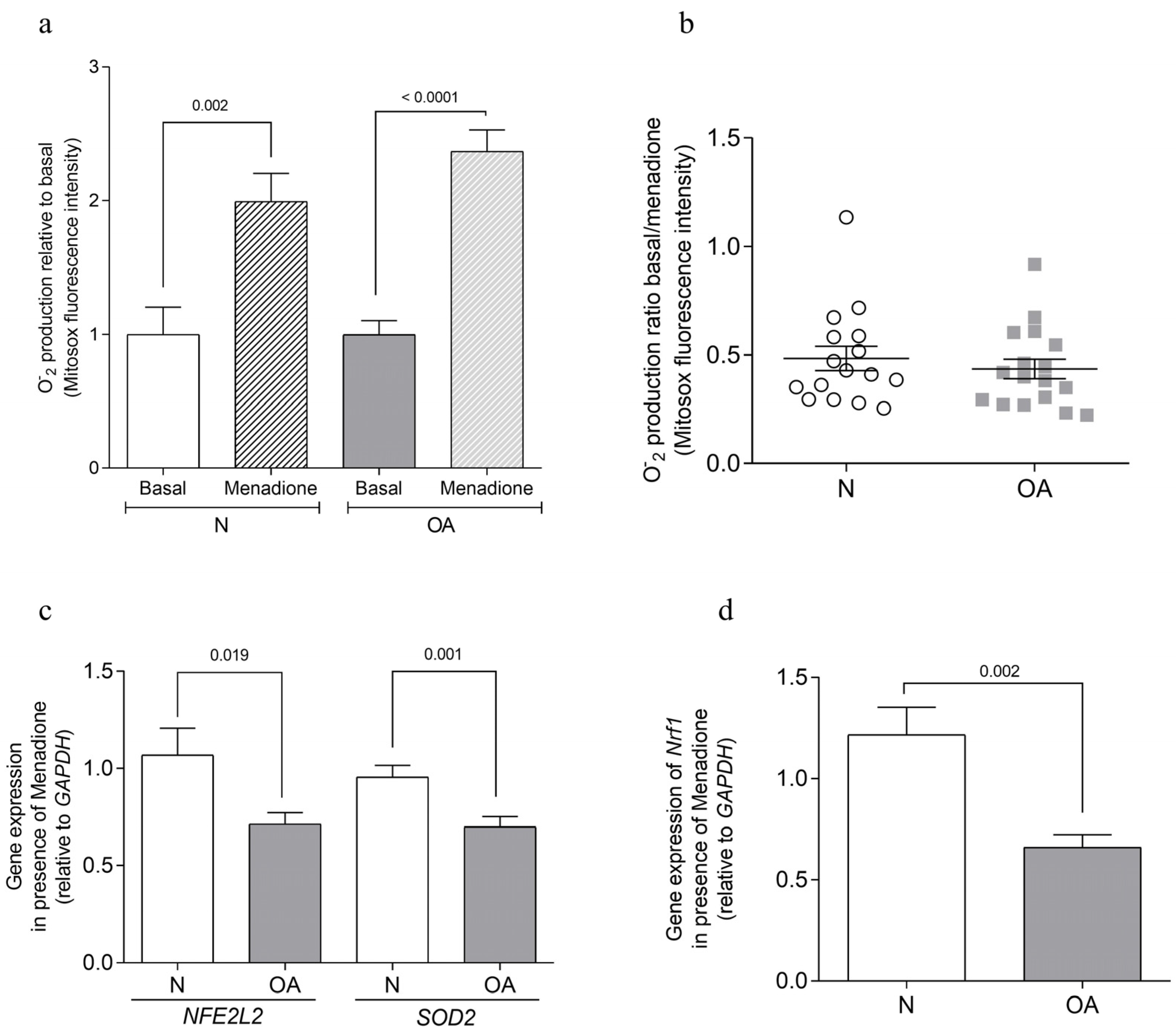
2.4. Analysis of Anion Superoxide Production
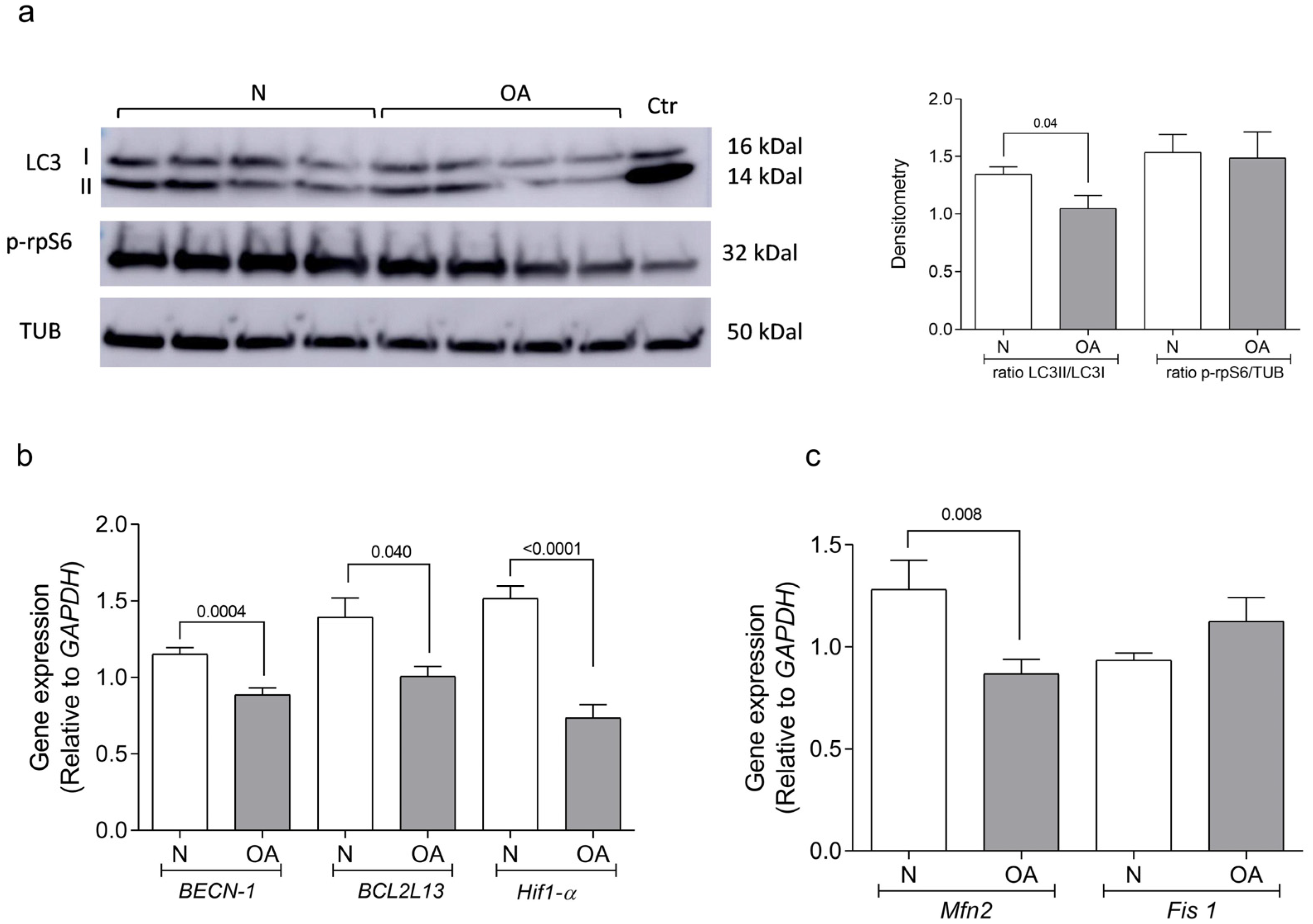
2.5. Autophagy Determination
2.6. Senescence Determination
2.7. Quantitative Real-Time PCR
2.8. Statistical Analysis
3. Results
3.1. Apoptosis Analysis
3.2. Autophagy Analysis
3.3. Senescence Analysis
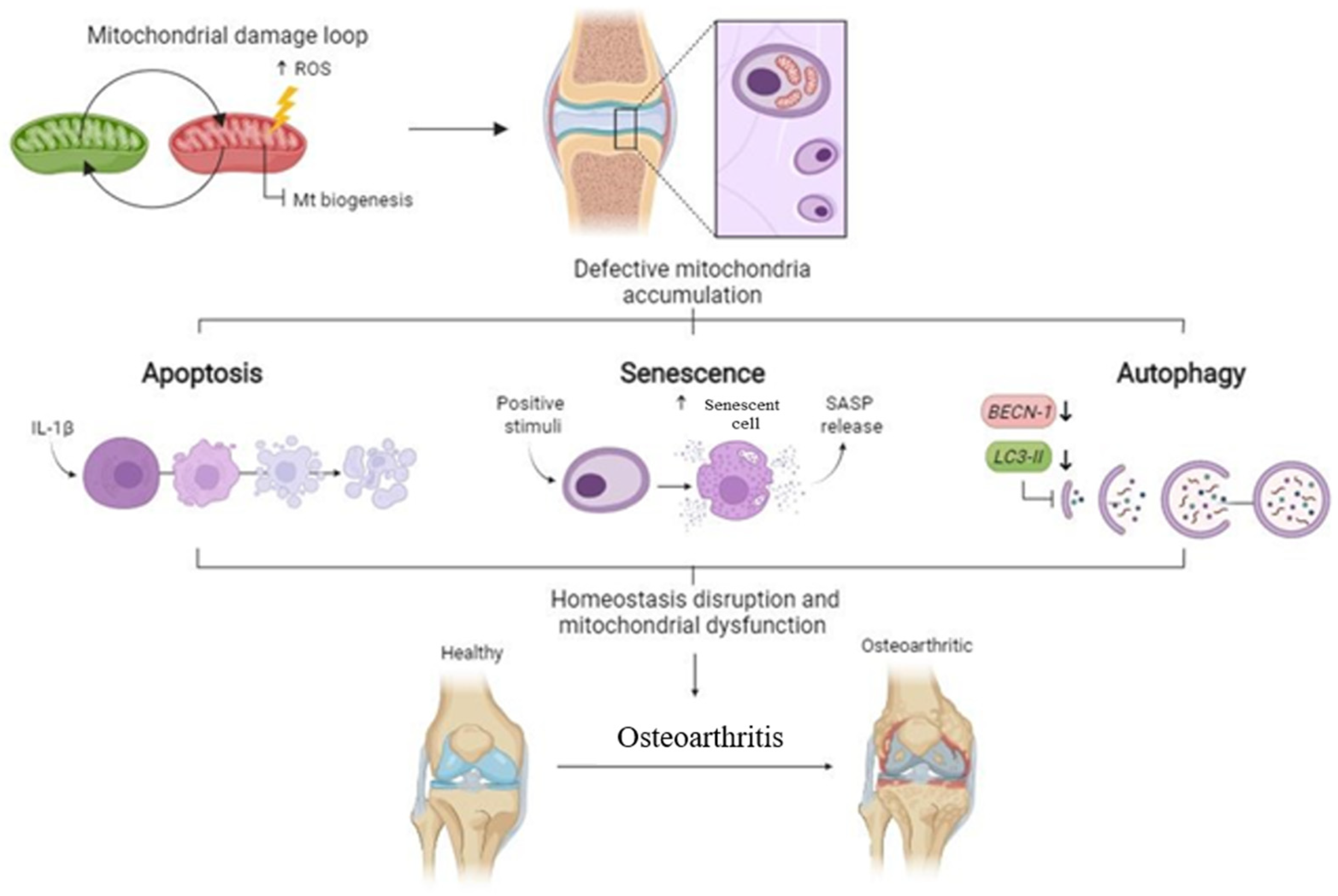
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bannuru, R.R.; Osani, M.C.; Vaysbrot, E.E.; Arden, N.K.; Bennell, K.; Bierma-Zeinstra, S.M.A.; Kraus, V.B.; Lohmander, L.S.; Abbott, J.H.; Bhandari, M.; et al. OARSI guidelines for the non-surgical management of knee, hip, and polyarticular osteoarthritis. Osteoarthr. Cartil. 2019, 27, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Kraus, V.B.; Blanco, F.J.; Englund, M.; Henrotin, Y.; Lohmander, L.S.; Losina, E.; Önnerfjord, P.; Persiani, S. OARSI Clinical Trials Recommendations: Soluble biomarker assessments in clinical trials in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J. Osteoarthritis and atherosclerosis in joint disease. Reumatol. Clin. 2018, 14, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Haudenschild, D.R.; Carlson, A.K.; Zignego, D.L.; Yik, J.H.N.; Hilmer, J.K.; June, R.K. Inhibition of early response genes prevents changes in global joint metabolomic profiles in mouse post-traumatic osteoarthritis. Osteoarthr. Cartil. 2019, 27, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.P.; Martel-Pelletier, J.; Abramson, S.B. Osteoarthritis, an inflammatory disease: Potential implication for the selection of new therapeutic targets. Arthritis Rheum. 2001, 44, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, J.A.; Collins, J.A.; Loeser, R.F. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic. Biol. Med. 2019, 132, 73–82. [Google Scholar] [CrossRef]
- Zhu, S.; Makosa, D.; Miller, B.; Griffin, T.M. Glutathione as a mediator of cartilage oxidative stress resistance and resilience during aging and osteoarthritis. Connect. Tissue Res. 2020, 61, 34–47. [Google Scholar] [CrossRef]
- Collins, J.A.; Wood, S.T.; Nelson, K.J.; Rowe, M.A.; Carlson, C.S.; Chubinskaya, S.; Poole, L.B.; Furdui, C.M.; Loeser, R.F. Oxidative Stress Promotes Peroxiredoxin Hyperoxidation and Attenuates Pro-survival Signaling in Aging Chondrocytes. J. Biol. Chem. 2016, 291, 6641–6654. [Google Scholar] [CrossRef]
- Loeser, R.F.; Collins, J.A.; Diekman, B.O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 412–420. [Google Scholar] [CrossRef]
- Zhao, Z.; Li, Y.; Wang, M.; Jin, Y.; Liao, W.; Zhao, Z.; Fang, J. Mitochondrial DNA haplogroups participate in osteoarthritis: Current evidence based on a meta-analysis. Clin. Rheumatol. 2020, 39, 1027–1037. [Google Scholar] [CrossRef]
- Fang, H.; Zhang, F.; Li, F.; Shi, H.; Ma, L.; Du, M.; You, Y.; Qiu, R.; Nie, H.; Shen, L.; et al. Mitochondrial DNA haplogroups modify the risk of osteoarthritis by altering mitochondrial function and intracellular mitochondrial signals. Biochim. Biophys. Acta 2016, 1862, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Rego-Pérez, I.; Fernández-Moreno, M.; Fernández-López, C.; Arenas, J.; Blanco, F.J. Mitochondrial DNA haplogroups: Role in the prevalence and severity of knee osteoarthritis. Arthritis Rheum. 2008, 58, 2387–2396. [Google Scholar] [CrossRef]
- Fernández-Moreno, M.; Soto-Hermida, A.; Vázquez-Mosquera, M.E.; Cortés-Pereira, E.; Relaño, S.; Hermida-Gómez, T.; Pértega, S.; Oreiro-Villar, N.; Fernández-López, C.; Garesse, R.; et al. Mitochondrial DNA haplogroups influence the risk of incident knee osteoarthritis in OAI and CHECK cohorts. A meta-analysis and functional study. Ann. Rheum. Dis. 2017, 76, 1114–1122. [Google Scholar] [CrossRef]
- Cortés-Pereira, E.; Fernández-Tajes, J.; Fernández-Moreno, M.; Vázquez-Mosquera, M.E.; Relaño, S.; Ramos-Louro, P.; Durán-Sotuela, A.; Dalmao-Fernández, A.; Oreiro, N.; Blanco, F.J.; et al. Differential Association of Mitochondrial DNA Haplogroups J and H with the Methylation Status of Articular Cartilage: Potential Role in Apoptosis and Metabolic and Developmental Processes. Arthritis Rheumatol. 2019, 71, 1191–1200. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Guitian, R.; Vázquez-Martul, E.; de Toro, F.J.; Galdo, F. Osteoarthritis chondrocytes die by apoptosis. A possible pathway for osteoarthritis pathology. Arthritis Rheum. 1998, 41, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Castrogiovanni, P.; Trovato, F.M.; Weinberg, A.M.; Al-Wasiyah, M.K.; Alqahtani, M.H.; Mobasheri, A. Biomarkers of Chondrocyte Apoptosis and Autophagy in Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 20560–20575. [Google Scholar] [CrossRef]
- Carroll, B.; Nelson, G.; Rabanal-Ruiz, Y.; Kucheryavenko, O.; Dunhill-Turner, N.A.; Chesterman, C.C.; Zahari, Q.; Zhang, T.; Conduit, S.E.; Mitchell, C.A.; et al. Persistent mTORC1 signaling in cell senescence results from defects in amino acid and growth factor sensing. J. Cell Biol. 2017, 216, 1949–1957. [Google Scholar] [CrossRef]
- Li, Y.S.; Zhang, F.J.; Zeng, C.; Luo, W.; Xiao, W.F.; Gao, S.G.; Lei, G.H. Autophagy in osteoarthritis. Jt. Bone Spine 2016, 83, 143–148. [Google Scholar] [CrossRef]
- Lotz, M.K.; Caramés, B. Autophagy and cartilage homeostasis mechanisms in joint health, aging and OA. Nat. Rev. Rheumatol. 2011, 7, 579–587. [Google Scholar] [CrossRef]
- Caramés, B.; Olmer, M.; Kiosses, W.B.; Lotz, M.K. The relationship of autophagy defects to cartilage damage during joint aging in a mouse model. Arthritis Rheumatol. 2015, 67, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- Caramés, B.; Taniguchi, N.; Otsuki, S.; Blanco, F.J.; Lotz, M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheumatol. 2010, 62, 791–801. [Google Scholar] [CrossRef]
- Blanco, F.J.; Rego, I.; Ruiz-Romero, C. The role of mitochondria in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.; Duan, M.; Liu, Y.; Wang, C.; Xie, J. Role of Mitochondria in Physiology of Chondrocytes and Diseases of Osteoarthritis and Rheumatoid Arthritis. Cartilage 2021, 13 (Suppl. 2), 1102s–1121s. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Rego-Pérez, I. Mitochondria and mitophagy: Biosensors for cartilage degradation and osteoarthritis. Osteoarthr. Cartil. 2018, 26, 989–991. [Google Scholar] [CrossRef] [PubMed]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef]
- Vizioli, M.G.; Liu, T.; Miller, K.N.; Robertson, N.A.; Gilroy, K.; Lagnado, A.B.; Perez-Garcia, A.; Kiourtis, C.; Dasgupta, N.; Lei, X.; et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020, 34, 428–445. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A.; Matta, C.; Zákány, R.; Musumeci, G. Chondrosenescence: Definition, hallmarks and potential role in the pathogenesis of osteoarthritis. Maturitas 2015, 80, 237–244. [Google Scholar] [CrossRef]
- Martin, J.A.; Brown, T.D.; Heiner, A.D.; Buckwalter, J.A. Chondrocyte senescence, joint loading and osteoarthritis. Clin. Orthop. Relat. Res. 2004, 427, S96–S103. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Palacín, M.; Zorzano, A. Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 2009, 89, 799–845. [Google Scholar] [CrossRef] [PubMed]
- Zorzano, A.; Liesa, M.; Sebastián, D.; Segalés, J.; Palacín, M. Mitochondrial fusion proteins: Dual regulators of morphology and metabolism. Semin. Cell Dev. Biol. 2010, 21, 566–574. [Google Scholar] [CrossRef]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Kaiser, W.J.; Bertrand, M.J.; Vandenabeele, P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol. Cell. Oncol. 2015, 2, e975093. [Google Scholar] [CrossRef] [PubMed]
- Dalmao-Fernández, A.; Lund, J.; Hermida-Gómez, T.; Vazquez-Mosquera, M.E.; Rego-Pérez, I.; Blanco, F.J.; Fernández-Moreno, M. Impaired Metabolic Flexibility in the Osteoarthritis Process: A Study on Transmitochondrial Cybrids. Cells 2020, 9, 809. [Google Scholar] [CrossRef] [PubMed]
- Dalmao-Fernández, A.; Hermida-Gómez, T.; Lund, J.; Vazquez-Mosquera, M.E.; Rego-Pérez, I.; Garesse, R.; Blanco, F.J.; Fernández-Moreno, M. Mitochondrial DNA from osteoarthritic patients drives functional impairment of mitochondrial activity: A study on transmitochondrial cybrids. Cytotherapy 2021, 23, 399–410. [Google Scholar] [CrossRef]
- Loor, G.; Kondapalli, J.; Schriewer, J.M.; Chandel, N.S.; Vanden Hoek, T.L.; Schumacker, P.T. Menadione triggers cell death through ROS-dependent mechanisms involving PARP activation without requiring apoptosis. Free Radic. Biol. Med. 2010, 49, 1925–1936. [Google Scholar] [CrossRef] [PubMed]
- Bohensky, J.; Shapiro, I.M.; Leshinsky, S.; Terkhorn, S.P.; Adams, C.S.; Srinivas, V. HIF-1 regulation of chondrocyte apoptosis: Induction of the autophagic pathway. Autophagy 2007, 3, 207–214. [Google Scholar] [CrossRef]
- Bohensky, J.; Leshinsky, S.; Srinivas, V.; Shapiro, I.M. Chondrocyte autophagy is stimulated by HIF-1 dependent AMPK activation and mTOR suppression. Pediatr. Nephrol. 2010, 25, 633–642. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, J.; Chu, J.; Yang, C.; Xiao, H.; Zhao, C.; Sun, Z.; Gao, X.; Chen, G.; Han, Z.; et al. MicroRNA-146a Induced by Hypoxia Promotes Chondrocyte Autophagy through Bcl-2. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 37, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Otsu, K.; Murakawa, T.; Yamaguchi, O. BCL2L13 is a mammalian homolog of the yeast mitophagy receptor Atg32. Autophagy 2015, 11, 1932–1933. [Google Scholar] [CrossRef] [PubMed]
- Murakawa, T.; Yamaguchi, O.; Hashimoto, A.; Hikoso, S.; Takeda, T.; Oka, T.; Yasui, H.; Ueda, H.; Akazawa, Y.; Nakayama, H.; et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat. Commun. 2015, 6, 7527. [Google Scholar] [CrossRef] [PubMed]
- Roach, H.I.; Aigner, T.; Kouri, J.B. Chondroptosis: A variant of apoptotic cell death in chondrocytes? Apoptosis Int. J. Program. Cell Death 2004, 9, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Lee, Y.J.; Seong, S.C.; Choe, K.W.; Song, Y.W. Apoptotic chondrocyte death in human osteoarthritis. J. Rheumatol. 2000, 27, 455–462. [Google Scholar] [PubMed]
- Hui, W.; Young, D.A.; Rowan, A.D.; Xu, X.; Cawston, T.E.; Proctor, C.J. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann. Rheum. Dis. 2016, 75, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, X.; Lotz, M.; Terkeltaub, R.; Liu-Bryan, R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor γ coactivator 1α. Arthritis Rheumatol. 2015, 67, 2141–2153. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Liu, S.; Li, J.; Tian, Y.; Xue, Y.; Liu, X. Parkin and Nrf2 prevent oxidative stress-induced apoptosis in intervertebral endplate chondrocytes via inducing mitophagy and anti-oxidant defenses. Life Sci. 2020, 243, 117244. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Abramov, A.Y. The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med. 2015, 88, 179–188. [Google Scholar] [CrossRef]
- Terkeltaub, R.; Johnson, K.; Murphy, A.; Ghosh, S. Invited review: The mitochondrion in osteoarthritis. Mitochondrion 2002, 1, 301–319. [Google Scholar] [CrossRef]
- Ruiz-Romero, C.; Calamia, V.; Mateos, J.; Carreira, V.; Martínez-Gomariz, M.; Fernández, M.; Blanco, F.J. Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: A decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol. Cell. Proteom. MCP 2009, 8, 172–189. [Google Scholar] [CrossRef]
- Gavriilidis, C.; Miwa, S.; von Zglinicki, T.; Taylor, R.W.; Young, D.A. Mitochondrial dysfunction in osteoarthritis is associated with down-regulation of superoxide dismutase 2. Arthritis Rheumatol. 2013, 65, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Almonte-Becerril, M.; Navarro-Garcia, F.; Gonzalez-Robles, A.; Vega-Lopez, M.A.; Lavalle, C.; Kouri, J.B. Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of Osteoarthritis within an experimental model. Apoptosis Int. J. Program. Cell Death 2010, 15, 631–638. [Google Scholar] [CrossRef]
- Vinatier, C.; Domínguez, E.; Guicheux, J.; Caramés, B. Role of the Inflammation-Autophagy-Senescence Integrative Network in Osteoarthritis. Front. Physiol. 2018, 9, 706. [Google Scholar] [CrossRef]
- Ryu, S.J.; Oh, Y.S.; Park, S.C. Failure of stress-induced downregulation of Bcl-2 contributes to apoptosis resistance in senescent human diploid fibroblasts. Cell Death Differ. 2007, 14, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Schipani, E.; Ryan, H.E.; Didrickson, S.; Kobayashi, T.; Knight, M.; Johnson, R.S. Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev. 2001, 15, 2865–2876. [Google Scholar] [CrossRef]
- Hu, S.; Zhang, C.; Ni, L.; Huang, C.; Chen, D.; Shi, K.; Jin, H.; Zhang, K.; Li, Y.; Xie, L.; et al. Stabilization of HIF-1α alleviates osteoarthritis via enhancing mitophagy. Cell Death Dis. 2020, 11, 481. [Google Scholar] [CrossRef]
- Filadi, R.; Pendin, D.; Pizzo, P. Mitofusin 2: From functions to disease. Cell Death Dis. 2018, 9, 330. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Fernández-Moreno, M. Mitochondrial biogenesis: A potential therapeutic target for osteoarthritis. Osteoarthr. Cartil. 2020, 28, 1003–1006. [Google Scholar] [CrossRef]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017, 36, 2280–2295. [Google Scholar] [CrossRef] [PubMed]
- Ock, S.A.; Knott, J.G.; Choi, I. Involvement of CDKN1A (p21) in cellular senescence in response to heat and irradiation stress during preimplantation development. Cell Stress Chaperones 2020, 25, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Coryell, P.R.; Diekman, B.O.; Loeser, R.F. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat. Rev. Rheumatol. 2021, 17, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Astrike-Davis, E.M.; Coryell, P.; Loeser, R.F. Targeting cellular senescence as a novel treatment for osteoarthritis. Curr. Opin. Pharmacol. 2022, 64, 102213. [Google Scholar] [CrossRef] [PubMed]
- Rego-Pérez, I.; Durán-Sotuela, A.; Ramos-Louro, P.; Blanco, F.J. Mitochondrial Genetics and Epigenetics in Osteoarthritis. Front. Genet. 2019, 10, 1335. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhang, Z.; Sheng, P.; Mobasheri, A. The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res. Rev. 2021, 66, 101249. [Google Scholar] [CrossRef]
- Loeser, R.F. Aging and osteoarthritis: The role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil. 2009, 17, 971–979. [Google Scholar] [CrossRef]
- Abate, M.; Festa, A.; Falco, M.; Lombardi, A.; Luce, A.; Grimaldi, A.; Zappavigna, S.; Sperlongano, P.; Irace, C.; Caraglia, M.; et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin. Cell Dev. Biol. 2020, 98, 139–153. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Symbol | Primer Fw (5′-3′) | Primer Rv (5′-3′) | UPL Probe |
|---|---|---|---|---|
| Nuclear factor erythroid 2 like 2 | NFE2L2 | gcaacaggacattgagcaag | tggacttggaaccatggtagt | #52 |
| Superoxide dismutase-2 | SOD2 | ctggacaaacctcagcccta | tgatggcttccagcaactc | #22 |
| Nuclear respiratory factor 1 | Nrf1 | ggggaaagaaagctgcaag | gtgcctgggtccatgaaa | #49 |
| Beclin-1 | BECN-1 | caggctgaggctgagagact | gctccagctgctgtcgtt | #69 |
| B-Cell Lymphoma 2 homology motifs like protein 13 | BCL2L13 | gacctcaacgcacagtacga | gagattgtacaggaccctcca | #68 |
| Cyclin Dependent Kinase Inhibitor 1A | CDKN1A | tcactgtcttgtacccttgtg | ggcgtttggagtggtagaaa | #32 |
| Hypoxia Inducible Factor 1 Subunit Alpha | Hif-1α | tggaatggagcaaaagacaa | tggtcagctgtggtaatcca | #3 |
| Mitofusin 2 | Mfn-2 | tcagctacactggctccaac | caaaggtcccagacagttcc | #83 |
| Mitochondrial Fusion 1 | Fis1 | ctgaacgagctggtgtctgt | gagcctgctgccttctca | #62 |
| Glyceraldehyde-3-Phosphate Dehydrogenase | GAPDH | gagtccactggcgtcttcac | gttcacacccatgacgaaca | #45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalmao-Fernández, A.; Hermida-Gómez, T.; Nogueira-Recalde, U.; Rego-Pérez, I.; Blanco-Garcia, F.J.; Fernández-Moreno, M. Mitochondrial Role on Cellular Apoptosis, Autophagy, and Senescence during Osteoarthritis Pathogenesis. Cells 2024, 13, 976. https://doi.org/10.3390/cells13110976
Dalmao-Fernández A, Hermida-Gómez T, Nogueira-Recalde U, Rego-Pérez I, Blanco-Garcia FJ, Fernández-Moreno M. Mitochondrial Role on Cellular Apoptosis, Autophagy, and Senescence during Osteoarthritis Pathogenesis. Cells. 2024; 13(11):976. https://doi.org/10.3390/cells13110976
Chicago/Turabian StyleDalmao-Fernández, Andrea, Tamara Hermida-Gómez, Uxia Nogueira-Recalde, Ignacio Rego-Pérez, Francisco J. Blanco-Garcia, and Mercedes Fernández-Moreno. 2024. "Mitochondrial Role on Cellular Apoptosis, Autophagy, and Senescence during Osteoarthritis Pathogenesis" Cells 13, no. 11: 976. https://doi.org/10.3390/cells13110976