
Myocardin-Related Transcription Factor Mediates Epithelial Fibrogenesis in Polycystic Kidney Disease

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. siRNA-Mediated Silencing
2.3. Real-Time Quantitative PCR (RT-qPCR)
2.4. Western Blot Analysis
2.5. Preparation of GST-Fusion Protein and Rho Activation Assay
2.6. Immunofluorescence Microscopy and Quantification
2.7. Next-Generation Sequencing Transcriptome Analysis
2.8. LLC-PK1 and Fibroblast Communication, Collagen Substrate Wrinkling Quantification
2.9. Animal Tissues and Patient Specimens
2.10. Immunohistochemistry and Quantification
2.11. RNAScope
2.12. Statistical Analysis
3. Results
3.1. PC1 or PC2 Downregulation Activates RhoA and Induces Nuclear Translocation of MRTF
3.2. PC1/2 Loss Elevates MRTF Expression
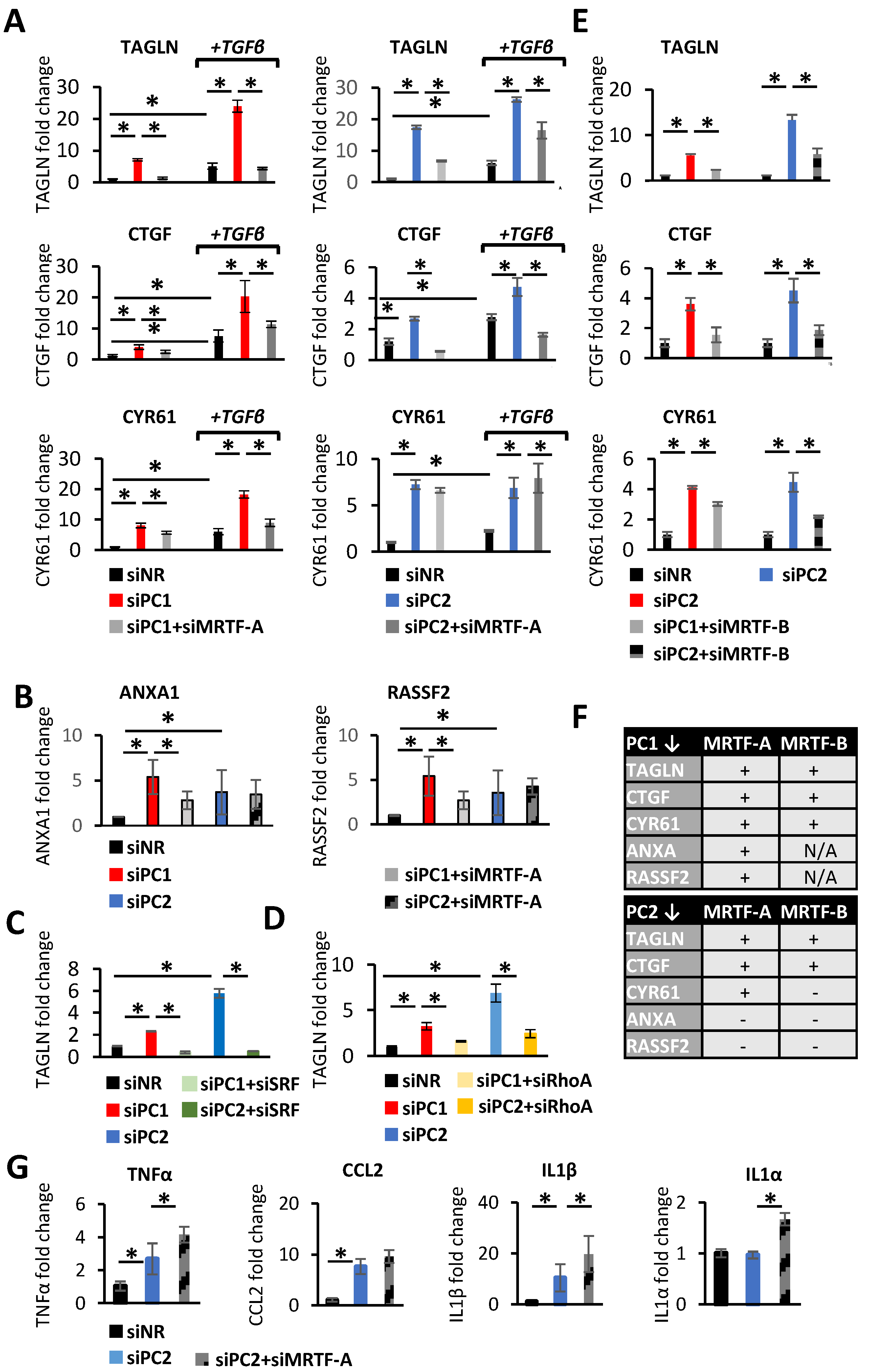
3.3. MRTF in Action upon PC Loss in the Epithelium: A Targeted Approach
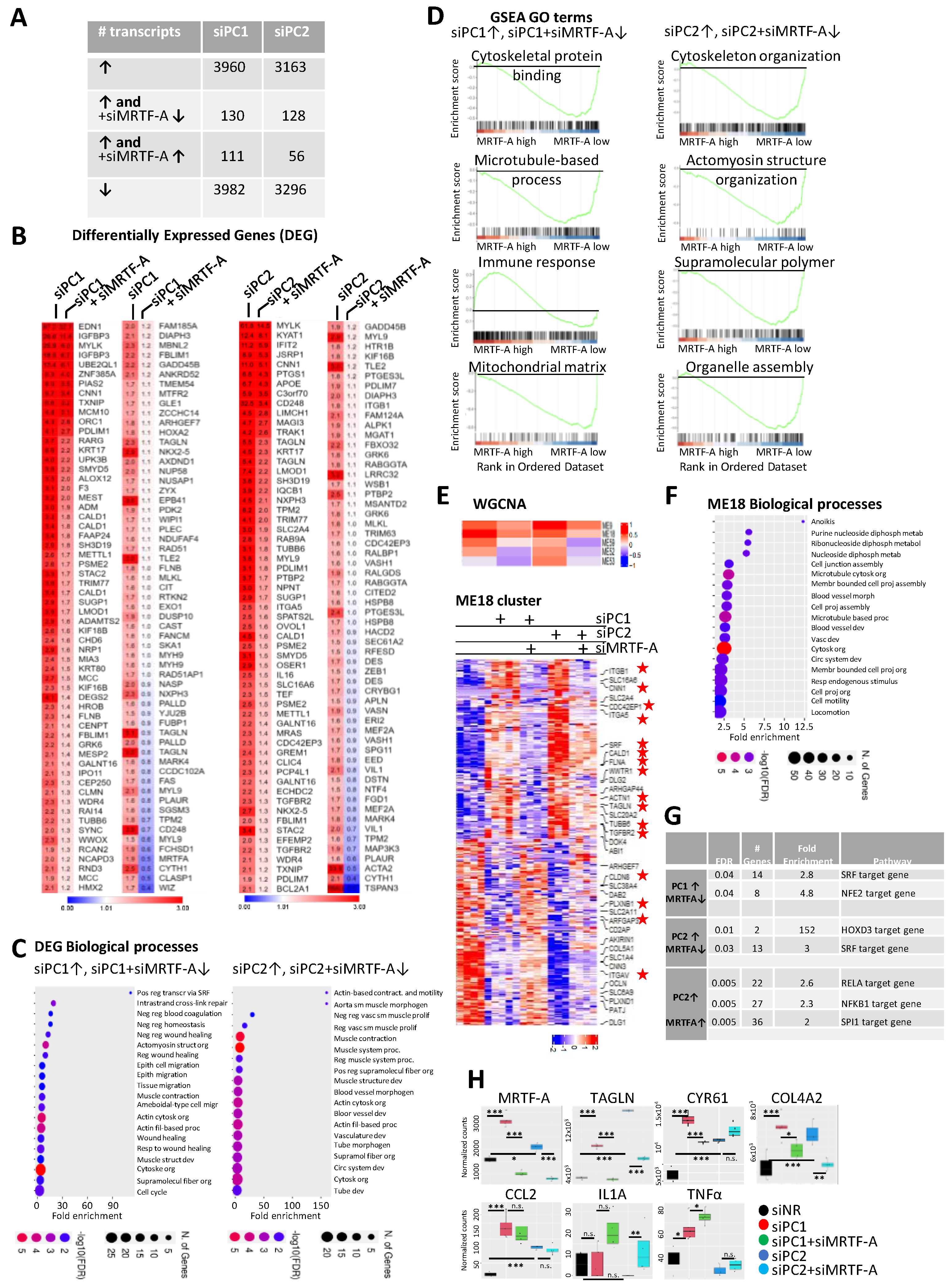
3.4. MRTF in Action upon PC Loss: A General Approach
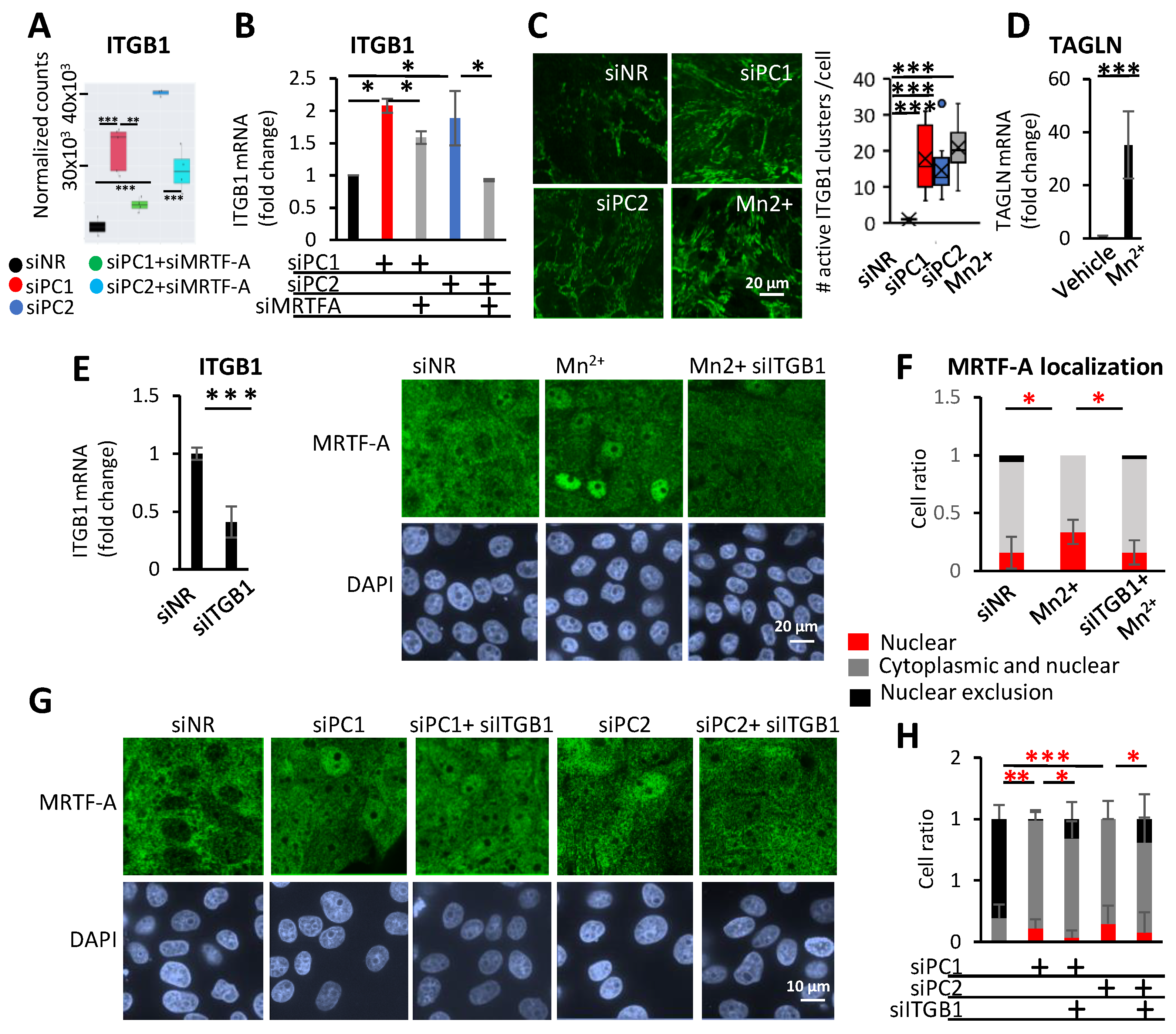
3.5. Integrin β1 (ITGB1) and MRTF-A Form a Feed-Forward Loop and Regulate Profibrotic Gene Expression
3.6. The Role of MRTF in Paracrine Epithelial–Mesenchymal Communication upon PC Loss
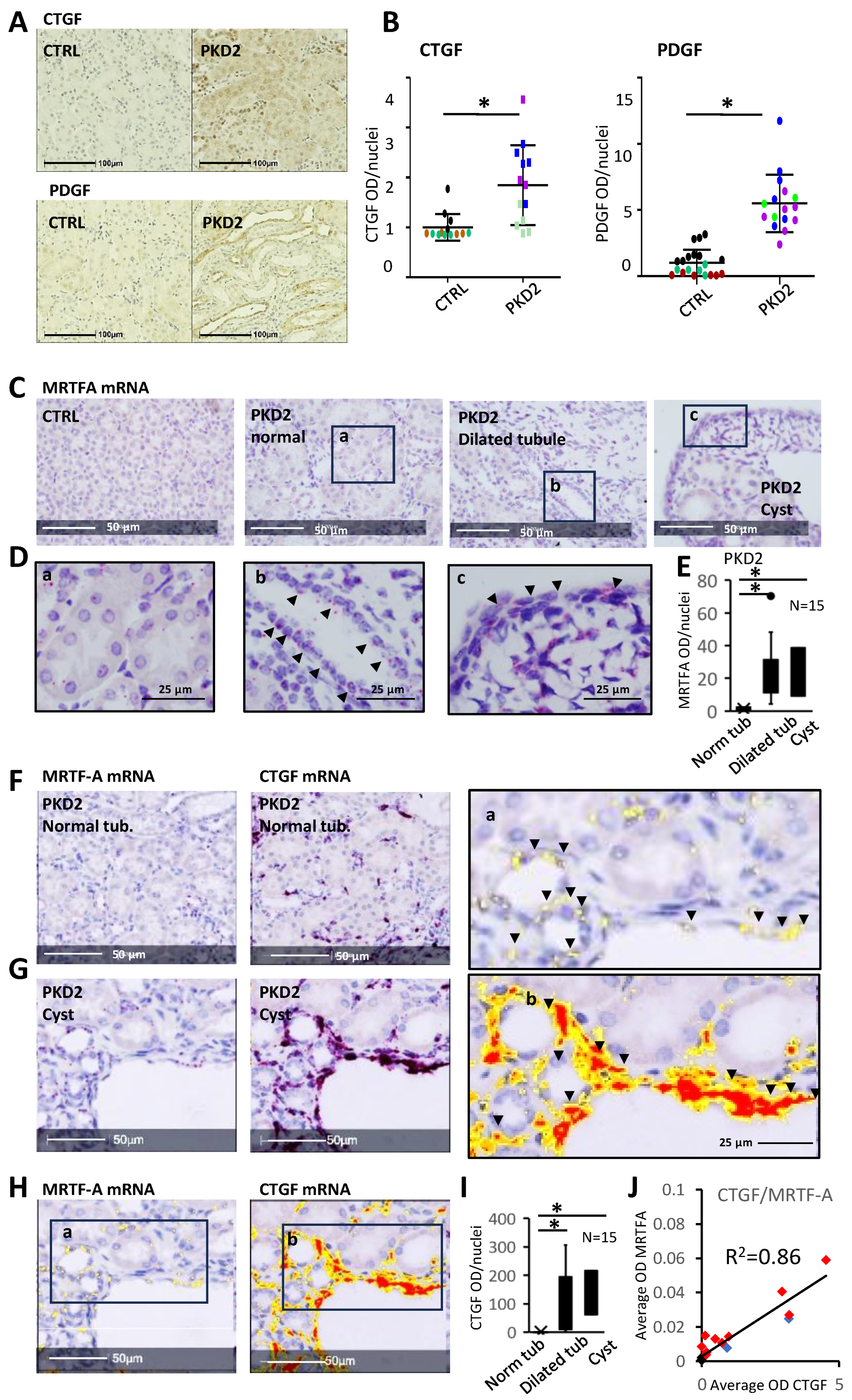
3.7. The MRTF Pathway Is Activated In Vivo in Various Forms of PKD
3.8. CTGF Expression Shows Spatial Correlation with Increased MRTF Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. RNASeq Data Analysis
References
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Primers 2018, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Feingold, J. Estimating prevalence in single-gene kidney diseases progressing to renal failure. Kidney Int. 2000, 58, 925–943. [Google Scholar] [CrossRef]
- Douguet, D.; Patel, A.; Honore, E. Structure and function of polycystins: Insights into polycystic kidney disease. Nat. Rev. Nephrol. 2019, 15, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Roy, S.; Li, L.; Ma, M. Polycystic kidney disease: Novel insights into polycystin function. Trends Mol. Med. 2023, 29, 268–281. [Google Scholar] [CrossRef]
- Gopalakrishnan, J.; Feistel, K.; Friedrich, B.M.; Grapin-Botton, A.; Jurisch-Yaksi, N.; Mass, E.; Mick, D.U.; Muller, R.U.; May-Simera, H.; Schermer, B.; et al. Emerging principles of primary cilia dynamics in controlling tissue organization and function. EMBO J. 2023, 42, e113891. [Google Scholar] [CrossRef]
- Kasahara, K.; Inagaki, M. Primary ciliary signaling: Links with the cell cycle. Trends Cell Biol. 2021, 31, 954–964. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Dynlacht, B.D. The regulation of cilium assembly and disassembly in development and disease. Development 2018, 145, dev151407. [Google Scholar] [CrossRef] [PubMed]
- Fragiadaki, M.; Macleod, F.M.; Ong, A.C.M. The Controversial Role of Fibrosis in Autosomal Dominant Polycystic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 8936. [Google Scholar] [CrossRef]
- Zhang, Y.; Reif, G.; Wallace, D.P. Extracellular matrix, integrins, and focal adhesion signaling in polycystic kidney disease. Cell Signal. 2020, 72, 109646. [Google Scholar] [CrossRef]
- Caplan, M.J. AMPK and Polycystic Kidney Disease Drug Development: An Interesting Off-Target Target. Front. Med. 2022, 9, 753418. [Google Scholar] [CrossRef]
- Chapin, H.C.; Caplan, M.J. The cell biology of polycystic kidney disease. J. Cell Biol. 2010, 191, 701–710. [Google Scholar] [CrossRef]
- Ferreira, F.M.; Watanabe, E.H.; Onuchic, L.F. Polycystins and Molecular Basis of Autosomal Dominant Polycystic Kidney Disease. In Polycystic Kidney Disease; Li, X., Ed.; Mayo Clinic: Brisbane, Australia, 2015. [Google Scholar]
- Marquez-Nogueras, K.M.; Vuchkovska, V.; Kuo, I.Y. Calcium signaling in polycystic kidney disease-cell death and survival. Cell Calcium. 2023, 112, 102733. [Google Scholar] [CrossRef]
- Onuchic, L.; Padovano, V.; Schena, G.; Rajendran, V.; Dong, K.; Shi, X.; Pandya, R.; Rai, V.; Gresko, N.P.; Ahmed, O.; et al. The C-terminal tail of polycystin-1 suppresses cystic disease in a mitochondrial enzyme-dependent fashion. Nat. Commun. 2023, 14, 1790. [Google Scholar] [CrossRef] [PubMed]
- Bialik, J.F.; Ding, M.; Speight, P.; Dan, Q.; Miranda, M.Z.; Di Ciano-Oliveira, C.; Kofler, M.M.; Rotstein, O.D.; Pedersen, S.F.; Szaszi, K.; et al. Profibrotic epithelial phenotype: A central role for MRTF and TAZ. Sci. Rep. 2019, 9, 4323. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.T.; Sanchez-Laorden, B.; Lopez-Blau, C.; De Frutos, C.A.; Boutet, A.; Arevalo, M.; Rowe, R.G.; Weiss, S.J.; Lopez-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Grgic, I.; Campanholle, G.; Bijol, V.; Wang, C.; Sabbisetti, V.S.; Ichimura, T.; Humphreys, B.D.; Bonventre, J.V. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012, 82, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Huang, S.; Park, J.; Park, Y.; Ko, Y.A.; Seasock, M.J.; Bryer, J.S.; Xu, X.X.; Song, W.C.; Palmer, M.; et al. Renal compartment-specific genetic variation analyses identify new pathways in chronic kidney disease. Nat. Med. 2018, 24, 1721–1731. [Google Scholar] [CrossRef]
- Liu, C.Y.; Chan, S.W.; Guo, F.; Toloczko, A.; Cui, L.; Hong, W. MRTF/SRF dependent transcriptional regulation of TAZ in breast cancer cells. Oncotarget 2016, 7, 13706–13716. [Google Scholar] [CrossRef]
- Liu, X.; Miao, J.; Wang, C.; Zhou, S.; Chen, S.; Ren, Q.; Hong, X.; Wang, Y.; Hou, F.F.; Zhou, L.; et al. Tubule-derived exosomes play a central role in fibroblast activation and kidney fibrosis. Kidney Int. 2020, 97, 1181–1195. [Google Scholar] [CrossRef]
- Tan, R.J.; Zhou, D.; Liu, Y. Signaling Crosstalk between Tubular Epithelial Cells and Interstitial Fibroblasts after Kidney Injury. Kidney Dis. 2016, 2, 136–144. [Google Scholar] [CrossRef]
- Zhou, D.; Fu, H.; Zhang, L.; Zhang, K.; Min, Y.; Xiao, L.; Lin, L.; Bastacky, S.I.; Liu, Y. Tubule-Derived Wnts Are Required for Fibroblast Activation and Kidney Fibrosis. J. Am. Soc. Nephrol. 2017, 28, 2322–2336. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Sebe, A.; Peterfi, Z.; Masszi, A.; Thirone, A.C.; Rotstein, O.D.; Nakano, H.; McCulloch, C.A.; Szaszi, K.; Mucsi, I.; et al. Cell contact-dependent regulation of epithelial-myofibroblast transition via the rho-rho kinase-phospho-myosin pathway. Mol. Biol. Cell 2007, 18, 1083–1097. [Google Scholar] [CrossRef]
- Fintha, A.; Gasparics, A.; Fang, L.; Erdei, Z.; Hamar, P.; Mozes, M.M.; Kokeny, G.; Rosivall, L.; Sebe, A. Characterization and role of SCAI during renal fibrosis and epithelial-to-mesenchymal transition. Am. J. Pathol. 2013, 182, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Liu, L.; Zhang, T.; Wu, X.; Zhang, T.; Xu, Y. MKL1 mediates TGF-beta-induced CTGF transcription to promote renal fibrosis. J. Cell Physiol. 2020, 235, 4790–4803. [Google Scholar] [CrossRef]
- Masszi, A.; Speight, P.; Charbonney, E.; Lodyga, M.; Nakano, H.; Szaszi, K.; Kapus, A. Fate-determining mechanisms in epithelial-myofibroblast transition: Major inhibitory role for Smad3. J. Cell Biol. 2010, 188, 383–399. [Google Scholar] [CrossRef]
- Rozycki, M.; Bialik, J.F.; Speight, P.; Dan, Q.; Knudsen, T.E.; Szeto, S.G.; Yuen, D.A.; Szaszi, K.; Pedersen, S.F.; Kapus, A. Myocardin-related Transcription Factor Regulates Nox4 Protein Expression: Linking Cytoskeletal Organization to Redox State. J. Biol. Chem. 2016, 291, 227–243. [Google Scholar] [CrossRef]
- Speight, P.; Kofler, M.; Szaszi, K.; Kapus, A. Context-dependent switch in chemo/mechanotransduction via multilevel crosstalk among cytoskeleton-regulated MRTF and TAZ and TGFbeta-regulated Smad3. Nat. Commun. 2016, 7, 11642. [Google Scholar] [CrossRef]
- Xu, H.; Wu, X.; Qin, H.; Tian, W.; Chen, J.; Sun, L.; Fang, M.; Xu, Y. Myocardin-Related Transcription Factor A Epigenetically Regulates Renal Fibrosis in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1648–1660. [Google Scholar] [CrossRef]
- Yamamura, Y.; Sakai, N.; Iwata, Y.; Lagares, D.; Hara, A.; Kitajima, S.; Toyama, T.; Miyagawa, T.; Ogura, H.; Sato, K.; et al. Myocardin-related transcription factor contributes to renal fibrosis through the regulation of extracellular microenvironment surrounding fibroblasts. FASEB J. 2023, 37, e23005. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.Z.; Lichner, Z.; Szaszi, K.; Kapus, A. MRTF: Basic Biology and Role in Kidney Disease. Int. J. Mol. Sci. 2021, 22, 6040. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, K.; Barrientos, T.; Pipes, G.C.; Li, S.; Olson, E.N. Muscle-specific signaling mechanism that links actin dynamics to serum response factor. Mol. Cell Biol. 2005, 25, 3173–3181. [Google Scholar] [CrossRef] [PubMed]
- Miralles, F.; Posern, G.; Zaromytidou, A.I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Vartiainen, M.K.; Guettler, S.; Larijani, B.; Treisman, R. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 2007, 316, 1749–1752. [Google Scholar] [CrossRef] [PubMed]
- Esnault, C.; Stewart, A.; Gualdrini, F.; East, P.; Horswell, S.; Matthews, N.; Treisman, R. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev. 2014, 28, 943–958. [Google Scholar] [CrossRef] [PubMed]
- Iwanciw, D.; Rehm, M.; Porst, M.; Goppelt-Struebe, M. Induction of connective tissue growth factor by angiotensin II: Integration of signaling pathways. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1782–1787. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, L.L.; Patenaude, C.A.; Smith, B.D.; Layne, M.D. Myocardin-related transcription factor-A complexes activate type I collagen expression in lung fibroblasts. J. Biol. Chem. 2011, 286, 44116–44125. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Mayanagi, T.; Sobue, K. Dual roles of myocardin-related transcription factors in epithelial mesenchymal transition via slug induction and actin remodeling. J. Cell Biol. 2007, 179, 1027–1042. [Google Scholar] [CrossRef]
- Small, E.M.; Thatcher, J.E.; Sutherland, L.B.; Kinoshita, H.; Gerard, R.D.; Richardson, J.A.; Dimaio, J.M.; Sadek, H.; Kuwahara, K.; Olson, E.N. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ. Res. 2010, 107, 294–304. [Google Scholar] [CrossRef]
- Gualdrini, F.; Esnault, C.; Horswell, S.; Stewart, A.; Matthews, N.; Treisman, R. SRF Co-factors Control the Balance between Cell Proliferation and Contractility. Mol. Cell 2016, 64, 1048–1061. [Google Scholar] [CrossRef] [PubMed]
- Lakhia, R.; Mishra, A.; Biggers, L.; Malladi, V.; Cobo-Stark, P.; Hajarnis, S.; Patel, V. Enhancer and super-enhancer landscape in polycystic kidney disease. Kidney Int. 2023, 103, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Song, X.; Wang, W.; Watnick, T.; Pei, Y.; Qian, F.; Pan, D. A RhoA-YAP-c-Myc signaling axis promotes the development of polycystic kidney disease. Genes Dev. 2018, 32, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Streets, A.J.; Prosseda, P.P.; Ong, A.C. Polycystin-1 regulates ARHGAP35-dependent centrosomal RhoA activation and ROCK signaling. JCI Insight 2020, 5, e135385. [Google Scholar] [CrossRef] [PubMed]
- Foster, C.T.; Gualdrini, F.; Treisman, R. Mutual dependence of the MRTF-SRF and YAP-TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev. 2017, 31, 2361–2375. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Hwang, D.; Lee, D.; Kim, J.H.; Kim, S.Y.; Lim, D.S. MRTF potentiates TEAD-YAP transcriptional activity causing metastasis. EMBO J. 2017, 36, 520–535. [Google Scholar] [CrossRef] [PubMed]
- Yu, O.M.; Miyamoto, S.; Brown, J.H. Myocardin-Related Transcription Factor A and Yes-Associated Protein Exert Dual Control in G Protein-Coupled Receptor- and RhoA-Mediated Transcriptional Regulation and Cell Proliferation. Mol. Cell Biol. 2016, 36, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.Z.; Bialik, J.F.; Speight, P.; Dan, Q.; Yeung, T.; Szaszi, K.; Pedersen, S.F.; Kapus, A. TGF-beta1 regulates the expression and transcriptional activity of TAZ protein via a Smad3-independent, myocardin-related transcription factor-mediated mechanism. J. Biol. Chem. 2017, 292, 14902–14920. [Google Scholar] [CrossRef]
- Song, X.; Di Giovanni, V.; He, N.; Wang, K.; Ingram, A.; Rosenblum, N.D.; Pei, Y. Systems biology of autosomal dominant polycystic kidney disease (ADPKD): Computational identification of gene expression pathways and integrated regulatory networks. Hum. Mol. Genet. 2009, 18, 2328–2343. [Google Scholar] [CrossRef]
- Burns, K.D.; Harris, R.C. Signaling and growth responses of LLC-PK1/Cl4 cells transfected with the rabbit AT1 ANG II receptor. Am. J. Physiol. 1995, 268 Pt 1, C925–C935. [Google Scholar] [CrossRef]
- Kakiashvili, E.; Speight, P.; Waheed, F.; Seth, R.; Lodyga, M.; Tanimura, S.; Kohno, M.; Rotstein, O.D.; Kapus, A.; Szaszi, K. GEF-H1 mediates tumor necrosis factor-alpha-induced Rho activation and myosin phosphorylation: Role in the regulation of tubular paracellular permeability. J. Biol. Chem. 2009, 284, 11454–11466. [Google Scholar] [CrossRef] [PubMed]
- Kofler, M.; Kapus, A. Nucleocytoplasmic Shuttling of the Mechanosensitive Transcription Factors MRTF and YAP/TAZ. Methods Mol. Biol. 2021, 2299, 197–216. [Google Scholar] [PubMed]
- Wipff, P.J.; Majd, H.; Acharya, C.; Buscemi, L.; Meister, J.J.; Hinz, B. The covalent attachment of adhesion molecules to silicone membranes for cell stretching applications. Biomaterials 2009, 30, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Hopp, K.; Ward, C.J.; Hommerding, C.J.; Nasr, S.H.; Tuan, H.F.; Gainullin, V.G.; Rossetti, S.; Torres, V.E.; Harris, P.C. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J. Clin. Investig. 2012, 122, 4257–4273. [Google Scholar] [CrossRef]
- Wu, G.; D’Agati, V.; Cai, Y.; Markowitz, G.; Park, J.H.; Reynolds, D.M.; Maeda, Y.; Le, T.C.; Hou, H., Jr.; Kucherlapati, R.; et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell 1998, 93, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Yook, Y.J.; Woo, Y.M.; Yang, M.H.; Ko, J.Y.; Kim, B.H.; Lee, E.J.; Chang, E.S.; Lee, M.J.; Lee, S.; Park, J.H. Differential Expression of PKD2-Associated Genes in Autosomal Dominant Polycystic Kidney Disease. Genom. Inform. 2012, 10, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Hassane, S.; Leonhard, W.N.; van der Wal, A.; Hawinkels, L.J.; Lantinga-van Leeuwen, I.S.; ten Dijke, P.; Breuning, M.H.; de Heer, E.; Peters, D.J. Elevated TGFbeta-Smad signalling in experimental Pkd1 models and human patients with polycystic kidney disease. J. Pathol. 2010, 222, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dai, Y.; Raman, A.; Daniel, E.; Metcalf, J.; Reif, G.; Pierucci-Alves, F.; Wallace, D.P. Overexpression of TGF-beta1 induces renal fibrosis and accelerates the decline in kidney function in polycystic kidney disease. Am. J. Physiol. Renal Physiol. 2020, 319, F1135–F1148. [Google Scholar] [CrossRef]
- Hayashi, K.; Murai, T.; Oikawa, H.; Masuda, T.; Kimura, K.; Muehlich, S.; Prywes, R.; Morita, T. A novel inhibitory mechanism of MRTF-A/B on the ICAM-1 gene expression in vascular endothelial cells. Sci. Rep. 2015, 5, 10627. [Google Scholar] [CrossRef]
- Wang, D.; Prakash, J.; Nguyen, P.; Davis-Dusenbery, B.N.; Hill, N.S.; Layne, M.D.; Hata, A.; Lagna, G. Bone morphogenetic protein signaling in vascular disease: Anti-inflammatory action through myocardin-related transcription factor A. J. Biol. Chem. 2012, 287, 28067–28077. [Google Scholar] [CrossRef]
- Padovano, V.; Podrini, C.; Boletta, A.; Caplan, M.J. Metabolism and mitochondria in polycystic kidney disease research and therapy. Nat. Rev. Nephrol. 2018, 14, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Podrini, C.; Cassina, L.; Boletta, A. Metabolic reprogramming and the role of mitochondria in polycystic kidney disease. Cell Signal. 2020, 67, 109495. [Google Scholar] [CrossRef] [PubMed]
- Doke, T.; Abedini, A.; Aldridge, D.L.; Yang, Y.W.; Park, J.; Hernandez, C.M.; Balzer, M.S.; Shrestra, R.; Coppock, G.; Rico, J.M.I.; et al. Single-cell analysis identifies the interaction of altered renal tubules with basophils orchestrating kidney fibrosis. Nat. Immunol. 2022, 23, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. Integrin beta1: A Mechanosignaling Sensor Essential for Connective Tissue Deposition by Fibroblasts. Adv. Wound Care 2013, 2, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Boctor, S.; Barisoni, L.M.; Gusella, G.L. Inactivation of integrin-beta1 prevents the development of polycystic kidney disease after the loss of polycystin-1. J. Am. Soc. Nephrol. 2015, 26, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Brandt, D.T.; Baarlink, C.; Kitzing, T.M.; Kremmer, E.; Ivaska, J.; Nollau, P.; Grosse, R. SCAI acts as a suppressor of cancer cell invasion through the transcriptional control of beta1-integrin. Nat. Cell Biol. 2009, 11, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Brandt, D.T.; Xu, J.; Steinbeisser, H.; Grosse, R. Regulation of myocardin-related transcriptional coactivators through cofactor interactions in differentiation and cancer. Cell Cycle 2009, 8, 2523–2527. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Li, A.; Simonsen, N.; Wilkins, J.A. Integrin activation by dithiothreitol or Mn2+ induces a ligand-occupied conformation and exposure of a novel NH2-terminal regulatory site on the beta1 integrin chain. J. Biol. Chem. 1998, 273, 7981–7987. [Google Scholar] [CrossRef]
- Yao, L.; Zhou, Y.; Li, J.; Wickens, L.; Conforti, F.; Rattu, A.; Ibrahim, F.M.; Alzetani, A.; Marshall, B.G.; Fletcher, S.V.; et al. Bidirectional epithelial-mesenchymal crosstalk provides self-sustaining profibrotic signals in pulmonary fibrosis. J. Biol. Chem. 2021, 297, 101096. [Google Scholar] [CrossRef]
- Schuster, R.; Younesi, F.; Ezzo, M.; Hinz, B. The Role of Myofibroblasts in Physiological and Pathological Tissue Repair. Cold Spring Harb. Perspect. Biol. 2023, 15, a041231. [Google Scholar] [CrossRef]
- Lee, H.S. Paracrine role for TGF-beta-induced CTGF and VEGF in mesangial matrix expansion in progressive glomerular disease. Histol. Histopathol. 2012, 27, 1131–1141. [Google Scholar]
- Rayego-Mateos, S.; Campillo, S.; Rodrigues-Diez, R.R.; Tejera-Munoz, A.; Marquez-Exposito, L.; Goldschmeding, R.; Rodriguez-Puyol, D.; Calleros, L.; Ruiz-Ortega, M. Interplay between extracellular matrix components and cellular and molecular mechanisms in kidney fibrosis. Clin. Sci. 2021, 135, 1999–2029. [Google Scholar] [CrossRef]
- Yin, Q.; Liu, H. Connective Tissue Growth Factor and Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 365–380. [Google Scholar] [PubMed]
- Sakai, N.; Chun, J.; Duffield, J.S.; Wada, T.; Luster, A.D.; Tager, A.M. LPA1-induced cytoskeleton reorganization drives fibrosis through CTGF-dependent fibroblast proliferation. FASEB J. 2013, 27, 1830–1846. [Google Scholar] [CrossRef] [PubMed]
- Anorga, S.; Overstreet, J.M.; Falke, L.L.; Tang, J.; Goldschmeding, R.G.; Higgins, P.J.; Samarakoon, R. Deregulation of Hippo-TAZ pathway during renal injury confers a fibrotic maladaptive phenotype. FASEB J. 2018, 32, 2644–2657. [Google Scholar] [CrossRef] [PubMed]
- Boutet, A.; De Frutos, C.A.; Maxwell, P.H.; Mayol, M.J.; Romero, J.; Nieto, M.A. Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. EMBO J. 2006, 25, 5603–5613. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Kikuta, T.; Kobayashi, T.; Inoue, T.; Kanno, Y.; Takigawa, M.; Sugaya, T.; Kopp, J.B.; Suzuki, H. Connective tissue growth factor expressed in tubular epithelium plays a pivotal role in renal fibrogenesis. J. Am. Soc. Nephrol. 2005, 16, 133–143. [Google Scholar] [CrossRef]
- Traykova-Brauch, M.; Schonig, K.; Greiner, O.; Miloud, T.; Jauch, A.; Bode, M.; Felsher, D.W.; Glick, A.B.; Kwiatkowski, D.J.; Bujard, H.; et al. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat. Med. 2008, 14, 979–984. [Google Scholar] [CrossRef]
- Hama, T.; Park, F. Heterotrimeric G protein signaling in polycystic kidney disease. Physiol. Genom. 2016, 48, 429–445. [Google Scholar] [CrossRef]
- Parnell, S.C.; Magenheimer, B.S.; Maser, R.L.; Rankin, C.A.; Smine, A.; Okamoto, T.; Calvet, J.P. The polycystic kidney disease-1 protein, polycystin-1, binds and activates heterotrimeric G-proteins in vitro. Biochem. Biophys. Res. Commun. 1998, 251, 625–631. [Google Scholar] [CrossRef]
- Suzuki, N.; Hajicek, N.; Kozasa, T. Regulation and physiological functions of G12/13-mediated signaling pathways. Neurosignals 2009, 17, 55–70. [Google Scholar] [CrossRef]
- Suzuki, N.; Nakamura, S.; Mano, H.; Kozasa, T. Galpha 12 activates Rho GTPase through tyrosine-phosphorylated leukemia-associated RhoGEF. Proc. Natl. Acad. Sci. USA 2003, 100, 733–738. [Google Scholar] [CrossRef]
- Yuasa, T.; Takakura, A.; Denker, B.M.; Venugopal, B.; Zhou, J. Polycystin-1L2 is a novel G-protein-binding protein. Genomics 2004, 84, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xu, J.X.; El-Jouni, W.; Lu, T.; Li, S.; Wang, Q.; Tran, M.; Yu, W.; Wu, M.; Barrera, I.E.; et al. Galpha12 is required for renal cystogenesis induced by Pkd1 inactivation. J. Cell Sci. 2016, 129, 3675–3684. [Google Scholar] [CrossRef] [PubMed]
- Guilluy, C.; Swaminathan, V.; Garcia-Mata, R.; O’Brien, E.T.; Superfine, R.; Burridge, K. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat. Cell Biol. 2011, 13, 722–727. [Google Scholar] [CrossRef]
- Raman, A.; Reif, G.A.; Dai, Y.; Khanna, A.; Li, X.; Astleford, L.; Parnell, S.C.; Calvet, J.P.; Wallace, D.P. Integrin-Linked Kinase Signaling Promotes Cyst Growth and Fibrosis in Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2017, 28, 2708–2719. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Estevez, B.; Xu, Z.; Kreutz, B.; Karginov, A.; Bai, Y.; Qian, F.; Norifumi, U.; Mosher, D.; Du, X. The interaction of Galpha13 with integrin beta1 mediates cell migration by dynamic regulation of RhoA. Mol. Biol. Cell 2015, 26, 3658–3670. [Google Scholar] [CrossRef]
- Tanner, G.A.; McQuillan, P.F.; Maxwell, M.R.; Keck, J.K.; McAteer, J.A. An in vitro test of the cell stretch-proliferation hypothesis of renal cyst enlargement. J. Am. Soc. Nephrol. 1995, 6, 1230–1241. [Google Scholar] [CrossRef]
- Gauer, S.; Holzmann, Y.; Kranzlin, B.; Hoffmann, S.C.; Gretz, N.; Hauser, I.A.; Goppelt-Struebe, M.; Geiger, H.; Obermuller, N. CTGF Is Expressed During Cystic Remodeling in the PKD/Mhm (cy/+) Rat Model for Autosomal-Dominant Polycystic Kidney Disease (ADPKD). J. Histochem. Cytochem. 2017, 65, 743–755. [Google Scholar] [CrossRef]
- Happe, H.; van der Wal, A.M.; Leonhard, W.N.; Kunnen, S.J.; Breuning, M.H.; de Heer, E.; Peters, D.J. Altered Hippo signalling in polycystic kidney disease. J. Pathol. 2011, 224, 133–142. [Google Scholar] [CrossRef]
- Raman, A.; Parnell, S.C.; Zhang, Y.; Reif, G.A.; Dai, Y.; Khanna, A.; Daniel, E.; White, C.; Vivian, J.L.; Wallace, D.P. Periostin overexpression in collecting ducts accelerates renal cyst growth and fibrosis in polycystic kidney disease. Am. J. Physiol. Renal Physiol. 2018, 315, F1695–F1707. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.P.; White, C.; Savinkova, L.; Nivens, E.; Reif, G.A.; Pinto, C.S.; Raman, A.; Parnell, S.C.; Conway, S.J.; Fields, T.A. Periostin promotes renal cyst growth and interstitial fibrosis in polycystic kidney disease. Kidney Int. 2014, 85, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Brigstock, D.R. Connective tissue growth factor (CCN2) induces adhesion of rat activated hepatic stellate cells by binding of its C-terminal domain to integrin alpha(v)beta(3) and heparan sulfate proteoglycan. J. Biol. Chem. 2004, 279, 8848–8855. [Google Scholar] [CrossRef] [PubMed]
- Gillan, L.; Matei, D.; Fishman, D.A.; Gerbin, C.S.; Karlan, B.Y.; Chang, D.D. Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(V)beta(3) and alpha(V)beta(5) integrins and promotes cell motility. Cancer Res. 2002, 62, 5358–5364. [Google Scholar] [PubMed]
- Orecchia, P.; Conte, R.; Balza, E.; Castellani, P.; Borsi, L.; Zardi, L.; Mingari, M.C.; Carnemolla, B. Identification of a novel cell binding site of periostin involved in tumour growth. Eur. J. Cancer 2011, 47, 2221–2229. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, N.; Tao, S.; Jamadar, A.; Sinha, S.; Howard, C.; Wallace, D.P.; Fields, T.A.; Leask, A.; Calvet, J.P.; Rao, R. Epithelial Vasopressin Type-2 Receptors Regulate Myofibroblasts by a YAP-CCN2-Dependent Mechanism in Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2020, 31, 1697–1710. [Google Scholar] [CrossRef] [PubMed]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F.; Chan, L.; De Freitas, K.; Bialik, J.F.; Majumder, S.; et al. YAP/TAZ Are Mechanoregulators of TGF-beta-Smad Signaling and Renal Fibrogenesis. J. Am. Soc. Nephrol. 2016, 27, 3117–3128. [Google Scholar] [CrossRef] [PubMed]
- Crider, B.J.; Risinger, G.M., Jr.; Haaksma, C.J.; Howard, E.W.; Tomasek, J.J. Myocardin-related transcription factors A and B are key regulators of TGF-beta1-induced fibroblast to myofibroblast differentiation. J. Investig. Dermatol. 2011, 131, 2378–2385. [Google Scholar] [CrossRef] [PubMed]
- Muehlich, S.; Rehm, M.; Ebenau, A.; Goppelt-Struebe, M. Synergistic induction of CTGF by cytochalasin D and TGFbeta-1 in primary human renal epithelial cells: Role of transcriptional regulators MKL1, YAP/TAZ and Smad2/3. Cell Signal. 2017, 29, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Cassina, L.; Chiaravalli, M.; Boletta, A. Increased mitochondrial fragmentation in polycystic kidney disease acts as a modifier of disease progression. FASEB J. 2020, 34, 6493–6507. [Google Scholar] [CrossRef]
- Menezes, L.F.; Germino, G.G. The pathobiology of polycystic kidney disease from a metabolic viewpoint. Nat. Rev. Nephrol. 2019, 15, 735–749. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analysis for RNA-Seq: Transcript-level estimates improve gene-level interferences. F1000Res 2015, 4, 1521. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-mediated genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Ihaka, R.; Gentelman, R.R. Language for data analysis and graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Reyes, A.; Huber, W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012, 22, 2008–2017. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| siRNA Sequences | |
| siPC1 | GCG CUG ACA GAG UUG GAC AUA (dT) (dT) |
| siPC2 | GAC CGU GAG AGA UAC CUU AAA (dT) (dT) |
| siRhoA | AGC AGG UAG AGU UGG CUU U (dT) (dT) |
| siMRTF-A | CCA AGG AGC UGA AGC CAA A (dT) (dT) |
| siMRTFB | CGA CAA ACA CCG UAG CAA A (dT) (dT) |
| siITGB1 | CUG AAG AAG UAG AGA UAA U (dT) (dT) |
| siSRF | GGA ACU GUG CUG AAG AGU A (dT) (dT) |
| siPC1 4331 | UGU CAA GCC GCG UGA AUA A (dT) (dT) |
| siPC2 1644 | CAA GAU UGA UGC AGU GAU A |
| qPCR Primer Sequences | |
| Porcine CTGF F | GTG AAG ACA TAC CGG GCT AAG |
| Porcine CTGF R | GAC ACT TGA ACT CCA CAG GAA |
| Porcine GAPDH F | GCA AAG TGG ACA TGG TCG CCA TCA |
| Porcine GAPDH R | AGC TTC CCA TTC TCA GCC TTG ACT |
| Porcine PPIA F | CGG GTC CTG GCA TCT TGT |
| Porcine PPIA R | TGG CAG TGC AAA TGA AAA ACT G |
| Porcine MRTF-A-E7F | TAT CCT GCC TGT GGA GTC CA |
| Porcine MRTF-A-E8R | ATA AGG CGT CAC TGC TGT CC |
| Porcine MRTFB-E6F | GCA GGC CAC TCA GAT GAA GT |
| Porcine MRTFB-E7R | GTC ACT GCT GTC CTC GTC AA |
| Porcine IFNB1-F | ATG AGC TAT GAT GTG CTT CGA TAC |
| Porcine IFNB1-R | GAA TTG TGG TGG TTG CAT AAT C |
| Porcine TNFA-F E2-3 | CCA ATG GCA GAG TGG GTA TG |
| Porcine TNFA-R E2-3 | CTG AAG AGG ACC TGG GAG TAG |
| Porcine IL1A-F E8-9 | GCA ACT TCC TGT GAC TCT AAG |
| Porcine IL1A-R E8-9 | GCG GCT GAT TTG AAG TAG TC |
| Porcine CCL2-F | CAG CAG CAA GTG TCC TAA AG |
| Porcine CCL2-R | TCC AGG TGG CTT ATG GAG TC |
| Porcine TAGLN E3 F | GAG CAG GTG GCT CAG TTC TT |
| Porcine TAGLN E4 R | CCA CGG TAG TGT CCA TCA TTC |
| Porcine CYR61 E3F | AAA GGC GGC TCC CTG AAG |
| Porcine CYR61 E4 R | GAC GTG GTT TGA ACG ATG C |
| Porcine RASSF1 E2 F | GAG TGG GAG ACA CCT GAC CT |
| Porcine RASSF1 E3 R | ATA CAG GAC GCA CCA GCT TC |
| Porcine ANXA 1 E5 F | TTG CTG AAA ACT CCA GCT CA |
| Porcine ANXA 1 E7 R | GCA AAG CCT TCT GAT AAT CTC C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lichner, Z.; Ding, M.; Khare, T.; Dan, Q.; Benitez, R.; Praszner, M.; Song, X.; Saleeb, R.; Hinz, B.; Pei, Y.; et al. Myocardin-Related Transcription Factor Mediates Epithelial Fibrogenesis in Polycystic Kidney Disease. Cells 2024, 13, 984. https://doi.org/10.3390/cells13110984
Lichner Z, Ding M, Khare T, Dan Q, Benitez R, Praszner M, Song X, Saleeb R, Hinz B, Pei Y, et al. Myocardin-Related Transcription Factor Mediates Epithelial Fibrogenesis in Polycystic Kidney Disease. Cells. 2024; 13(11):984. https://doi.org/10.3390/cells13110984
Chicago/Turabian StyleLichner, Zsuzsanna, Mei Ding, Tarang Khare, Qinghong Dan, Raquel Benitez, Mercédesz Praszner, Xuewen Song, Rola Saleeb, Boris Hinz, York Pei, and et al. 2024. "Myocardin-Related Transcription Factor Mediates Epithelial Fibrogenesis in Polycystic Kidney Disease" Cells 13, no. 11: 984. https://doi.org/10.3390/cells13110984