
Biallelic Variants in MNS1 Are Associated with Laterality Defects and Respiratory Involvement

 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Samples
2.2. Genomic Studies
2.3. Genes Included in Targeted PCD Gene Panel Sequencing
2.4. Immunofluorescence Analysis of Human Respiratory Cells
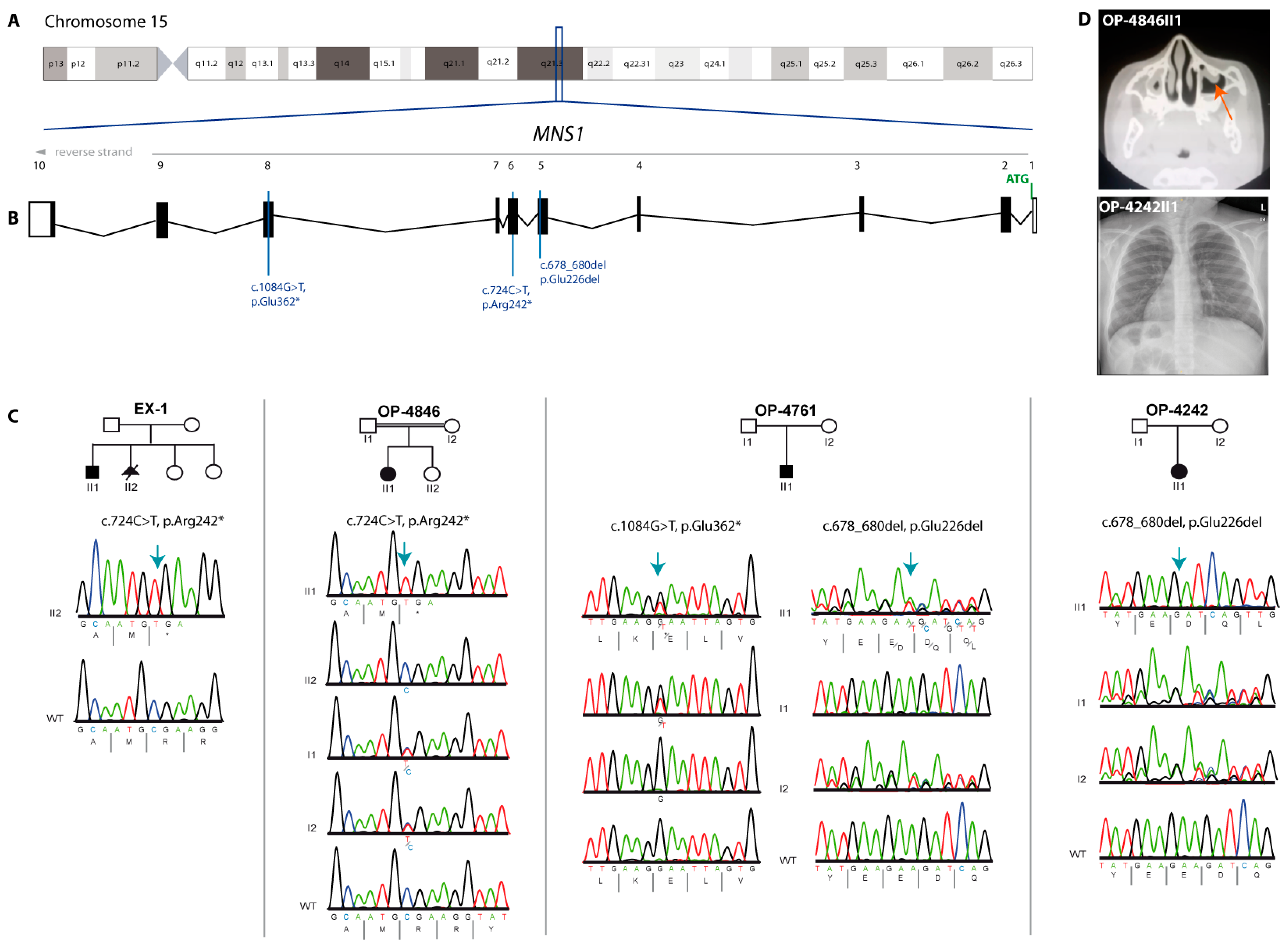
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wallmeier, J.; Nielsen, K.G.; Kuehni, C.E.; Lucas, J.S.; Leigh, M.W.; Zariwala, M.A.; Omran, H. Motile ciliopathies. Nat. Rev. Dis. Prim. 2020, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Horani, A.; Ferkol, T.W. Primary ciliary dyskinesia and associated sensory ciliopathies. Expert Rev. Respir. Med. 2016, 10, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Nöthe-Menchen, T.; Wallmeier, J.; Pennekamp, P.; Höben, I.M.; Olbrich, H.; Loges, N.T.; Raidt, J.; Dougherty, G.W.; Hjeij, R.; Dworniczak, B.; et al. Randomization of Left-Right Asymmetry and Congenital Heart Defects. Circ. Genom. Precis. Med. 2019, 12, e002686. [Google Scholar] [CrossRef] [PubMed]
- Fisch, C.; Dupuis-Williams, P. Ultrastructure of cilia and flagella—Back to the future! Biol. Cell. 2011, 103, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Porter, M.E.; Sale, W.S. The 9 + 2 axoneme anchors multiple inner arm dyneins and a network of kinases and phosphatases that control motility. J. Cell Biol. 2000, 151, F37–F42. [Google Scholar] [CrossRef] [PubMed]
- Best, S.; Shoemark, A.; Rubbo, B.; Patel, M.P.; Fassad, M.R.; Dixon, M.; Rogers, A.V.; Hirst, R.A.; Rutman, A.; Ollosson, S.; et al. Risk factors for situs defects and congenital heart disease in primary ciliary dyskinesia. Thorax 2019, 74, 203–205. [Google Scholar] [CrossRef] [PubMed]
- Fliegauf, M.; Benzing, T.; Omran, H. When cilia go bad: Cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007, 8, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Gorham, G.W.; Merselis, J.G., Jr. Kartagener’s triad: A family study. Bull. Johns Hopkins Hosp. 1959, 104, 11–16. [Google Scholar] [PubMed]
- Brueckner, M. Cilia propel the embryo in the right direction. Am. J. Med. Genet. 2001, 101, 339–344. [Google Scholar] [CrossRef]
- Essner, J.J.; Vogan, K.J.; Wagner, M.K.; Tabin, C.J.; Yost, H.J.; Brueckner, M. Conserved function for embryonic nodal cilia. Nature 2002, 418, 37–38. [Google Scholar] [CrossRef]
- Lucas, J.S.; Barbato, A.; Collins, S.A.; Goutaki, M.; Behan, L.; Caudri, D.; Kuehni, C.E. European Respiratory Society Guidelines for the Diagnosis of Primary Ciliary Dyskinesia. Eur. Respir. J. 2017, 49, 1601090. [Google Scholar] [CrossRef] [PubMed]
- Hjeij, R.; Aprea, I.; Poeta, M.; Nöthe-Menchen, T.; Bracht, D.; Raidt, J.; Honecker, B.I.; Dougherty, G.W.; Olbrich, H.; Schwartz, O.; et al. Pathogenic variants in CLXN encoding the outer dynein arm docking-associated calcium-binding protein calaxin cause primary ciliary dyskinesia. Genet. Med. 2023, 25, 100798. [Google Scholar] [CrossRef] [PubMed]
- Frommer, A.; Hjeij, R.; Loges, N.T.; Edelbusch, C.; Jahnke, C.; Raidt, J.; Werner, C.; Wallmeier, J.; Große-Onnebrink, J.; Olbrich, H.; et al. Immunofluorescence Analysis and Diagnosis of Primary Ciliary Dyskinesia with Radial Spoke Defects. Am. J. Respir. Cell Mol. Biol. 2015, 53, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Cindrić, S.; Dougherty, G.W.; Olbrich, H.; Hjeij, R.; Loges, N.T.; Amirav, I.; Philipsen, M.C.; Marthin, J.K.; Nielsen, K.G.; Sutharsan, S.; et al. SPEF2- and HYDIN-Mutant cilia lack the central pair-associated protein SPEF2, aiding primray ciliary dyskinesia diagnostics. Am. J. Respir. Cell Mol. Biol. 2020, 62, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Hirst, R.A.; Rutman, A.; Williams, G.; O’Callaghan, C. Ciliated air-liquid cultures as an aid to diagnostic testing of primary ciliary dyskinesia. Chest 2010, 138, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, V.; Hjeij, R.; Vij, S.; Loges, N.T.; Wallmeier, J.; Koerner-Rettberg, C.; Werner, C.; Thamilselvam, S.K.; Boey, A.; Choksi, S.P.; et al. Mutations in CCDC11, which encodes a coiled-coil containing ciliary protein, causes situs inversus due to dysmotility of monocilia in the left-right organizer. Hum. Mutat. 2015, 36, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Sigg, M.A.; Menchen, T.; Lee, C.; Johnson, J.; Jungnickel, M.K.; Choksi, S.P.; Garcia, G.; Busengdal, H.; Dougherty, G.W.; Pennekamp, P.; et al. Evolutionary proteomics uncovers ancient associations of cilia with signaling pathways. Dev. Cell. 2017, 43, 744–762.e11. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, G.W.; Mizuno, K.; Nöthe-Menchen, T.; Ikawa, Y.; Boldt, K.; Ta-Shma, A.; Aprea, I.; Minegishi, K.; Pang, Y.P.; Pennekamp, P.; et al. CFAP45 deficiency causes situs abnormalities and asthenospermia by disrupting an axonemal adenine nucleotide homeostasis module. Nat. Commun. 2020, 11, 5520. [Google Scholar] [CrossRef] [PubMed]
- Ta-Shma, A.; Perles, Z.; Yaacov, B.; Werner, M.; Frumkin, A.; Rein, A.J.; Elpeleg, O. A human laterality disorder associated with a homozygous WDR16 deletion. Eur. J. Hum. Genet. 2015, 23, 1262–1265. [Google Scholar] [CrossRef]
- Leslie, J.S.; Hjeij, R.; Vivante, A.; Bearce, E.A.; Dyer, L.; Wang, J.; Rawlins, L.; Kennedy, J.; Ubeyratna, N.; Fasham, J.; et al. Biallelic DAW1 variants cause a motile ciliopathy characterized by laterality defects and subtle ciliary beating abnormalities. Genet. Med. 2022, 24, 2249–2261. [Google Scholar] [CrossRef]
- Ta-Shma, A.; Hjeij, R.; Perles, Z.; Dougherty, G.W.; Abu Zahira, I.; Letteboer, S.J.F.; Antony, D.; Darwish, A.; Mans, D.A.; Spittler, S.; et al. Homozygous loss-of-function mutations in MNS1 cause laterality defects and likely male infertility. PLoS Genet. 2018, 14, e1007602. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, L.E.; Blackburn, K.; Radde, K.M.; Moyer, M.B.; Schlatzer, D.M.; Moseley, A.; Boucher, R.C. A proteomic analysis of human cilia: Identification of novel components. Mol. Cell Proteom. 2002, 1, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Inagaki, H.; Naruge, T.; Tabata, S.; Tomida, T.; Yamaguchi, A.; Yoshikuni, M.; Nagahama, Y.; Hotta, Y. cDNA cloning and functional characterization of a meiosis-specific protein (MNS1) with apparent nuclear association. Chromosome Res. 1994, 2, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yang, F.; Leu, N.A.; Jeremy Wang, P. MNS1 is essential for spermiogenesis and motile ciliary functions in mice. PLoS Genet. 2012, 8, e1002516. [Google Scholar] [CrossRef] [PubMed]
- Leslie, J.S.; Rawlins, L.E.; Chioza, B.A.; Olubodun, O.R.; Salter, C.G.; Fasham, J.; Jones, H.F.; Cross, H.E.; Lam, S.; Harlalka, G.V.; et al. MNS1 variant associated with situs inversus and male infertility. Eur. J. Hum. Genet. 2020, 28, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, W.L.; Tu, C.F.; Meng, L.L.; Hu, T.-Y.; Du, J.; Lin, G.; Nie, H.-C.; Tan, Y.-Q. A novel homozygous frameshift mutation in MNS1 associated with severe oligoasthenoteratozoospermia in humans. Asian J. Androl. 2021, 23, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Hjeij, R.; Onoufriadis, A.; Watson, C.M.; Slagle, C.E.; Klena, N.T.; Dougherty, G.W.; Kurkowiak, M.; Loges, N.T.; Diggle, C.P.; Morante, N.F.C.; et al. CCDC151 Mutations Cause Primary Ciliary Dyskinesia by Disruption of the Outer Dynein Arm Docking Complex Formation. Am. J. Hum. Genet. 2014, 95, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Horani, A.; Ferkol, T.W. Understanding Primary Ciliary Dyskinesia and Other Ciliopathies. J. Pediatr. 2021, 230, 15–22.e11. [Google Scholar] [CrossRef]
- Leigh, M.W.; Pittman, J.E.; Carson, J.L.; Ferkol, T.W.; Dell, S.D.; Davis, S.D.; Knowles, M.R.; Zariwala, M.A. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet. Med. 2009, 11, 473–487. [Google Scholar] [CrossRef]
- Aprea, I.; Raidt, J.; Höben, I.M.; Loges, N.T.; Nöthe-Menchen, T.; Pennekamp, P.; Omran, H. Defects in the cytoplasmic assembly of axonemal dynein arms cause morphological abnormalities and dysmotility in sperm cells leading to male infertility. PLoS Genet. 2021, 17, e1009306. [Google Scholar] [CrossRef]
- Xu, C.; Yang, B.; Lei, C.; Yang, D.; Ding, S.; Lu, C.; Wang, L.; Guo, T.; Wang, R.; Luo, H. Novel compound heterozygous variants in CCDC40 associated with primary ciliary dyskinesia and multiple morphological abnormalities of the sperm flagella. Pharmgenom. Pers. Med. 2022, 15, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Olbrich, H.; Häffner, K.; Kispert, A.; Völkel, A.; Volz, A.; Sasmaz, G.; Reinhardt, R.; Hennig, S.; Lehrach, H.; Konietzko, N.; et al. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 2002, 30, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Onoufriadis, A.; Paff, T.; Antony, D.; Shoemark, A.; Micha, D.; Kuyt, B.; Schmidts, M.; Petridi, S.; Dankert-Roelse, J.E.; Haarman, E.G.; et al. Splice-site mutations in the axonemal outer dynein arm docking complex gene CCDC114 cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2013, 92, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Stoyanova, M.; Rademacher, G.; Dutcher, S.K.; Brown, A.; Zhang, R. Structure of the decorated ciliary doublet microtubule. Cell 2019, 179, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Gui, M.; Farley, H.; Anujan, P.; Anderson, J.R.; Maxwell, D.W.; Whitchurch, J.B.; Botsch, J.J.; Qiu, T.; Meleppattu, S.; Singh, S.K.; et al. De novo identification of mammalian ciliary motility proteins using cryo-EM. Cell 2021, 184, 5791–5806.e19. [Google Scholar] [CrossRef] [PubMed]
- Ide, T.; Twan, W.K.; Lu, H.; Ikawa, Y.; Lim, L.X.; Henninger, N.; Nishimura, H.; Takaoka, H.; Narasimhan, V.; Yan, X.; et al. CFAP53 regulates mammalian cilia-type motility patterns through differential localization and recruitment of axonemal dynein components. PLoS Genet. 2020, 16, e1009232. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| ID | Sex | Age | Ethnicity | MNS1 Genotype | Cons | Laterality Defect | Congenital Heart Disease | RDS | Rhino-Sinusitis | Chronic Wet Cough | Bronchiectasis |
|---|---|---|---|---|---|---|---|---|---|---|---|
| EX-1II1 | M | 10y | Afghan | p.(Arg242*)/p.(Arg242*) | Yes | Heterotaxy asplenia | Yes | No | No | No | No |
| EX-1II2 | M | Fetus | Afghan | p.(Arg242*)/p.(Arg242*) | Yes | Situs inversus | No | NK | NK | NK | NK |
| OP-4846II1 | F | 7y | Egypt | p.(Arg242*)/p.(Arg242*) | Yes | Situs inversus | No | Yes | Yes; bilateral maxillary and ethmoidal sinusitis | Yes | No |
| OP-4761II1 | M | 2y | Kosovo | p.(Glu362*)/p.(Glu226del) | NK | Situs inversus | No | Yes; transient tachypnoe | Yes | No | NK |
| OP-4242II1 | F | 15y | Kosovo | p.(Glu226del)/p.(Glu226del) | No | Situs inversus | No | No | No | No | NK |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hjeij, R.; Leslie, J.; Rizk, H.; Dworniczak, B.; Olbrich, H.; Raidt, J.; Bode, S.F.N.; Gardham, A.; Stals, K.; Al-Haggar, M.; et al. Biallelic Variants in MNS1 Are Associated with Laterality Defects and Respiratory Involvement. Cells 2024, 13, 1017. https://doi.org/10.3390/cells13121017
Hjeij R, Leslie J, Rizk H, Dworniczak B, Olbrich H, Raidt J, Bode SFN, Gardham A, Stals K, Al-Haggar M, et al. Biallelic Variants in MNS1 Are Associated with Laterality Defects and Respiratory Involvement. Cells. 2024; 13(12):1017. https://doi.org/10.3390/cells13121017
Chicago/Turabian StyleHjeij, Rim, Joseph Leslie, Hoda Rizk, Bernd Dworniczak, Heike Olbrich, Johanna Raidt, Sebastian Felix Nepomuk Bode, Alice Gardham, Karen Stals, Mohammad Al-Haggar, and et al. 2024. "Biallelic Variants in MNS1 Are Associated with Laterality Defects and Respiratory Involvement" Cells 13, no. 12: 1017. https://doi.org/10.3390/cells13121017
APA StyleHjeij, R., Leslie, J., Rizk, H., Dworniczak, B., Olbrich, H., Raidt, J., Bode, S. F. N., Gardham, A., Stals, K., Al-Haggar, M., Osman, E., Crosby, A., Eldesoky, T., Baple, E., & Omran, H. (2024). Biallelic Variants in MNS1 Are Associated with Laterality Defects and Respiratory Involvement. Cells, 13(12), 1017. https://doi.org/10.3390/cells13121017