

Digital Pathology Identifies Associations between Tissue Inflammatory Biomarkers and Multiple Sclerosis Outcomes

 , , , and
, , , and
Abstract

1. Introduction
2. Methods
2.1. Tissue Resource and Accompanying Clinical and Demographic Data
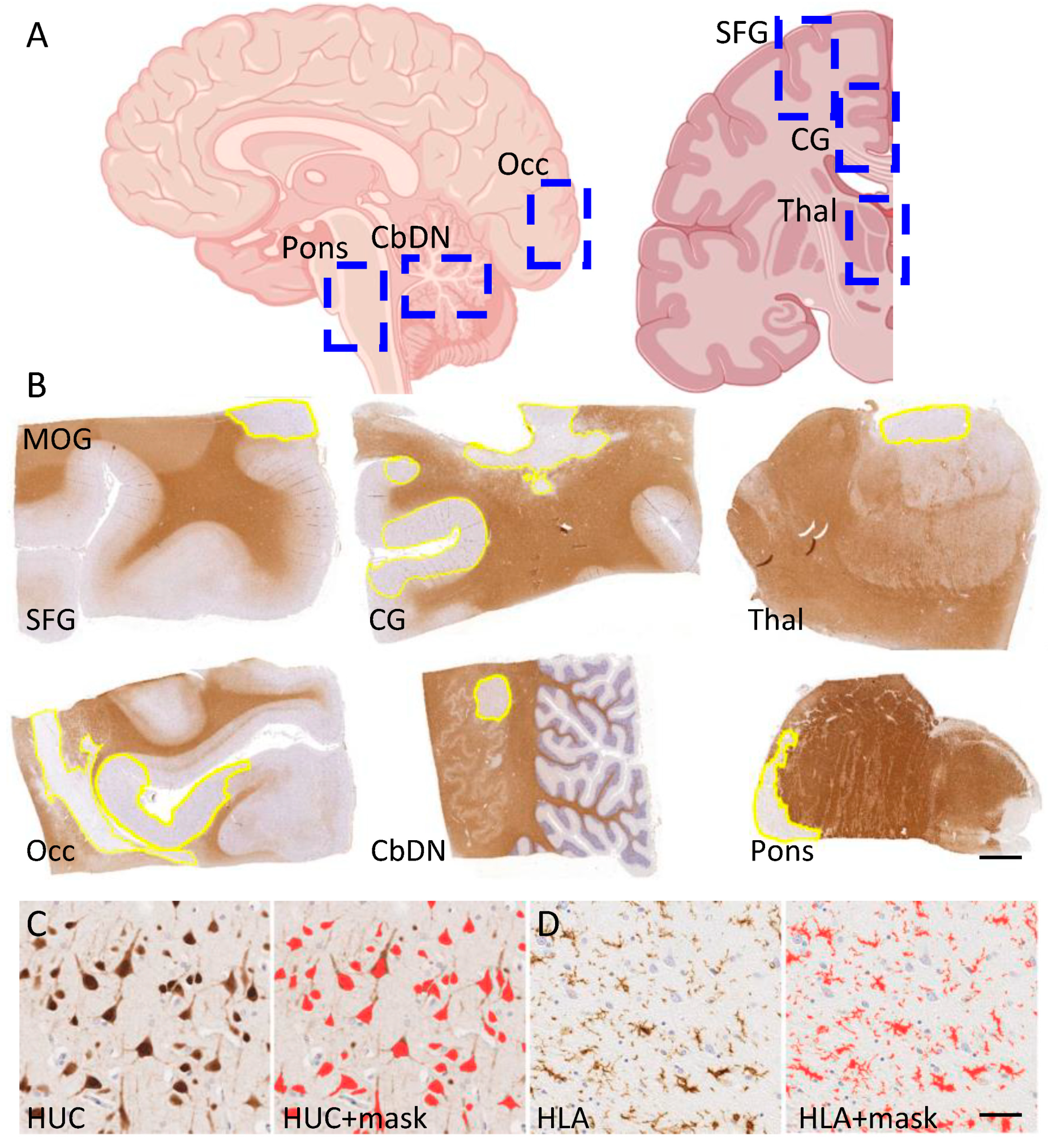
2.2. Tissue Staining
2.3. Lesion Classification
2.4. Digital Pathology and the Quantification of Demyelination, Inflammation and Neuron Density
2.5. Data Handling and Statistical Analysis
3. Results
3.1. Selecting a Representative Cohort of Progressive MS
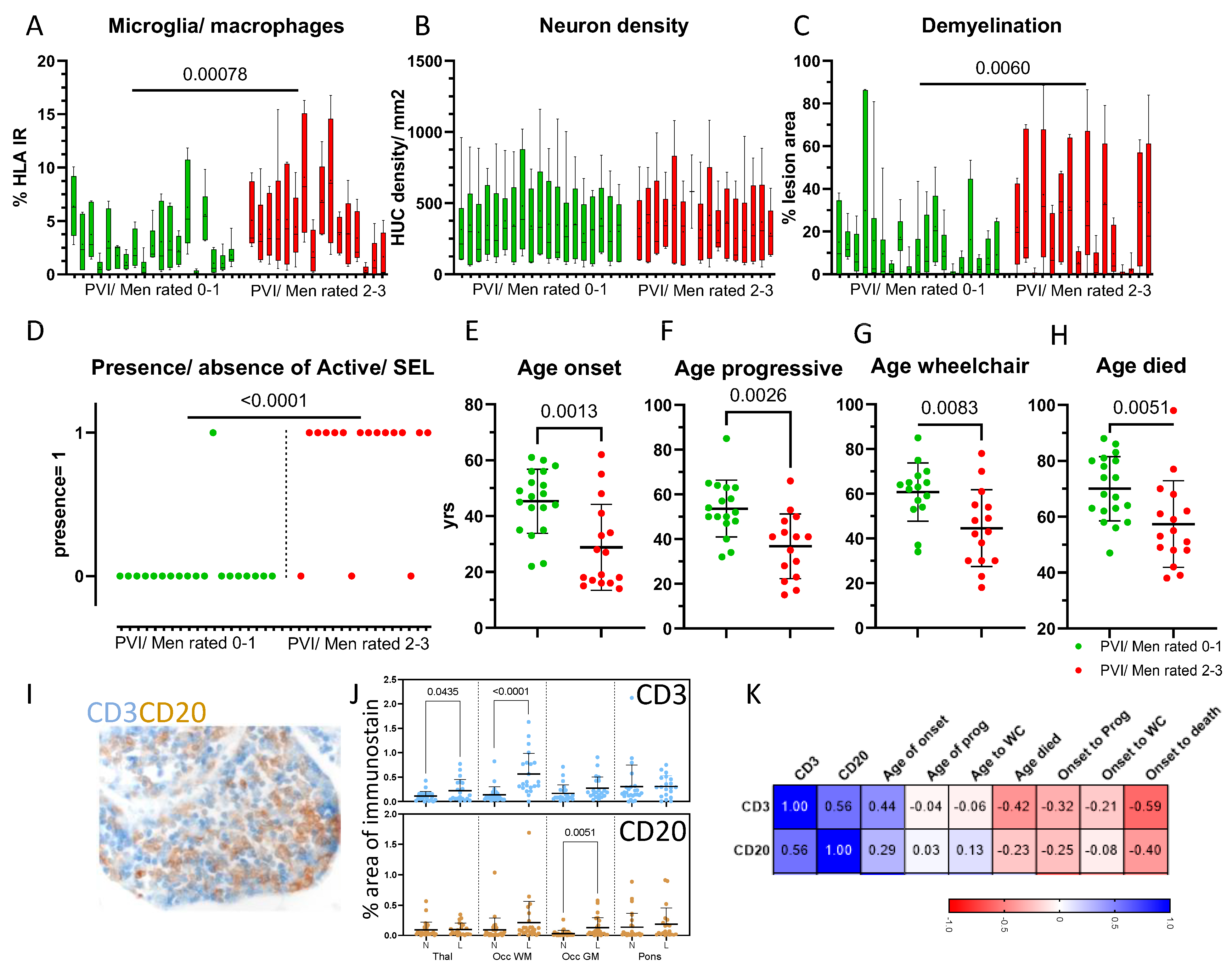
3.2. Progressive MS Is Characterised by a Highly Variable Extent of Inflammation, Demyelination and Neuron Density
3.3. The Extent of Activated Microglia/Macrophages Was Elevated across All Sampled Blocks
3.4. The Association between Microglial/Macrophage Activation and Other Pathological Variables Was Weak
3.5. Phenotyping Cases on the Basis of Compartmentalised Inflammation Reveals Groups That Differ by Disease Outcomes
3.6. Meningeal Inflammation
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lucchinetti, C.; Bruck, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of Multiple Sclerosis Lesions: Implications for the Pathogenesis of Demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Lassmann, H. Pathogenic Mechanisms Associated with Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2019, 9, 3116. [Google Scholar] [CrossRef] [PubMed]
- Knowles, S.; Middleton, R.; Cooze, B.; Farkas, I.; Leung, Y.; Allen, K.; Winslade, M.; Owen, D.R.; Magliozzi, R.; Reynolds, R.; et al. Comparing the Pathology, Clinical and Demographic Characteristics of Younger and Older-onset Multiple Sclerosis. Ann. Neurol. 2023, 95, 471–486. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, E.F.; Zanghì, P.A.; Chisari, C.G.; Lo Fermo, S.; Zappia, M. Late-Onset and Young-Onset Relapsing-Remitting Multiple Sclerosis: Evidence from a Retrospective Long-Term Follow-up Study. Eur. J. Neurol. 2018, 25, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Scalfari, A.; Lederer, C.; Daumer, M.; Nicholas, R.; Ebers, G.C.; Muraro, P.A. The relationship of age with the clinical phenotype in multiple sclerosis. Mult. Scler. 2016, 13, 1750–1758. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sorensen, P.S.; Thompson, A.J.; Wolinsky, I.S.; Balcer, L.J.; Banwell, B.; Barkhoff, F.; et al. Defining the Clinical Course of Multiple Sclerosis: The 2013 Revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Confavreux, C.; Vukusic, S. Natural History of Multiple Sclerosis: A Unifying Concept. Brain 2006, 129, 606–616. [Google Scholar] [CrossRef]
- Pitt, D.; Lo, C.H.; Gauthier, S.A.; Hickman, R.A.; Longbrake, E.; Airas, L.M.; Mao-Draayer, Y.; Riley, C.; DeJager, P.L.; Wesely, S.; et al. Toward Precision Phenotyping of Multiple Sclerosis. Neurol. Neuroimmunol. NeuroInflamm. 2022, 9, e200025. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Moccia, M.; Coetzee, T.; Cohen, J.A.; Correale, J.; Graves, J.A.; Marrie, R.A.; Montalban, X.; Wee Yong, V.; Thompson, A.J.; et al. Multiple Sclerosis Progression: Time for a New Mechanism-Driven Framework. Lancet Neurol. 2023, 1, 78–88. [Google Scholar] [CrossRef]
- Luchetti, S.; Fransen, N.L.; Van Eden, C.G.; Ramaglia, V.; Mason, M.; Huitinga, I. Progressive Multiple Sclerosis Patients Show Substantial Lesion Activity That Correlates with Clinical Disease Severity and Sex: A Retrospective Autopsy Cohort Analysis. Acta Neuropathol. 2018, 135, 511–528. [Google Scholar] [CrossRef]
- Kutzelnigg, A.; Lucchinetti, C.F.; Stadelmann, C.; Brück, W.; Rauschka, H.; Bergmann, M.; Schmidbauer, M.; Parisi, J.E.; Lassmann, H. Cortical Demyelination and Diffuse White Matter Injury in Multiple Sclerosis. Brain 2005, 128, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.; Roncaroli, F.; Nicholas, R.; Radotra, B.; Gveric, G.; Howell, O.W. The Neuropathological Basis of Clinical Progression in Multiple Sclerosis. Acta Neuropathol. 2011, 122, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Reali, C.; Magliozzi, R.; Roncaroli, F.; Nicholas, R.; Howell, O.W.; Reynolds, R. B Cell Rich Meningeal Inflammation Associates with Increased Spinal Cord Pathology in Multiple Sclerosis. Brain Pathol. 2020, 30, 779–793. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Howell, O.W.; Calabrese, M.; Reynolds, R. Meningeal Inflammation as a Driver of Cortical Grey Matter Pathology and Clinical Progression in Multiple Sclerosis. Nat. Rev. Neurol. 2023, 8, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.A.; Kappos, K.; Giovannoni, G.; Plavina, T.; Koulinska, I.; Edwards, M.R.; Kieseier, B.; de Moor, C.; Sotirchos, E.S.; Fisher, E.; et al. Measuring Treatment Response to Advance Precision Medicine for Multiple Sclerosis. Ann. Clin. Transl. Neurol. 2021, 8, 2166–2173. [Google Scholar] [CrossRef] [PubMed]
- Buscarinu, M.; Roberta, R.; Morena, E.; Romano, C.; Bellucci, G.; Marrone, A.; Bigi, R.; Salvetti, M.; Ristori, G. Late-Onset MS: Disease Course and Safety-Efficacy of DMTS. Front. Neurol. 2022, 13, 829331. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, G.; Lang, S.; Wolff, R.; Duffy, S.; Hyde, R.; Kinter, E.; Wakeford, C.; Sormani, M.P.; Kleijnen, J. A Systematic Review and Mixed Treatment Comparison of Pharmaceutical Interventions for Multiple Sclerosis. Neurol. Ther. 2022, 9, 359–374. [Google Scholar] [CrossRef] [PubMed]
- Hartung, H.-P.; Cree, B.A.C.; Barnett, M.; Meuth, S.G.; Bar-Or, A.; Steinman, L. Bioavailable Central Nervous System Disease-Modifying Therapies for Multiple Sclerosis. Front. Immunol. 2023, 14, 1290666. [Google Scholar] [CrossRef]
- Scalco, R.; Hamsafar, Y.; White, C.L.; Schneider, J.A.; Reichard, R.R.; Prokop, S.; Perrin, R.J.; Nelson, P.T.; Mooney, S.; Lieberman, A.P.; et al. The Status of Digital Pathology and Associated Infrastructure within Alzheimer’s Disease Centres. J. Neuropathol. Exp. Neurol. 2023, 82, 202–211. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.; et al. QuPath: Open-Source Software for Digital Pathology Image Analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef]
- Humphries, M.P.; Maxwell, P.; Salto-Tellez, M. QuPath: The Global Impact of an Open-Source Digital Pathology System. Comput. Struct. Biotechnol. J. 2021, 19, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Bennett, A.; Buchman, A.S.; Boyle, P.A.; Barnes, L.L.; Wilson, R.S.; Schneider, J.A. Religious Orders Study and Rush Memory and Aging Project. J. Alzheimer’s Dis. 2018, 64, S161–S189. [Google Scholar] [CrossRef] [PubMed]
- Cooze, B.J.; Dickerson, M.; Loganathan, R.; Watkins, L.M.; Grounds, E.; Pearson, B.R.; Bevan, R.J.; Morgan, B.P.; Magliozzi, R.; Reynolds, R.; et al. The Association between Neurodegeneration and Local Complement Activation in the Thalamus to Progressive Multiple Sclerosis Outcome. Brain Pathol. 2022, 32, e13054. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bosch, A.M.R.; Hümmert, S.; Steyer, A.; Ruhwedel, T.; Jörg Hamann, J.; Smolders, J.; Nave, K.-A.; Stadelmann, C.; Kole, M.H.P.; Mobius, W.; et al. Ultrastructural Axon–Myelin Unit Alterations in Multiple Sclerosis Correlate with Inflammation. Ann. Neurol. 2023, 93, 856–870. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; Watkins, L.M.; Hawkins, K.; Santiago, G.; Demetriou, C.; Naughton, M.; Dittmer, M.; Rees, M.I.; Fitzgerald, D.; Morgan, B.P.; et al. Complement Activation and Increased Anaphylatoxin Receptor Expression Are Associated with Cortical Grey Matter Lesions and the Compartmentalised Inflammatory Response of Multiple Sclerosis. Front. Cell. Neurosci. 2023, 17, 1094106. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Brück, W.; Lassmann, H. An Updated Histological Classification System for Multiple Sclerosis Lesions. Acta Neuropathol. 2017, 133, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Zeis, T.; Howell, O.W.; Reynolds, R.; Schaeren-Wiemers, N. Molecular Pathology of Multiple Sclerosis Lesions Reveals a Heterogeneous Expression Pattern of Genes Involved in Oligodendrogliogenesis. Exp. Neurol. 2018, 305, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Moccia, M.; Haider, L.; Eshaghi, A.; van de Pavert, S.H.P.; Morra, B.V.; Patel, A.; Wheeler-Kingshott, C.A.M.; Barkhof, F.; Ciccarelli, O.B. Cells in the CNS at Postmortem Are Associated with Worse Outcome and Cell Types in Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2021, 10, e1108. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Singh, S.; Metz, I.; Amor, S.; Van Der Valk, P.; Stadelmann, C.; Brück, W. Microglial Nodules in Early Multiple Sclerosis White Matter Are Associated with Degenerating Axons. Acta Neuropathol. 2013, 125, 595–608. [Google Scholar] [CrossRef]
- Singhal, T.; Weiner, H.; Bakshi, R. TSPO-PET Imaging to Assess Cerebral Microglial Activation in Multiple Sclerosis. Semin. Neurol. 2017, 37, 546–557. [Google Scholar] [CrossRef]
- Sucksdorff, M.; Matilainen, M.; Tuisku, J.; Polvinen, E.; Vuorimaa, A.; Rokka, J.; Nylund, M.; Rissanen, E.; Airas, L. Brain TSPO-PET Predicts Later Disease Progression Independent of Relapses in Multiple Sclerosis. Brain 2020, 143, 3318–3330. [Google Scholar] [CrossRef]
- Nutma, E.; Gebro, E.; Marzin, M.C.; Van Der Valk, P.; Matthews, P.M.; Owen, D.R.; Amor, S. Activated Microglia Do Not Increase 18 kDa Translocator Protein (TSPO) Expression in the Multiple Sclerosis Brain. Glia 2021, 69, 2447–2458. [Google Scholar] [CrossRef]
- Ontaneda, D.; Raza, P.C.; Mahajan, K.R.; Arnold, D.L.; Dwyer, M.G.; Gauthier, S.A.; Greve, D.N.; Harrison, D.M.; Henry, R.G.; Li, D.K.B.; et al. Deep Grey Matter Injury in Multiple Sclerosis: A NAIMS Consensus Statement. Brain 2021, 144, 1974–1984. [Google Scholar] [CrossRef]
- Misin, O.; Matilainen, M.; Nylund, M.; Honkonen, E.; Rissanen, E.; Sucksdorff, M.; Airas, L. Innate Immune Cell-Related Pathology in the Thalamus Signals a Risk for Disability Progression in Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1182. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nicholas, R.; Magliozzi, R.; Marastoni, D.; Howell, O.W.; Roncaroli, F.; Muraro, P.; Reynolds, R.; Friede, T. High Levels of Perivascular Inflammation and Active Demyelinating Lesions at Time of Death Associated with Rapidly Progressive Multiple Sclerosis Disease Course: A Retrospective Postmortem Cohort Study. Ann. Neurol. 2024, 95, 706–719. [Google Scholar] [CrossRef]
- Amato, M.P.; Fonderico, M.; Portaccio, E.; Pastò, L.; Razzolini, L.; Prestipino, E.; Bellinvia, A.; Tudisco, L.; Fratangelo, R.; Comi, G.; et al. Disease-Modifying Drugs Can Reduce Disability Progression in Relapsing Multiple Sclerosis. Brain 2020, 143, 3013–3024. [Google Scholar] [CrossRef]
- Fisniku, L.K.; Brex, P.A.; Altmann, D.R.; Miszkiel, K.A.; Benton, C.E.; Lanyon, R.; Thompson, A.J.; Miller, D.H. Disability and T2 MRI Lesions: A 20-Year Follow-up of Patients with Relapse Onset of Multiple Sclerosis. Brain 2008, 131, 808–817. [Google Scholar] [CrossRef]
- Barkhof, F. The Clinico-Radiological Paradox in Multiple Sclerosis Revisited. Curr. Opin. Neurol. 2002, 15, 239–245. [Google Scholar] [CrossRef]
- University of California; San Francisco MS-EPIC Team; Cree, B.A.; Hollenbach, J.A.; Bove, R.; Kirkish, G.; Sacco, S.; Caverzasi, E.; Bischof, A.; Gundel, T.; et al. Silent Progression in Disease Activity–Free Relapsing Multiple Sclerosis. Ann. Neurol. 2019, 85, 653–666. [Google Scholar] [CrossRef]
- Calabrese, M.; Poretto, V.; Favaretto, A.; Alessio, S.; Bernardi, V.; Romualdi, C.; Rinaldi, F.; Perini, P.; Gallo, P. Cortical Lesion Load Associates with Progression of Disability in Multiple Sclerosis. Brain 2012, 135, 2952–2961. [Google Scholar] [CrossRef] [PubMed]
- Absinta, M.A.; Vuolo, L.; Rao, A.; Nair, N.; Sati, P.; Cortese, I.C.M.; Ohayon, J.; Wu, T.; Meani, A.; Filippi, M.; et al. Gadolinium-Based MRI Characterization of Leptomeningeal Inflammation in Multiple Sclerosis. Neurology 2015, 85, 18–28. [Google Scholar] [CrossRef]
- Ransohoff, R.M. Multiple Sclerosis: Role of Meningeal Lymphoid Aggregates in Progression Independent of Relapse Activity. Trends Immunol. 2023, 44, 266–275. [Google Scholar] [CrossRef]
- Haider, L.; Zrzavy, T.; Hametner, S.; Höftberger, R.; Bagnato, F.; Grabner, G.; Trattnig, S.; Pfeifenbring, S.; Brück, W.; Lassmann, H. The Topography of Demyelination and Neurodegeneration in the Multiple Sclerosis Brain. Brain 2016, 139, 807–815. [Google Scholar] [CrossRef]
- Griffiths, L.; Reynolds, R.; Evans, R.; Bevan, R.J.; Rees, M.I.; Gveric, D.; Neal, J.W.; Howell, O.W. Substantial subpial cortical demyelination in progressive multiple sclerosis: Have we underestimated the extent of cortical pathology. Neuroimmunol. NeuroInflamm. 2020, 7, 51–67. [Google Scholar] [CrossRef]
- Siller, N.; Kuhle, J.; Muthuraman, M.; Barro, C.; Uphaus, T.; Groppa, S.; Kappos, L.; Bittner, S. Serum neurofilament light chain is a biomarker of acute and chronic neuronal damage in early Multiple sclerosis. Mult. Scle. R. 2019, 5, 678–686. [Google Scholar] [CrossRef]
- Schneider, R.; Bellenberg, B.; Gisevius, B.; Hirschberg, S.; Sankowsk, I.R.; Prinz, M.; Gold, R.; Lukas, C.; Haghikia, A. Chitinase-3-like1 and neurofilament light chain in CSF and CNS atrophy in MS. Neurol. Neuroimmunol. 2020, 10, e906. [Google Scholar] [CrossRef]
- Bose, G.; Healy, B.C.; Saxena, S.; Saleh, F.; Paul, A.; Barro, C.; Hrishikesh, A.L.; Polgar-Turcsanyi, M.; Anderson, M.; Glanz, B.I.; et al. Early Neurofilament Light and Glial Fibrillary Acidic Protein Levels Improve Predictive Models of Multiple Sclerosis Outcomes. Mult. Scler. Relat. Disord. 2023, 74, 104695. [Google Scholar] [CrossRef]
- Fox, R.J.; Cree, B.; de Seze, J.; Gold, R.; Hartung, H.-P.; Jeffery, D.; Kappos, L.; Montalban, X.; Weinstock-Guttman, B.; Singh, C.M.; et al. Temporal Relationship between serum Neurofilament light chain and Radiological disease activity in patients with multiple sclerosis. Neurology 2024, 14, e209357. [Google Scholar] [CrossRef]
- Talat, F.; Abelatty, S.; Ragaie, C.; Dahshan, A. Chitinase-3-like 1 protein in CSF: A novel biomarker for progression in patients with multiple sclerosis. Neurol. Sci. 2023, 44, 3243–3252. [Google Scholar] [CrossRef]
- Cross, A.H.; Gelfand, J.M.; Thebault, S.; Bennett, J.L.; Von Büdingen, C.; Cameron, B.; Carruthers, R.; Edwards, K.; Fallis, R.; Gerstein, R.; et al. Emerging Cerebrospinal Fluid Biomarkers of Disease Activity and Progression in Multiple Sclerosis. JAMA Neurol. 2024, 81, 373. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Fransen, N.L.; Touil, H.; Michailidou, I.; Huitinga, I.; Gommerman, J.L.; Amit Bar-Or, A.; Ramaglia, V. Accumulation of Meningeal Lymphocytes Correlates with White Matter Lesion Activity in Progressive Multiple Sclerosis. JCI Insight 2022, 7, e151683. [Google Scholar] [CrossRef]
- Komori, M.; Blake, A.; Greenwood, M.; Lin, Y.C.; Kosa, P.; Ghazali, D.; Winokur, P.; Natrajan, M.S.; Wuest, S.C.; Romm, E.; et al. Cerebrospinal Fluid Markers Reveal Intrathecal Inflammation in Progressive Multiple Sclerosis. Ann. Neurol. 2015, 78, 3–20. [Google Scholar] [CrossRef]
- Magliozzi, R.; Howell, O.W.; Nicholas, R.; Cruciani, C.; Castellaro, M.; Romualdi, C.; Rossi, S.; Pitteri, M.; Benedetti, M.D.; Gajofatto, A.; et al. Inflammatory Intrathecal Profiles and Cortical Damage in Multiple Sclerosis. Ann. Neurol. 2018, 83, 739–755. [Google Scholar] [CrossRef]
- Kosa, P.; Barbour, C.; Varosanec, M.; Wichman, A.; Sandford, M.; Greenwood, M.; Bielekova, B. Molecular Models of Multiple Sclerosis Severity Identify Heterogeneity of Pathogenic Mechanisms. Nat. Commun. 2022, 1, 7670. [Google Scholar] [CrossRef]
- Coerver, E.; Janssens, S.; Ahmed, A.; Wessels, M.; van Kempen, Z.; Jasperse, B.; Barkhof, F.; Koch, M.; Mostert, J.; Utidehagg, B.; et al. Association between Age and Inflammatory Disease Activity on Magnetic Resonance Imaging in Relapse Onset Multiple Sclerosis during Long-term Follow-up. Eur. J. Neurol. 2023, 30, 2385–2392. [Google Scholar] [CrossRef]
- Lucchinetti, C.F.; Popescu, B.F.; Bunyan, R.F.; Moll, N.M.; Roemer, S.F.; Lassmann, H.; Brück, W.; Parisi, J.E.; Scheithauer, B.W.; Giannini, C.; et al. Inflammatory cortical demyelination in early multiple sclerosis. N. Engl. J. Med. 2011, 8, 2188–2197. [Google Scholar] [CrossRef]
- Lauerer, M.; McGinnis, J.; Bussas, M.; El Husseini, M.; Pongratz, V.; Engl, C.; Wuschek, A.; Berthele, A.; Riederer, I.; Kirschke, J.S.; et al. Prognostic Value of Spinal Cord Lesion Measures in Early Relapsing-Remitting Multiple Sclerosis. J. Neurol. Neurosurg. Psychiatry 2024, 95, 37–43. [Google Scholar] [CrossRef]
- Patrikios, P.C.; Stadelmann, A.; Kutzelnigg, H.; Rauschka, M.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Bruck, W.; Lucchinetti, C.; Lassmann, H. Remyelination Is Extensive in a Subset of Multiple Sclerosis Patients. Brain 2006, 129, 3165–3172. [Google Scholar] [CrossRef]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of ‘Homeostatic’ Microglia and Patterns of Their Activation in Active Multiple Sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef]
- Möck, E.E.; Honkonen, E.; Airas, L. Synaptic Loss in Multiple Sclerosis: A Systematic Review of Human Post-Mortem Studies. Front. Neurol. 2021, 12, 782599. [Google Scholar] [CrossRef]
- Kapasi, A.; Poirier, J.; Hedayat, A.; Scherlek, A.; Mondal, S.; Wu, T.; Gibbons, J.; Barnes, L.L.; Bennett, D.A.; Leurgans, S.E.; et al. High-Throughput Digital Quantification of Alzheimer Disease Pathology and Associated Infrastructure in Large Autopsy Studies. J. Neuropathol. Exp. Neurol. 2023, 82, 976–986. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n | Sex (Female) | Onset | 2yr-RR | Age Prog | Age WC | Age Died | Disease Duration |
|---|---|---|---|---|---|---|---|
| 35 | 28 | 41 (14–62) | 1 (0–7) | 48 (15–85) | 54.5 (18–85) | 63 (38–98) | 27 (3–49) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cooze, B.; Neal, J.; Vineed, A.; Oliveira, J.C.; Griffiths, L.; Allen, K.H.; Hawkins, K.; Yadanar, H.; Gerhards, K.; Farkas, I.; et al. Digital Pathology Identifies Associations between Tissue Inflammatory Biomarkers and Multiple Sclerosis Outcomes. Cells 2024, 13, 1020. https://doi.org/10.3390/cells13121020
Cooze B, Neal J, Vineed A, Oliveira JC, Griffiths L, Allen KH, Hawkins K, Yadanar H, Gerhards K, Farkas I, et al. Digital Pathology Identifies Associations between Tissue Inflammatory Biomarkers and Multiple Sclerosis Outcomes. Cells. 2024; 13(12):1020. https://doi.org/10.3390/cells13121020
Chicago/Turabian StyleCooze, Benjamin, James Neal, Alka Vineed, J. C. Oliveira, Lauren Griffiths, K. H. Allen, Kristen Hawkins, Htoo Yadanar, Krisjanis Gerhards, Ildiko Farkas, and et al. 2024. "Digital Pathology Identifies Associations between Tissue Inflammatory Biomarkers and Multiple Sclerosis Outcomes" Cells 13, no. 12: 1020. https://doi.org/10.3390/cells13121020
APA StyleCooze, B., Neal, J., Vineed, A., Oliveira, J. C., Griffiths, L., Allen, K. H., Hawkins, K., Yadanar, H., Gerhards, K., Farkas, I., Reynolds, R., & Howell, O. (2024). Digital Pathology Identifies Associations between Tissue Inflammatory Biomarkers and Multiple Sclerosis Outcomes. Cells, 13(12), 1020. https://doi.org/10.3390/cells13121020