
Deletion of the Murine Ortholog of the Human 9p21.3 Locus Leads to Insulin Resistance and Obesity in Hypercholesterolemic Mice

 , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Insulin Tolerance Test, Glucose Tolerance Test, and Insulin Secretion Test
2.3. Blood Glucose, Cell Count, and Lipid Analysis
2.4. Metabolic Rate
2.5. Histology
2.6. Cell Culture and Seahorse
2.7. Gene Expression Analysis
2.8. Statistical Analysis
3. Results
3.1. Deletion of the 9p21.3 Risk Interval Ortholog Leads to Obesity and Enlargement of Adipocytes during Aging in Ldlr−/−ApoB100/100 Mice on a Standard Diet
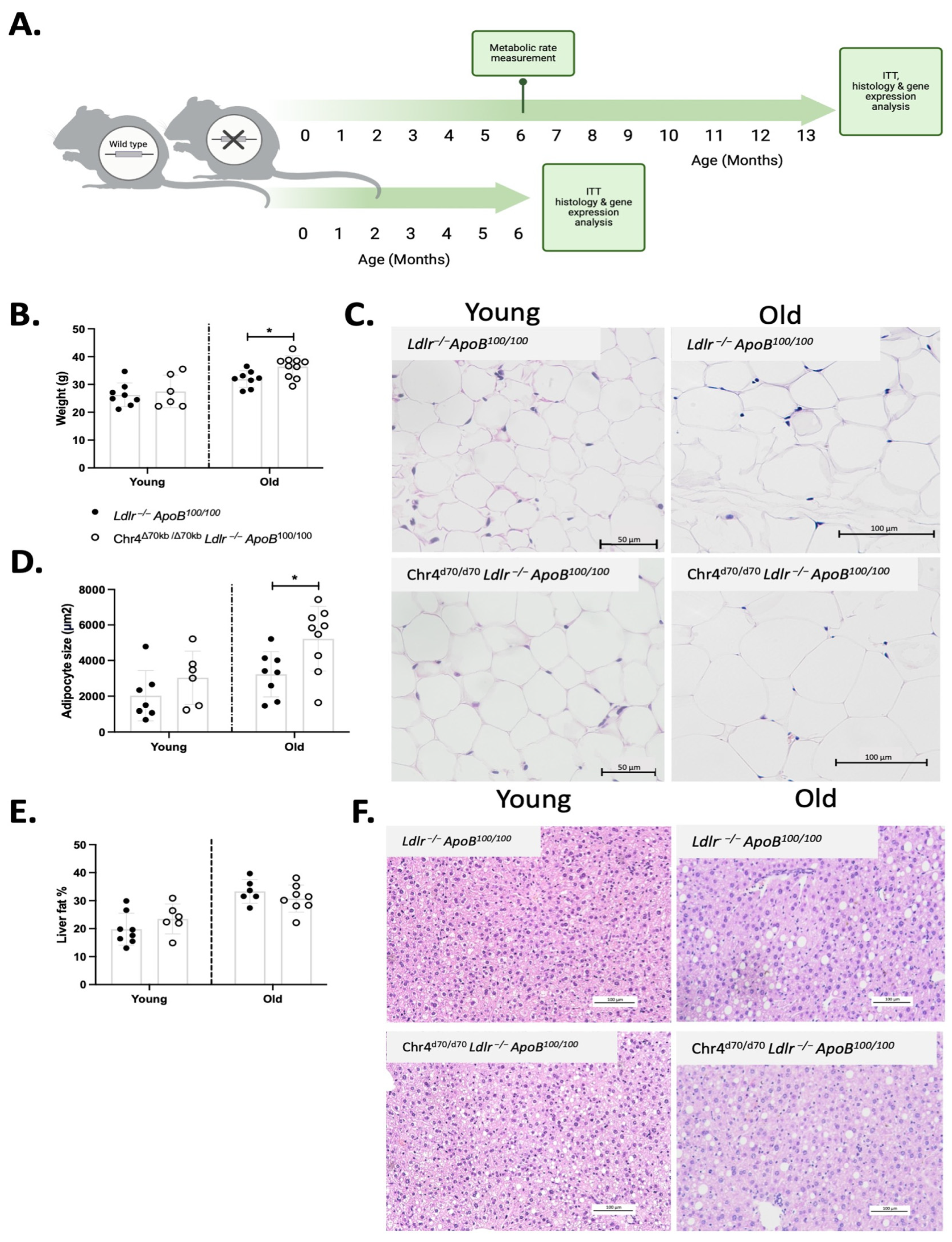
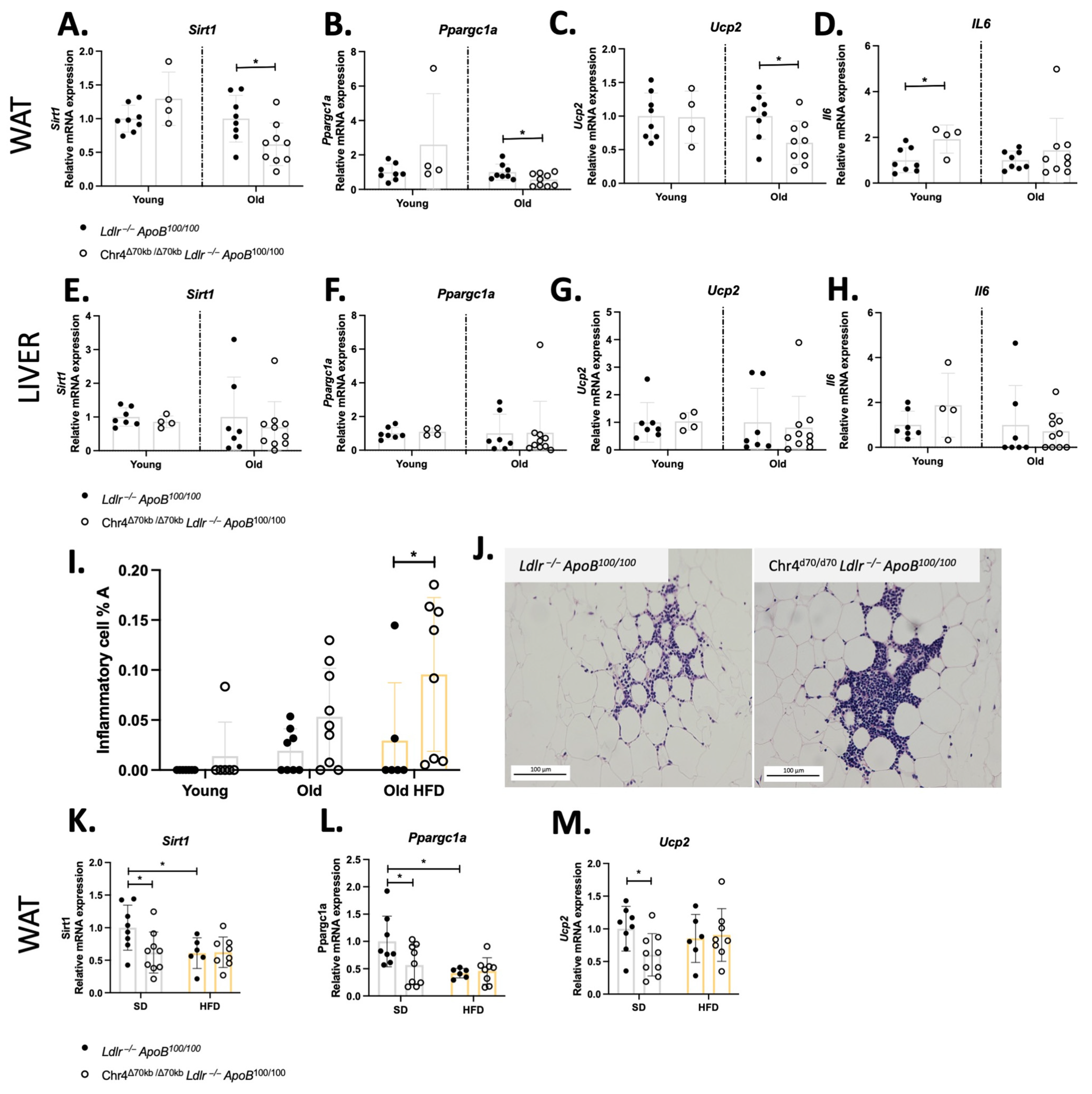
3.2. Chr4Δ70/Δ70 Leads to Inhibition of Sirt1-Ppargc1a-Ucp2 Axis and Promotes Inflammation in White Adipose Tissue in Ldlr−/−ApoB100/100 Mice
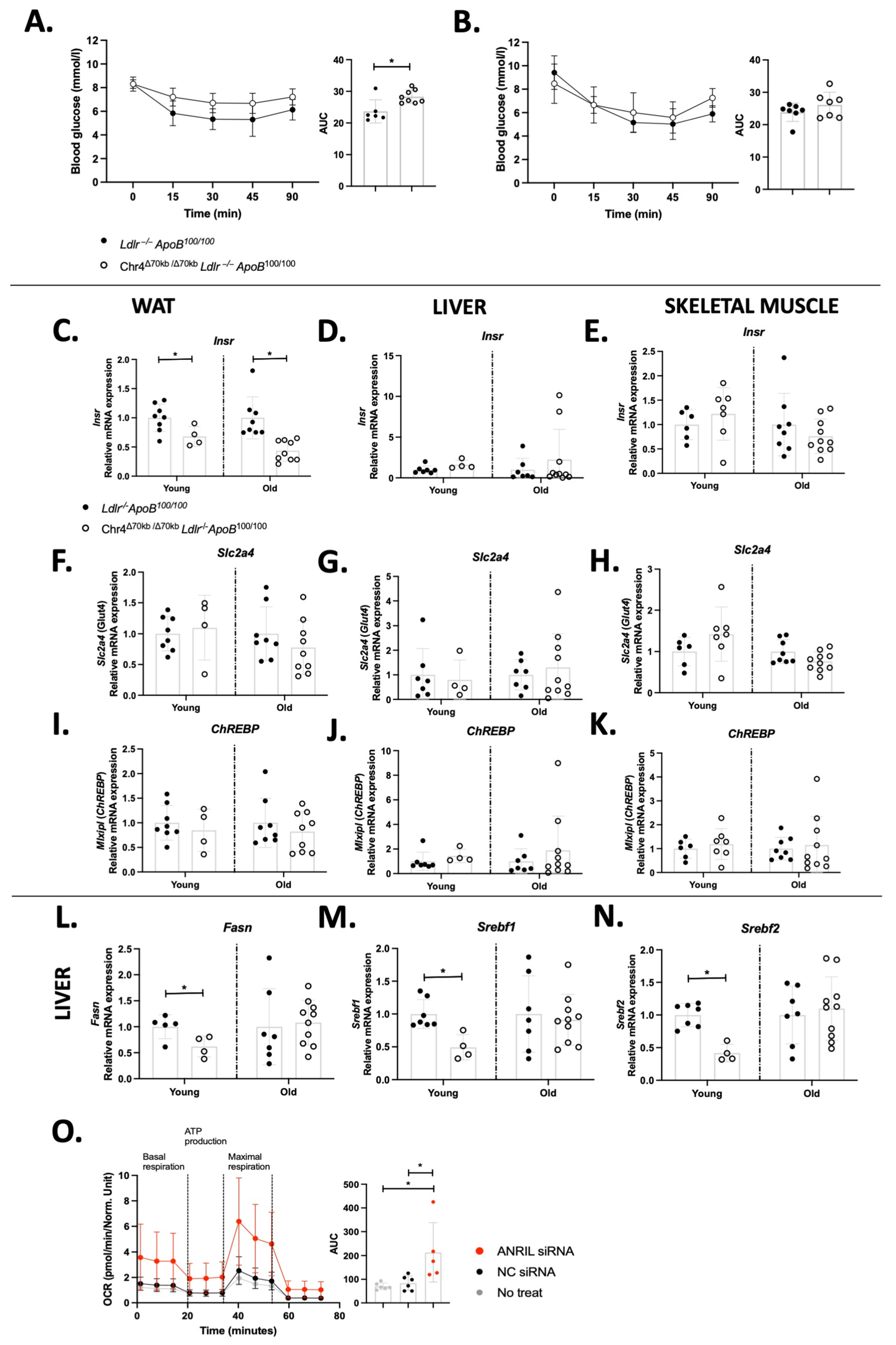
3.3. Chr4Δ70/Δ70 Downregulates the Expression of Insulin Receptor Specifically in White Adipose Tissue in ApoB100/100 Mice
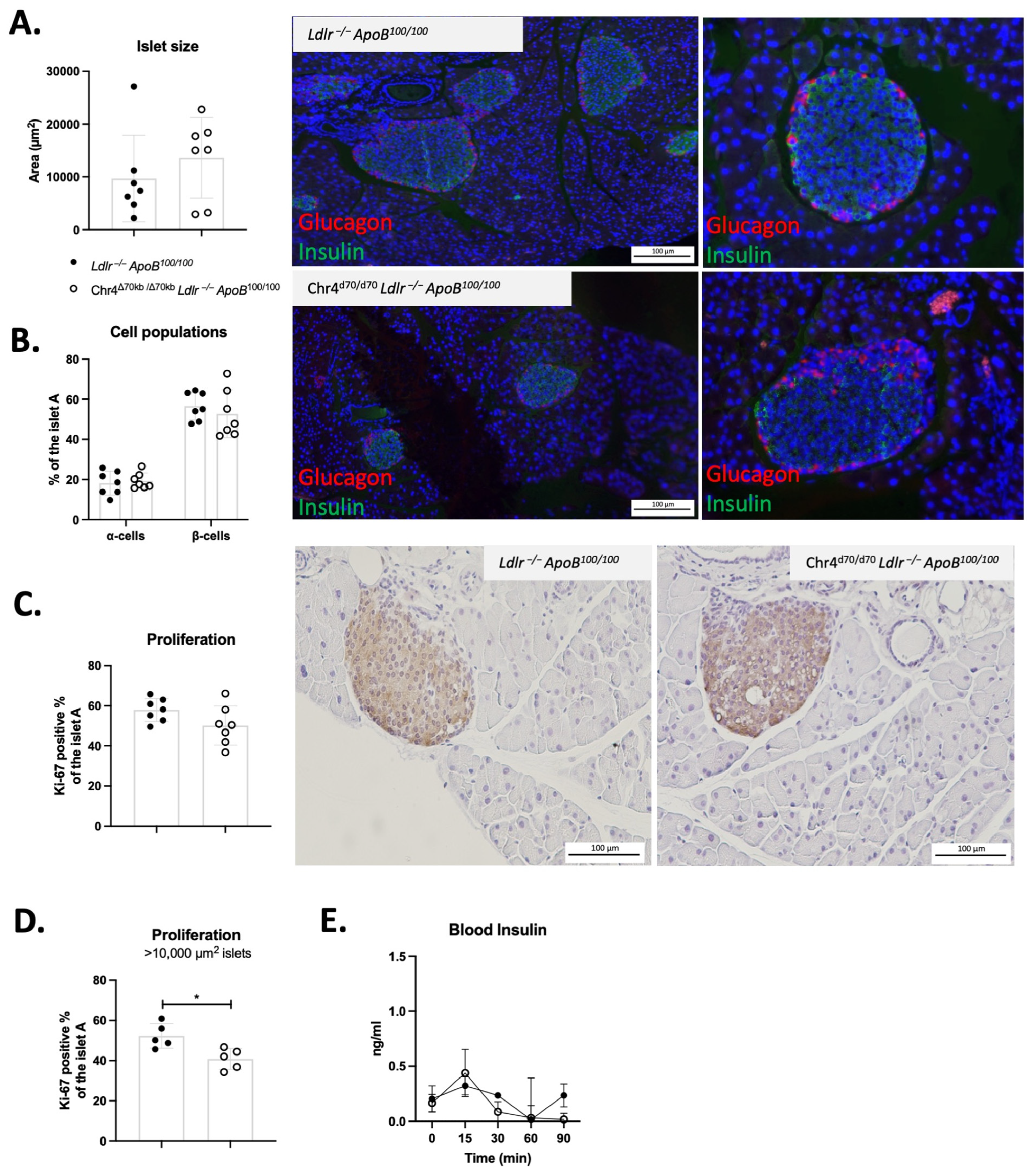
3.4. Chr4Δ70/Δ70 Reduces Cell Proliferation in Pancreatic Islets in Young Ldlr−/−ApoB100/100 Mice
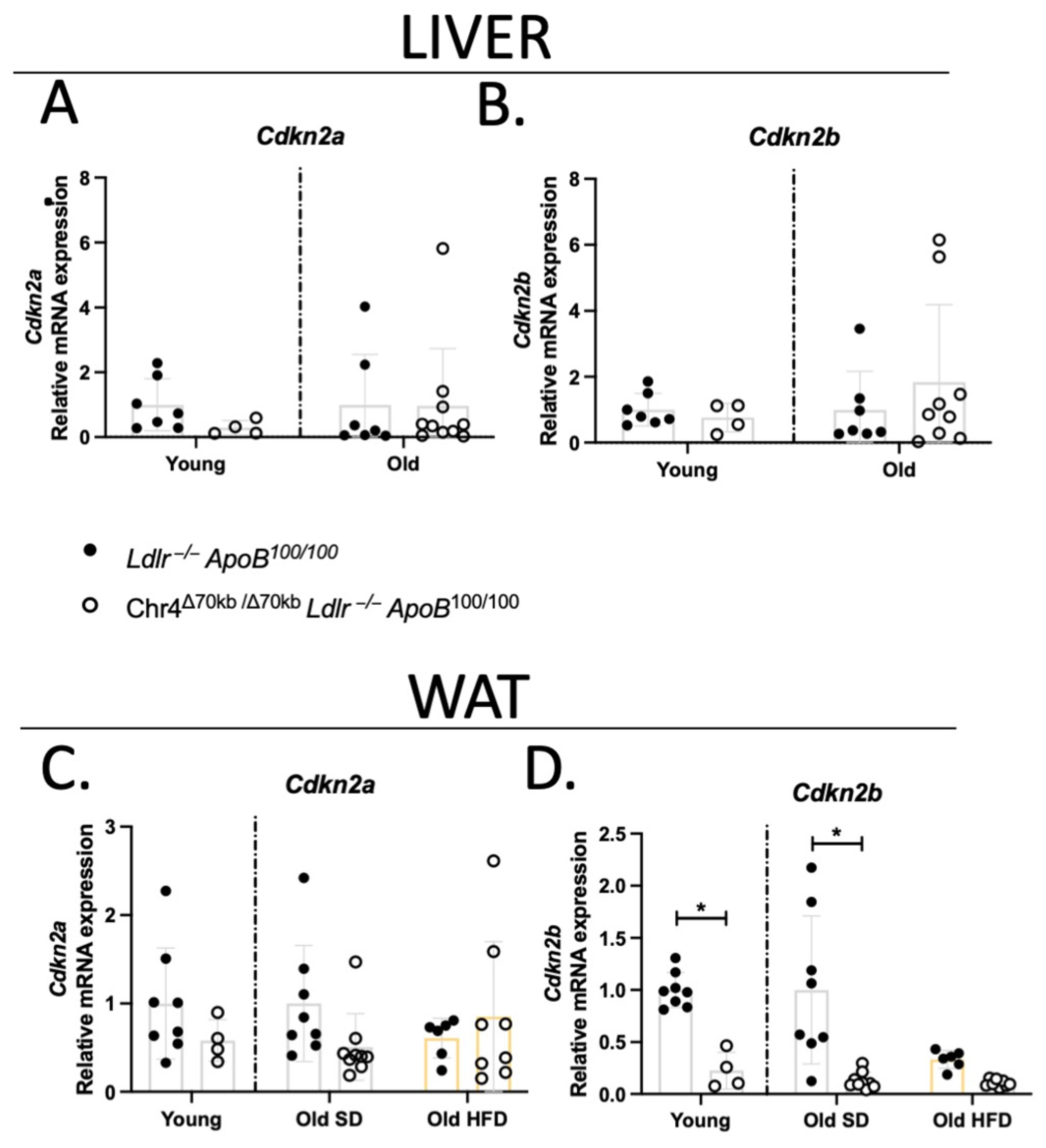
3.5. Chr4Δ70/Δ70 Downregulates Cdkn2b Specifically in White Adipose Tissue in Ldlr−/−ApoB100/100 Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ong, K.L.; Stafford, L.K.; McLaughlin, S.A.; Boyko, E.J.; Vollset, S.E.; Smith, A.E.; Dalton, B.E.; Duprey, J.; Cruz, J.A.; Hagins, H.; et al. Global, Regional, and National Burden of Diabetes from 1990 to 2021, with Projections of Prevalence to 2050: A Systematic Analysis for the Global Burden of Disease Study 2021. Lancet 2023, 402, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Ruze, R.; Liu, T.; Zou, X.; Song, J.; Chen, Y.; Xu, R.; Yin, X.; Xu, Q. Obesity and Type 2 Diabetes Mellitus: Connections in Epidemiology, Pathogenesis, and Treatments. Front. Endocrinol. 2023, 14, 1161521. [Google Scholar] [CrossRef]
- Rohm, T.V.; Meier, D.T.; Olefsky, J.M.; Donath, M.Y. Inflammation in Obesity, Diabetes, and Related Disorders. Immunity 2022, 55, 31–55. [Google Scholar] [CrossRef]
- Guerre-Millo, M.; Guesnet, P.; Guichard, C.; Durand, G.; Lavau, M. Alteration in Membrane Lipid Order and Composition in Metabolically Hyperactive Fatty Rat Adipocytes. Lipids 1994, 29, 205–209. [Google Scholar] [CrossRef]
- Le Lay, S.; Ferré, P.; Dugail, I. Adipocyte Cholesterol Balance in Obesity. Biochem. Soc. Trans. 2004, 32, 103–106. [Google Scholar] [CrossRef]
- The Myocardial Infarction Genetics Consortium; Shea, J.; Agarwala, V.; Philippakis, A.A.; Maguire, J.; Banks, E.; DePristo, M.; Thomson, B.; Guiducci, C.; Onofrio, R.C.; et al. Comparing Strategies to Fine-Map the Association of Common SNPs at Chromosome 9p21 with Type 2 Diabetes and Myocardial Infarction. Nat. Genet. 2011, 43, 801–805. [Google Scholar] [CrossRef]
- Li, W.-Q.; Pfeiffer, R.M.; Hyland, P.L.; Shi, J.; Gu, F.; Wang, Z.; Bhattacharjee, S.; Luo, J.; Xiong, X.; Yeager, M.; et al. Genetic Polymorphisms in the 9p21 Region Associated with Risk of Multiple Cancers. Carcinogenesis 2014, 35, 2698–2705. [Google Scholar] [CrossRef]
- Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; de Bakker, P.I.W.; Chen, H.; Roix, J.J.; Kathiresan, S.; Hirschhorn, J.N.; Daly, M.J.; et al. Genome-Wide Association Analysis Identifies Loci for Type 2 Diabetes and Triglyceride Levels. Science 2007, 316, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Silander, K.; Tang, H.; Myles, S.; Jakkula, E.; Timpson, N.J.; Cavalli-Sforza, L.; Peltonen, L. Worldwide Patterns of Haplotype Diversity at 9p21.3, a Locus Associated with Type 2 Diabetes and Coronary Heart Disease. Genome Med. 2009, 1, 51. [Google Scholar] [CrossRef] [PubMed]
- Broadbent, H.M.; Peden, J.F.; Lorkowski, S.; Goel, A.; Ongen, H.; Green, F.; Clarke, R.; Collins, R.; Franzosi, M.G.; Tognoni, G.; et al. Susceptibility to Coronary Artery Disease and Diabetes Is Encoded by Distinct, Tightly Linked SNPs in the ANRIL Locus on Chromosome 9p. Hum. Mol. Genet. 2008, 17, 806–814. [Google Scholar] [CrossRef]
- Holdt, L.M.; Beutner, F.; Scholz, M.; Gielen, S.; Gäbel, G.; Bergert, H.; Schuler, G.; Thiery, J.; Teupser, D. ANRIL Expression Is Associated with Atherosclerosis Risk at Chromosome 9p21. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Jarinova, O.; Stewart, A.F.R.; Roberts, R.; Wells, G.; Lau, P.; Naing, T.; Buerki, C.; McLean, B.W.; Cook, R.C.; Parker, J.S.; et al. Functional Analysis of the Chromosome 9p21.3 Coronary Artery Disease Risk Locus. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Folkersen, L.; Kyriakou, T.; Goel, A.; Peden, J.; Mälarstig, A.; Paulsson-Berne, G.; Hamsten, A.; Hugh Watkins on behalf of the PROCARDIS consortia; Franco-Cereceda, A.; Gabrielsen, A.; et al. Relationship between CAD Risk Genotype in the Chromosome 9p21 Locus and Gene Expression. Identification of Eight New ANRIL Splice Variants. PLoS ONE 2009, 4, e7677. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sanoff, H.K.; Cho, H.; Burd, C.E.; Torrice, C.; Mohlke, K.L.; Ibrahim, J.G.; Thomas, N.E.; Sharpless, N.E. INK4/ARF Transcript Expression Is Associated with Chromosome 9p21 Variants Linked to Atherosclerosis. PLoS ONE 2009, 4, e5027. [Google Scholar] [CrossRef] [PubMed]
- Samani, N.J.; Erdmann, J.; Hall, A.S.; Hengstenberg, C.; Mangino, M.; Mayer, B.; Dixon, R.J.; Meitinger, T.; Braund, P.; Wichmann, H.-E.; et al. Genomewide Association Analysis of Coronary Artery Disease. N. Engl. J. Med. 2007, 357, 443–453. [Google Scholar] [CrossRef] [PubMed]
- McPherson, R.; Pertsemlidis, A.; Kavaslar, N.; Stewart, A.; Roberts, R.; Cox, D.R.; Hinds, D.A.; Pennacchio, L.A.; Tybjaerg-Hansen, A.; Folsom, A.R.; et al. A Common Allele on Chromosome 9 Associated with Coronary Heart Disease. Science 2007, 316, 1488–1491. [Google Scholar] [CrossRef] [PubMed]
- Helgadottir, A.; Thorleifsson, G.; Manolescu, A.; Gretarsdottir, S.; Blondal, T.; Jonasdottir, A.; Jonasdottir, A.; Sigurdsson, A.; Baker, A.; Palsson, A.; et al. A Common Variant on Chromosome 9p21 Affects the Risk of Myocardial Infarction. Science 2007, 316, 1491–1493. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Liu, Y.; Li, H.; Peng, Y.; Luo, Z. Effect of 9p21.3 (lncRNA and CDKN2A/2B) Variant on Lipid Profile. Front. Cardiovasc. Med. 2022, 9, 946289. [Google Scholar] [CrossRef] [PubMed]
- Ou, M.; Li, X.; Zhao, S.; Cui, S.; Tu, J. Long Non-Coding RNA CDKN2B-AS1 Contributes to Atherosclerotic Plaque Formation by Forming RNA-DNA Triplex in the CDKN2B Promoter. EBioMedicine 2020, 55, 102694. [Google Scholar] [CrossRef]
- Wenzl, F.A.; Mengozzi, A.; Mohammed, S.A.; Pugliese, N.R.; Mongelli, A.; Gorica, E.; Ambrosini, S.; Riederer, P.; Fischer, P.; Hinterberger, M.; et al. Circulating Long Noncoding RNA Signatures Associate with Incident Diabetes in Older Adults: A Prospective Analysis From the VITA Cohort Study. Diabetes Care 2023, 46, 1239–1244. [Google Scholar] [CrossRef]
- MacMillan, H.J.; Kong, Y.; Calvo-Roitberg, E.; Alonso, L.C.; Pai, A.A. High-Throughput Analysis of ANRIL circRNA Isoforms in Human Pancreatic Islets. Sci. Rep. 2022, 12, 7745. [Google Scholar] [CrossRef]
- Svensson, P.-A.; Wahlstrand, B.; Olsson, M.; Froguel, P.; Falchi, M.; Bergman, R.N.; McTernan, P.G.; Hedner, T.; Carlsson, L.M.S.; Jacobson, P. CDKN2B Expression and Subcutaneous Adipose Tissue Expandability: Possible Influence of the 9p21 Atherosclerosis Locus. Biochem. Biophys. Res. Commun. 2014, 446, 1126–1131. [Google Scholar] [CrossRef] [PubMed]
- Rabhi, N.; Hannou, S.A.; Gromada, X.; Salas, E.; Yao, X.; Oger, F.; Carney, C.; Lopez-Mejia, I.C.; Durand, E.; Rabearivelo, I.; et al. Cdkn2a Deficiency Promotes Adipose Tissue Browning. Mol. Metab. 2018, 8, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Zhu, Y.; May, D.; Afzal, V.; Gong, E.; Attanasio, C.; Blow, M.J.; Cohen, J.C.; Rubin, E.M.; Pennacchio, L.A. Targeted Deletion of the 9p21 Non-Coding Coronary Artery Disease Risk Interval in Mice. Nature 2010, 464, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Ye, J.; Nanda, V.; Wang, Y.; Flores, A.M.; Jarr, K.-U.; Tsantilas, P.; Guo, L.; Finn, A.V.; Virmani, R.; et al. Knockout of the Murine Ortholog to the Human 9p21 Coronary Artery Disease Locus Leads to Smooth Muscle Cell Proliferation, Vascular Calcification, and Advanced Atherosclerosis. Circulation 2020, 141, 1274–1276. [Google Scholar] [CrossRef]
- Kettunen, S.; Ruotsalainen, A.-K.; Örd, T.; Suoranta, T.; Heikkilä, J.; Kaikkonen, M.U.; Laham-Karam, N.; Ylä-Herttuala, S. Deletion of the Murine Ortholog of Human 9p21.3 Locus Promotes Atherosclerosis by Increasing Macrophage Proinflammatory Activity. Front. Cardiovasc. Med. 2023, 10, 1113890. [Google Scholar] [CrossRef]
- Young, S.; Veniant, M.; Beigneux, A.; Bensadoun, A.; Fong, L. Lipoprotein Size and Susceptibility to Atherosclerosis—Insights from Genetically Modified Mouse Models. Curr. Drug Targets 2008, 9, 174–189. [Google Scholar] [CrossRef]
- Small, L.; Ehrlich, A.; Iversen, J.; Ashcroft, S.P.; Trošt, K.; Moritz, T.; Hartmann, B.; Holst, J.J.; Treebak, J.T.; Zierath, J.R.; et al. Comparative Analysis of Oral and Intraperitoneal Glucose Tolerance Tests in Mice. Mol. Metab. 2022, 57, 101440. [Google Scholar] [CrossRef]
- Weir, J.B.D.V. New Methods for Calculating Metabolic Rate with Special Reference to Protein Metabolism. J. Physiol. 1949, 109, 1–9. [Google Scholar] [CrossRef]
- Ragan, D.K.; Bankson, J.A. Two-point Dixon Technique Provides Robust Fat Suppression for Multi-mouse Imaging. J. Magn. Reson. Imaging 2010, 31, 510–514. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Agilent. Agilent Technologies Agilent Seahorse XF Cell Mito Stress Test Kit; User Guide; Agilent Technologies: Santa Clara, CA, USA, 2008. [Google Scholar]
- Divakaruni, A.S.; Paradyse, A.; Ferrick, D.A.; Murphy, A.N.; Jastroch, M. Analysis and Interpretation of Microplate-Based Oxygen Consumption and pH Data. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 547, pp. 309–354. ISBN 978-0-12-801415-8. [Google Scholar]
- Schmittgen, T.D.; Livak, K.J. Analyzing Real-Time PCR Data by the Comparative CT Method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Congrains, A.; Kamide, K.; Oguro, R.; Yasuda, O.; Miyata, K.; Yamamoto, E.; Kawai, T.; Kusunoki, H.; Yamamoto, H.; Takeya, Y.; et al. Genetic Variants at the 9p21 Locus Contribute to Atherosclerosis through Modulation of ANRIL and CDKN2A/B. Atherosclerosis 2012, 220, 449–455. [Google Scholar] [CrossRef]
- Cunnington, M.S.; Santibanez Koref, M.; Mayosi, B.M.; Burn, J.; Keavney, B. Chromosome 9p21 SNPs Associated with Multiple Disease Phenotypes Correlate with ANRIL Expression. PLoS Genet. 2010, 6, e1000899. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Sharma, R.B.; Ly, S.; Stamateris, R.E.; Jesdale, W.M.; Alonso, L.C. CDKN2A/B T2D Genome-Wide Association Study Risk SNPs Impact Locus Gene Expression and Proliferation in Human Islets. Diabetes 2018, 67, 872–884. [Google Scholar] [CrossRef]
- GISSI Prevenzione Investigators and SIBioC-GISSI Prevenzione Group; Gori, F.; Specchia, C.; Pietri, S.; Crociati, L.; Barlera, S.; Franciosi, M.; Nicolucci, A.; Signorini, S.; Brambilla, P.; et al. Common Genetic Variants on Chromosome 9p21 Are Associated with Myocardial Infarction and Type 2 Diabetes in an Italian Population. BMC Med. Genet. 2010, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Zeggini, E.; Weedon, M.N.; Lindgren, C.M.; Frayling, T.M.; Elliott, K.S.; Lango, H.; Timpson, N.J.; Perry, J.R.B.; Rayner, N.W.; Freathy, R.M.; et al. Replication of Genome-Wide Association Signals in UK Samples Reveals Risk Loci for Type 2 Diabetes. Science 2007, 316, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.; Murray, R.; Cheong, C.; Teh, A.L.; Clarke-Harris, R.; Barton, S.; Costello, P.; Garratt, E.; Cook, E.; Titcombe, P.; et al. ANRIL Promoter DNA Methylation: A Perinatal Marker for Later Adiposity. EBioMedicine 2017, 19, 60–72. [Google Scholar] [CrossRef]
- Oraha, J.; Enriquez, R.F.; Herzog, H.; Lee, N.J. Sex-Specific Changes in Metabolism during the Transition from Chow to High-Fat Diet Feeding Are Abolished in Response to Dieting in C57BL/6J Mice. Int. J. Obes. 2022, 46, 1749–1758. [Google Scholar] [CrossRef]
- Hong, J.; Stubbins, R.E.; Smith, R.R.; Harvey, A.E.; Núñez, N.P. Differential Susceptibility to Obesity between Male, Female and Ovariectomized Female Mice. Nutr. J. 2009, 8, 11. [Google Scholar] [CrossRef]
- Mayoral, R.; Osborn, O.; McNelis, J.; Johnson, A.M.; Oh, D.Y.; Izquierdo, C.L.; Chung, H.; Li, P.; Traves, P.G.; Bandyopadhyay, G.; et al. Adipocyte SIRT1 Knockout Promotes PPARγ Activity, Adipogenesis and Insulin Sensitivity in Chronic-HFD and Obesity. Mol. Metab. 2015, 4, 378–391. [Google Scholar] [CrossRef]
- Liang, F.; Kume, S.; Koya, D. SIRT1 and Insulin Resistance. Nat. Rev. Endocrinol. 2009, 5, 367–373. [Google Scholar] [CrossRef]
- Chalkiadaki, A.; Guarente, L. High-Fat Diet Triggers Inflammation-Induced Cleavage of SIRT1 in Adipose Tissue to Promote Metabolic Dysfunction. Cell Metab. 2012, 16, 180–188. [Google Scholar] [CrossRef]
- Kurylowicz, A.; Owczarz, M.; Polosak, J.; Jonas, M.I.; Lisik, W.; Jonas, M.; Chmura, A.; Puzianowska-Kuznicka, M. SIRT1 and SIRT7 Expression in Adipose Tissues of Obese and Normal-Weight Individuals Is Regulated by microRNAs but Not by Methylation Status. Int. J. Obes. 2016, 40, 1635–1642. [Google Scholar] [CrossRef]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-κB-Dependent Transcription and Cell Survival by the SIRT1 Deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]
- Yoshizaki, T.; Milne, J.C.; Imamura, T.; Schenk, S.; Sonoda, N.; Babendure, J.L.; Lu, J.-C.; Smith, J.J.; Jirousek, M.R.; Olefsky, J.M. SIRT1 Exerts Anti-Inflammatory Effects and Improves Insulin Sensitivity in Adipocytes. Mol. Cell. Biol. 2009, 29, 1363–1374. [Google Scholar] [CrossRef]
- Arcidiacono, B.; Chiefari, E.; Foryst-Ludwig, A.; Currò, G.; Navarra, G.; Brunetti, F.S.; Mirabelli, M.; Corigliano, D.M.; Kintscher, U.; Britti, D.; et al. Obesity-Related Hypoxia via miR-128 Decreases Insulin-Receptor Expression in Human and Mouse Adipose Tissue Promoting Systemic Insulin Resistance. eBioMedicine 2020, 59, 102912. [Google Scholar] [CrossRef]
- Shen, S.; Shen, M.; Kuang, L.; Yang, K.; Wu, S.; Liu, X.; Wang, Y.; Wang, Y. SIRT1/SREBPs-Mediated Regulation of Lipid Metabolism. Pharmacol. Res. 2024, 199, 107037. [Google Scholar] [CrossRef]
- Shimano, H.; Sato, R. SREBP-Regulated Lipid Metabolism: Convergent Physiology—Divergent Pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef]
- Buchner, D.A.; Yazbek, S.N.; Solinas, P.; Burrage, L.C.; Morgan, M.G.; Hoppel, C.L.; Nadeau, J.H. Increased Mitochondrial Oxidative Phosphorylation in the Liver Is Associated with Obesity and Insulin Resistance. Obesity 2011, 19, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Takamura, T.; Misu, H.; Matsuzawa-Nagata, N.; Sakurai, M.; Ota, T.; Shimizu, A.; Kurita, S.; Takeshita, Y.; Ando, H.; Honda, M.; et al. Obesity Upregulates Genes Involved in Oxidative Phosphorylation in Livers of Diabetic Patients. Obesity 2008, 16, 2601–2609. [Google Scholar] [CrossRef]
- Jurczyk, A.; Bortell, R.; Alonso, L.C. Human β-Cell Regeneration: Progress, Hurdles, and Controversy. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 102–108. [Google Scholar] [CrossRef]
- Kong, Y.; Sharma, R.B.; Nwosu, B.U.; Alonso, L.C. Islet Biology, the CDKN2A/B Locus and Type 2 Diabetes Risk. Diabetologia 2016, 59, 1579–1593. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blood Values | Young Mice 1 | Aged Mice | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ldlr−/−ApoB100/100 | Chr4Δ70/Δ70Ldlr−/−ApoB100/100 | Ref. | Ldlr−/−ApoB100/100 | Chr4Δ70/Δ70Ldlr−/−ApoB100/100 | |||||||||||
| Glucose (mmol/L) | 6.59 | ± | 0.62 (n = 7) | 7.35 | ± | 0.58 (n = 6) * p = 0.046 | (23) | 7.36 | ± | 0.43 | (n = 8) | 8.03 | ± | 1.03 | (n = 9) |
| Cholesterol (mmol/L) | 7.47 | ± | 1.60 (n = 8) | 6.82 | ± | 2.38 (n = 6) | (23) | 6.98 | ± | 1.14 | (n = 8) | 6.94 | ± | 2.64 | (n = 9) |
| Triglycerides (mmol/L) | 1.37 | ± | 0.52 (n = 8) | 1.18 | ± | 0.40 (n = 6) | (23) | 1.68 | ± | 0.42 | (n = 8) | 1.70 | ± | 0.46 | (n = 9) |
| WBCB (x10E09 cells/L) | 11.96 | ± | 4.69 (n = 7) | 13.73 | ± | 2.58 (n = 5) | (23) | 14.91 | ± | 12.17 | (n = 8) | 8.29 | ± | 2.55 | (n = 9) |
| %NEUT (%) | 8.37 | ± | 1.46 (n = 7) | 8.56 | ± | 2.14 (n = 5) | (23) | 7.26 | ± | 3.74 | (n = 8) | 9.42 | ± | 1.77 | (n = 9) |
| %LYM (%) | 78.87 | ± | 3.78 (n = 7) | 80.52 | ± | 6.35 (n = 5) | (23) | 82.71 | ± | 5.76 | (n = 8) | 80.19 | ± | 3.15 | (n = 9) |
| %MONO (%) | 1.53 | ± | 0.25 (n = 7) | 2.02 | ± | 0.85 (n = 5) | (23) | 3.21 | ± | 2.96 | (n = 8) | 3.40 | ± | 1.11 | (n = 9) |
| %EOS (%) | 4.6 | ± | 1.64 (n = 7) | 2.52 | ± | 0.76 (n = 5) * p = 0.026 | (23) | 3.81 | ± | 1.96 | (n = 8) | 4.36 | ± | 2.16 | (n = 9) |
| %BASO (%) | 6.27 | ± | 3.80 (n = 7) | 5.72 | ± | 7.56 (n = 5) | (23) | 2.61 | ± | 5.59 | (n = 8) | 0.57 | ± | 1.04 | (n = 9) |
| %Retic (%) | 0.39 | ± | 0.11 (n = 7) | 0.72 | ± | 0.84 (n = 5) | (23) | 4.28 | ± | 1.64 | (n = 8) | 3.37 | ± | 1.27 | (n = 9) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kettunen, S.; Suoranta, T.; Beikverdi, S.; Heikkilä, M.; Slita, A.; Räty, I.; Ylä-Herttuala, E.; Öörni, K.; Ruotsalainen, A.-K.; Ylä-Herttuala, S. Deletion of the Murine Ortholog of the Human 9p21.3 Locus Leads to Insulin Resistance and Obesity in Hypercholesterolemic Mice. Cells 2024, 13, 983. https://doi.org/10.3390/cells13110983
Kettunen S, Suoranta T, Beikverdi S, Heikkilä M, Slita A, Räty I, Ylä-Herttuala E, Öörni K, Ruotsalainen A-K, Ylä-Herttuala S. Deletion of the Murine Ortholog of the Human 9p21.3 Locus Leads to Insulin Resistance and Obesity in Hypercholesterolemic Mice. Cells. 2024; 13(11):983. https://doi.org/10.3390/cells13110983
Chicago/Turabian StyleKettunen, Sanna, Tuisku Suoranta, Sadegh Beikverdi, Minja Heikkilä, Anna Slita, Iida Räty, Elias Ylä-Herttuala, Katariina Öörni, Anna-Kaisa Ruotsalainen, and Seppo Ylä-Herttuala. 2024. "Deletion of the Murine Ortholog of the Human 9p21.3 Locus Leads to Insulin Resistance and Obesity in Hypercholesterolemic Mice" Cells 13, no. 11: 983. https://doi.org/10.3390/cells13110983