
CCT and Cullin1 Regulate the TORC1 Pathway to Promote Dendritic Arborization in Health and Disease
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Drosophila Husbandry and Stocks
2.2. Immunohistochemical Analysis
2.3. Live Confocal Imaging, Neural Reconstructions, and Morphometric Analyses
2.4. Live Multichannel Neural Reconstructions
2.5. mHTT Aggregate Inclusion Body Analysis
2.6. Experimental Design and Statistical Analyses
3. Results
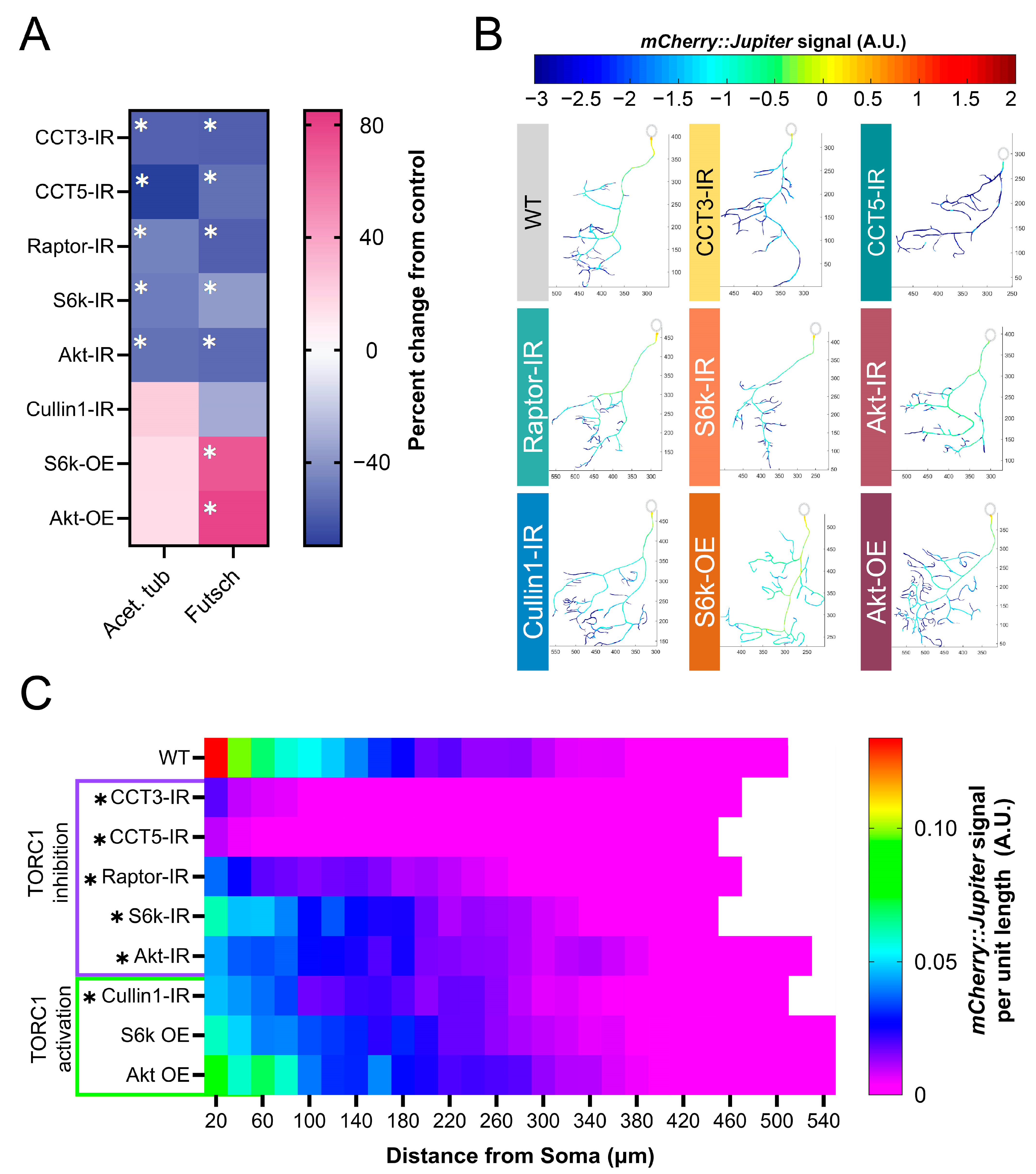
3.1. CCT LOF and Disruption of TORC1 Pathway Genes Results in Dendritic Hypotrophy
3.2. TORC1 Hyperactivation Results in Dendritic Hypertrophy
3.3. CCT Regulates Raptor Levels In Vivo
3.4. Cullin1 Regulates Dendritic Arborization through TORC1
3.5. TORC1 Pathway Disruption Results in Loss of Stable Microtubules
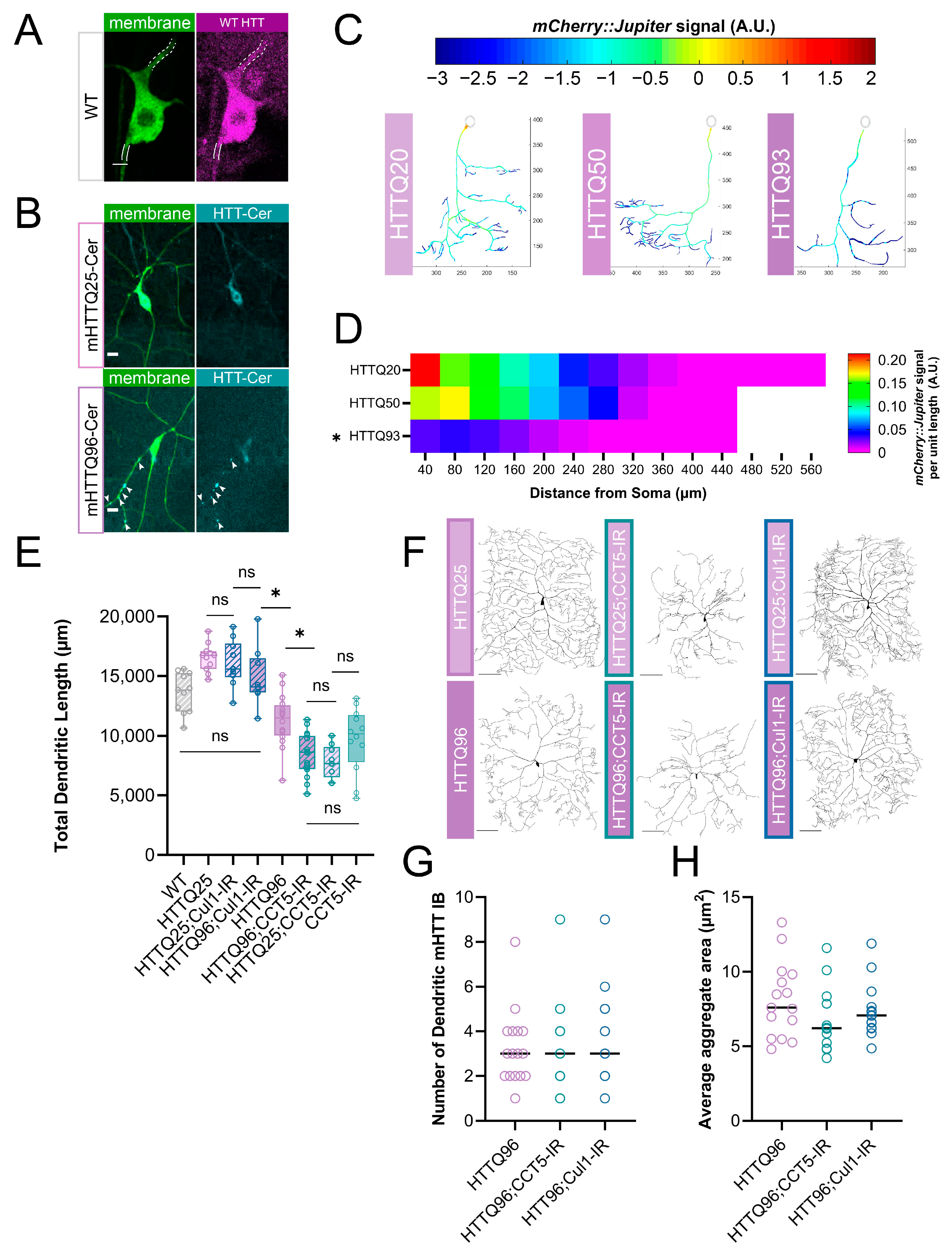
3.6. Mutant Huntingtin Expression Leads to Repeat-Length-Dependent Reduction in Branch Complexity and Underlying Microtubule Loss
3.7. Cullin1 LOF Rescues mHTT-Mediated Gross Morphological Phenotype, While CCT LOF Shows No Additive Effect
4. Discussion
4.1. A TORC1 Cytosolic Network Regulates Dendritic Development and the Underlying MT Cytoskeleton
4.2. TORC1 Cytosolic Network and mHTT Interact in the Regulation of Dendritic Arbors
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dewey, E.H. The No-Breakfast Plan and the Fasting-Cure, 4th ed.; The Health Culture Co.: New York, NY, USA, 1900. [Google Scholar]
- Miles, W.R.; Root, H.F. Psychologic Tests Applied to Diabetic Patients. Arch. Intern. Med. 1922, 30, 767–777. [Google Scholar] [CrossRef]
- Moheet, A.; Mangia, S.; Seaquist, E. Impact of diabetes on cognitive function and brain structure. Ann. N. Y. Acad. Sci. 2015, 1353, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Raizada, M.K. Localization of insulin-like immunoreactivity in the neurons from primary cultures of rat brain. Exp. Cell Res. 1983, 143, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Weyhenmeyer, J.A.; Fellows, R.E. Presence of immunoreactive insulin in neurons cultured from fetal rat brain. Cell. Mol. Neurobiol. 1983, 3, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Eisenberg, J.; Yang, J. Human Blood—Brain Barrier Insulin Receptor. J. Neurochem. 1985, 44, 1771–1778. [Google Scholar] [CrossRef]
- Craft, S. The Role of Metabolic Disorders in Alzheimer Disease and Vascular Dementia: Two Roads Converged. Arch. Neurol. 2009, 66, 300–305. [Google Scholar] [CrossRef]
- Schulingkamp, R.J.; Pagano, T.C.; Hung, D.; Raffa, R.B. Insulin receptors and insulin action in the brain: Review and clinical implications. Neurosci. Biobehav. Rev. 2000, 24, 855–872. [Google Scholar] [CrossRef]
- Wu, J.; Zhou, S.-L.; Pi, L.-H.; Shi, X.-J.; Ma, L.-R.; Chen, Z.; Qu, M.-L.; Li, X.; Nie, S.-D.; Liao, D.-F.; et al. High glucose induces formation of tau hyperphosphorylation via Cav-1-mTOR pathway: A potential molecular mechanism for diabetes-induced cognitive dysfunction. Oncotarget 2017, 8, 40843–40856. [Google Scholar] [CrossRef] [PubMed]
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef]
- Burillo, J.; Marqués, P.; Jiménez, B.; González-Blanco, C.; Benito, M.; Guillén, C. Insulin Resistance and Diabetes Mellitus in Alzheimer’s Disease. Cells 2021, 10, 1236. [Google Scholar] [CrossRef]
- Das, R.; Bhattacharjee, S.; Patel, A.A.; Harris, J.M.; Bhattacharya, S.; Letcher, J.M.; Clark, S.G.; Nanda, S.; Iyer, E.P.R.; Ascoli, G.A.; et al. Dendritic cytoskeletal architecture is modulated by combinatorial transcriptional regulation in Drosophila melanogaster. Genetics 2017, 207, 1401–1421. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Ding, Z.Y.; Cheng, Y.J.; Chien, C.T.; Huang, M.L. An Efficient Screen for Cell-Intrinsic Factors Identifies the Chaperonin CCT and Multiple Conserved Mechanisms as Mediating Dendrite Morphogenesis. Front. Cell. Neurosci. 2020, 14, 311. [Google Scholar] [CrossRef] [PubMed]
- Liou, A.K.; Willison, K.R. Elucidation of the subunit orientation in CCT (chaperonin containing TCP1) from the subunit composition of CCT micro-complexes. EMBO J. 1997, 16, 4311–4316. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Han, W.; Liu, C.; Zang, Y.; Li, J.; Wang, F.; Wang, Y.; Cong, Y. An ensemble of cryo-EM structures of TRiC reveal its conformational landscape and subunit specificity. Proc. Natl. Acad. Sci. USA 2019, 116, 19513–19522. [Google Scholar] [CrossRef] [PubMed]
- Thulasiraman, V.; Yang, C.-F.; Frydman, J. In Vivo Newly Translated Polypeptides Are Sequestered in a Protected Folding Environment. EMBO J. 1999, 18, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Grantham, J.; Brackley, K.I.; Willison, K.R. Substantial CCT activity is required for cell cycle progression and cytoskeletal organization in mammalian cells. Exp. Cell Res. 2006, 312, 2309–2324. [Google Scholar] [CrossRef] [PubMed]
- Brackley, K.I.; Grantham, J. Activities of the chaperonin containing TCP-1 (CCT): Implications for cell cycle progression and cytoskeletal organisation. Cell Stress Chaperones 2009, 14, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Willison, K.R. The substrate specificity of eukaryotic cytosolic chaperonin CCT. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170192. [Google Scholar] [CrossRef] [PubMed]
- Gestaut, D.; Zhao, Y.; Park, J.; Ma, B.; Leitner, A.; Collier, M.; Pintilie, G.; Roh, S.-H.; Chiu, W.; Frydman, J. Structural visualization of the tubulin folding pathway directed by human chaperonin TRiC/CCT. Cell 2022, 185, 4770–4787. [Google Scholar] [CrossRef]
- Cuéllar, J.; Ludlam, W.G.; Tensmeyer, N.C.; Aoba, T.; Dhavale, M.; Santiago, C.; Bueno-Carrasco, M.T.; Mann, M.J.; Plimpton, R.L.; Makaju, A.; et al. Structural and functional analysis of the role of the chaperonin CCT in mTOR complex assembly. Nat. Commun. 2019, 10, 2865. [Google Scholar] [CrossRef]
- Kim, A.-R.; Choi, K.-W. TRiC/CCT chaperonins are essential for organ growth by interacting with insulin/TOR signaling in Drosophila. Oncogene 2019, 38, 4739–4754. [Google Scholar] [CrossRef]
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002, 16, 1472–1487. [Google Scholar] [CrossRef]
- Kumar, V.; Zhang, M.-X.; Swank, M.W.; Kunz, J.; Wu, G.-Y. Regulation of Dendritic Morphogenesis by Ras–PI3K–Akt–mTOR and Ras–MAPK Signaling Pathways. J. Neurosci. 2005, 25, 11288–11299. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, J.; Spangler, S.; Seeburg, D.P.; Hoogenraad, C.C.; Sheng, M. Control of dendritic arborization by the phosphoinositide-3’-kinase-Akt-mammalian target of rapamycin pathway. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 11300–11312. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Chung, J. Discrete functions of rictor and raptor in cell growth regulation in Drosophila. Biochem. Biophys. Res. Commun. 2007, 357, 1154–1159. [Google Scholar] [CrossRef] [PubMed]
- Urbanska, M.; Gozdz, A.; Swiech, L.J.; Jaworski, J. Mammalian Target of Rapamycin Complex 1 (mTORC1) and 2 (mTORC2) Control the Dendritic Arbor Morphology of Hippocampal Neurons. J. Biol. Chem. 2012, 287, 30240. [Google Scholar] [CrossRef] [PubMed]
- Thomanetz, V.; Angliker, N.; Cloëtta, D.; Lustenberger, R.M.; Schweighauser, M.; Oliveri, F.; Suzuki, N.; Rüegg, M.A. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J. Cell Biol. 2013, 201, 293. [Google Scholar] [CrossRef] [PubMed]
- Shimono, K.; Fujishima, K.; Nomura, T.; Ohashi, M.; Usui, T.; Kengaku, M.; Toyoda, A.; Uemura, T. An evolutionarily conserved protein CHORD regulates scaling of dendritic arbors with body size. Sci. Rep. 2014, 4, 4415. [Google Scholar] [CrossRef] [PubMed]
- Skalecka, A.; Liszewska, E.; Bilinski, R.; Gkogkas, C.; Khoutorsky, A.; Malik, A.R.; Sonenberg, N.; Jaworski, J. mTOR Kinase is Needed for the Development and Stabilization of Dendritic Arbors in Newly Born Olfactory Bulb Neurons. Dev. Neurobiol. 2016, 76, 1308–1327. [Google Scholar] [CrossRef]
- Kosillo, P.; Doig, N.M.; Ahmed, K.M.; Agopyan-Miu, A.H.C.W.; Wong, C.D.; Conyers, L.; Threlfell, S.; Magill, P.J.; Bateup, H.S. Tsc1-mTORC1 signaling controls striatal dopamine release and cognitive flexibility. Nat. Commun. 2019, 10, 5426. [Google Scholar] [CrossRef] [PubMed]
- Kosillo, P.; Ahmed, K.M.; Aisenberg, E.E.; Karalis, V.; Roberts, B.M.; Cragg, S.J.; Bateup, H.S. Dopamine neuron morphology and output are differentially controlled by mTORC1 and mTORC2. eLife 2022, 11, e75398. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.J.L.; Li, S.; Lim, E.K.H.; Wang, Y.; Wang, C.; Zhang, H.; Kirilly, D.; Wu, C.; Liou, Y.C.; Wang, H.; et al. A Cullin1-Based SCF E3 Ubiquitin Ligase Targets the InR/PI3K/TOR Pathway to Regulate Neuronal Pruning. PLoS Biol. 2013, 11, e1001657. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.M.; Joachimiak, L.A.; Tan, Z.; Tomlinson, A.; Housman, D.E.; Glabe, C.G.; Potkinj, S.G.; Frydman, J.; Thompson, L.M. Exogenous delivery of chaperonin subunit fragment ApiCCT1 modulates mutant Huntingtin cellular phenotypes. Proc. Natl. Acad. Sci. USA 2013, 110, 3077–3082. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Tecedor, L.; Chen, Y.H.; Mas Monteys, A.; Sowada, M.J.; Thompson, L.M.; Davidson, B.L. Reinstating aberrant mTORC1 activity in Huntington’s disease mice improves disease phenotypes HHS Public Access. Neuron 2015, 85, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Noormohammadi, A.; Khodakarami, A.; Gutierrez-Garcia, R.; Lee, H.J.; Koyuncu, S.; König, T.; Schindler, C.; Saez, I.; Fatima, A.; Dieterich, C.; et al. Somatic increase of CCT8 mimics proteostasis of human pluripotent stem cells and extends C. elegans lifespan. Nat. Commun. 2016, 7, 13649. [Google Scholar] [CrossRef]
- Shen, K.; Calamini, B.; Fauerbach, J.A.; Ma, B.; Shahmoradian, S.H.; Serrano Lachapel, I.L.; Chiu, W.; Lo, D.C.; Frydman, J. Control of the structural landscape and neuronal proteotoxicity of mutant Huntingtin by domains flanking the polyQ tract. eLife 2016, 5, e18065. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Garcia Oroz, L.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’kane, C.J.; et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet. 2006, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- King, M.A.; Hands, S.; Hafiz, F.; Mizushima, N.; Tolkovsky, A.M.; Wyttenbach, A. Rapamycin Inhibits Polyglutamine Aggregation Independently of Autophagy by Reducing Protein Synthesis. Mol. Pharmacol. 2008, 73, 1052–1063. [Google Scholar] [CrossRef]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P.; et al. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Grueber, W.B.; Jan, L.Y.; Jan, Y.N. Tiling of the Drosophila epidermis by multidendritic sensory neurons. Development 2002, 129, 2867–2878. [Google Scholar] [CrossRef] [PubMed]
- Sulkowski, M.J.; Iyer, S.C.; Kurosawa, M.S.; Iyer, E.P.R.; Cox, D.N. Turtle Functions Downstream of Cut in Differentially Regulating Class Specific Dendrite Morphogenesis in Drosophila. PLoS ONE 2011, 6, e22611. [Google Scholar] [CrossRef]
- Iyer, E.P.R.; Iyer, S.C.; Sullivan, L.; Wang, D.; Meduri, R.; Graybeal, L.L.; Cox, D.N. Functional Genomic Analyses of Two Morphologically Distinct Classes of Drosophila Sensory Neurons: Post-Mitotic Roles of Transcription Factors in Dendritic Patterning. PLoS ONE 2013, 8, e72434. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.C.; Iyer, E.P.R.; Meduri, R.; Rubaharan, M.; Kuntimaddi, A.; Karamsetty, M.; Cox, D.N. Cut, via CrebA, transcriptionally regulates the COPII secretory pathway to direct dendrite development in Drosophila. J. Cell Sci. 2013, 126, 4732–4745. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.G.; Graybeal, L.L.; Bhattacharjee, S.; Thomas, C.; Bhattacharya, S.; Cox, D.N. Basal autophagy is required for promoting dendritic terminal branching in drosophila sensory neurons. PLoS ONE 2018, 13, e0206743. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, T.A.; Blackman, A.V.; Oyrer, J.; Jayabal, S.; Chung, A.J.; Watt, A.J.; Sjöström, P.J.; van Meyel, D.J. Neuronal morphometry directly from bitmap images. Nat. Methods 2014, 11, 982–984. [Google Scholar] [CrossRef] [PubMed]
- Arshadi, C.; Günther, U.; Eddison, M.; Harrington, K.I.S.; Ferreira, T.A. SNT: A unifying toolbox for quantification of neuronal anatomy. Nat. Methods 2021, 18, 374–377. [Google Scholar] [CrossRef]
- Nanda, S.; Bhattacharjee, S.; Cox, D.N.; Ascoli, G.A. An imaging analysis protocol to trace, quantify, and model multi-signal neuron morphology. STAR Protoc. 2021, 2, 100567. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Lottes, E.N.; Nanda, S.; Golshir, A.; Patel, A.A.; Ascoli, G.A.; Cox, D.N. PP2A phosphatase regulates cell-type specific cytoskeletal organization to drive dendrite diversity. Front. Mol. Neurosci. 2022, 15, 926567. [Google Scholar] [CrossRef]
- Feng, L.; Zhao, T.; Kim, J. neuTube 1.0: A New Design for Efficient Neuron Reconstruction Software Based on the SWC Format. eNeuro 2015, 2, ENEURO.0049-14.2014. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Zhong, F.L.; Venteicher, A.S.; Meng, Z.; Veenstra, T.D.; Frydman, J.; Artandi, S.E. Proteostatic control of telomerase function through TRiC-mediated folding of TCAB1. Cell 2014, 159, 1389–1403. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Fang, F.; Florio, J.B.; Rockenstein, E.; Masliah, E.; Mobley, W.C.; Rissman, R.A.; Wu, C. T-complex protein 1-ring complex enhances retrograde axonal transport by modulating tau phosphorylation. Traffic 2018, 19, 840–853. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Ruiz, O.; Zapata, A.; Shan, L.; Zhang, Y.; Tomac, A.C.; Malik, N.; de la Cruz, F.; Bäckman, C.M. Selective Deletion of PTEN in Dopamine Neurons Leads to Trophic Effects and Adaptation of Striatal Medium Spiny Projecting Neurons. PLoS ONE 2009, 4, e7027. [Google Scholar] [CrossRef] [PubMed]
- Kanaoka, Y.; Onodera, K.; Watanabe, K.; Hayashi, Y.; Usui, T.; Uemura, T.; Hattori, Y. Inter-organ Wingless/Ror/Akt signaling regulates nutrient-dependent hyperarborization of somatosensory neurons. eLife 2023, 12, e79461. [Google Scholar] [CrossRef] [PubMed]
- Tam, S.; Geller, R.; Spiess, C.; Frydman, J. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat. Cell Biol. 2006, 8, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Llorca, O. Eukaryotic chaperonin CCT stabilizes actin and tubulin folding intermediates in open quasi-native conformations. EMBO J. 2000, 19, 5971–5979. [Google Scholar] [CrossRef]
- Swiech, L.; Blazejczyk, M.; Urbanska, M.; Pietruszka, P.; Dortland, B.R.; Malik, A.R.; Wulf, P.S.; Hoogenraad, C.C.; Jaworski, J. Cellular/Molecular CLIP-170 and IQGAP1 Cooperatively Regulate Dendrite Morphology. J. Neurosci. 2011, 31, 4555–4568. [Google Scholar] [CrossRef]
- Hummel, T.; Krukkert, K.; Roos, J.; Davis, G.; Klämbt, C. Drosophila Futsch/22C10 Is a MAP1B-like Protein Required for Dendritic and Axonal Development. Neuron 2000, 26, 357–370. [Google Scholar] [CrossRef]
- Pawson, C.; Eaton, B.A.; Davis, G.W. Formin-Dependent Synaptic Growth: Evidence That Dlar Signals via Diaphanous to Modulate Synaptic Actin and Dynamic Pioneer Microtubules. J. Neurosci. 2008, 28, 11111–11123. [Google Scholar] [CrossRef]
- Weiner, A.T.; Lanz, M.C.; Goetschius, D.J.; Hancock, W.O.; Rolls, M.M. Kinesin-2 and Apc function at dendrite branch points to resolve microtubule collisions. Cytoskeleton 2016, 73, 35–44. [Google Scholar] [CrossRef]
- Eshun-Wilson, L.; Zhang, R.; Portran, D.; Nachury, M.V.; Toso, D.B.; Löhr, T.; Vendruscolo, M.; Bonomi, M.; Fraser, J.S.; Nogales, E. Effects of α-tubulin acetylation on microtubule structure and stability. Proc. Natl. Acad. Sci. USA 2019, 116, 10366–10371. [Google Scholar] [CrossRef] [PubMed]
- McKinstry, S.U.; Karadeniz, Y.B.; Worthington, A.K.; Hayrapetyan, V.Y.; Ilcim Ozlu, M.; Serafin-Molina, K.; Christopher Risher, W.; Ustunkaya, T.; Dragatsis, I.; Zeitlin, S.; et al. Huntingtin is required for normal excitatory synapse development in cortical and striatal circuits. J. Neurosci. 2014, 34, 9455–9472. [Google Scholar] [CrossRef] [PubMed]
- Barnat, M.; Le Friec, J.; Benstaali, C.; Humbert, S. Huntingtin-Mediated Multipolar-Bipolar Transition of Newborn Cortical Neurons Is Critical for Their Postnatal Neuronal Morphology. Neuron 2017, 93, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Krench, M.; Littleton, J.T. Modeling huntington disease in Drosophila: Insights into axonal transport defects and modifiers of toxicity. Fly 2013, 7, 229–236. [Google Scholar] [CrossRef]
- Bertrand, M.; Decoville, M.; Meudal, H.; Birman, S.; Landon, C. Metabolomic nuclear magnetic resonance studies at presymptomatic and symptomatic stages of huntington’s disease on a drosophila model. J. Proteome Res. 2020, 19, 4034–4045. [Google Scholar] [CrossRef]
- Vernizzi, L.; Paiardi, C.; Licata, G.; Vitali, T.; Santarelli, S.; Raneli, M.; Manelli, V.; Rizzetto, M.; Gioria, M.; Pasini, M.E.; et al. Glutamine Synthetase 1 Increases Autophagy Lysosomal Degradation of Mutant Huntingtin Aggregates in Neurons, Ameliorating Motility in a Drosophila Model for Huntington’s Disease. Cells 2020, 9, 196. [Google Scholar] [CrossRef] [PubMed]
- Khyati; Malik, I.; Agrawal, N.; Kumar, V. Melatonin and curcumin reestablish disturbed circadian gene expressions and restore locomotion ability and eclosion behavior in Drosophila model of Huntington’s disease. Chronobiol. Int. 2021, 38, 61–78. [Google Scholar] [CrossRef]
- Subhan, I.; Siddique, Y.H. Modulation of Huntington’s disease in Drosophila. CNS Neurol. Disord.-Drug Targets 2021, 20, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Heldebrant, M.P.; Perez-Terzic, C.M.; Bortolon, R.; Kovtun, I.V.; Badger, J.D.; Terzic, A.; Estévez, A.; Windebank, A.J.; Dyer, R.B.; et al. Microtubule destabilization and nuclear entry are sequential steps leading to toxicity in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 12171–12176. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Galaz-Montoya, J.G.; Schmid, M.F.; Cong, Y.; Ma, B.; Spiess, C.; Frydman, J.; Ludtke, S.J.; Chiu, W. TRiC’s tricks inhibit huntingtin aggregation. eLife 2013, 2013, 710. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, X.Q.; Han, E.; Hu, Y.; Paik, P.; Ding, Z.; Overman, J.; Lau, A.L.; Shahmoradian, S.H.; Chiu, W.; et al. TRiC subunits enhance BDNF axonal transport and rescue striatal atrophy in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2016, 113, E5655–E5664. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, A.; Kubota, H.; Pack, C.-G.; Matsumoto, G.; Hirayama, S.; Takahashi, Y.; Kimura, H.; Kinjo, M.; Morimoto, R.I.; Nagata, K. Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat. Cell Biol. 2006, 8, 1163–1169. [Google Scholar] [CrossRef]
- Sergeeva, O.A.; Tran, M.T.; Haase-Pettingell, C.; King, J.A. Biochemical characterization of mutants in chaperonin proteins CCT4 and CCT5 associated with hereditary sensory neuropathy. J. Biol. Chem. 2014, 289, 27470–27480. [Google Scholar] [CrossRef]
- Sarkar, S.; Ravikumar, B.; Floto, R.A.; Rubinsztein, D.C. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009, 16, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Pryor, W.M.; Biagioli, M.; Shahani, N.; Swarnkar, S.; Huang, W.C.; Page, D.T.; MacDonald, M.E.; Subramaniam, S. Huntingtin promotes mTORC1 signaling in the pathogenesis of Huntington’s disease. Sci. Signal. 2014, 7, ra103. [Google Scholar] [CrossRef] [PubMed]
- Vinayagram, A.; Kulkarni, M.M.; Sopko, R.; Sun, X.; Hu, Y.; Nand, A.; Villalta, C.; Moghimi, A.; Yang, X.; Mohr, S.E.; et al. An Integrative Analysis of the InR/PI3K/Akt Network Identifies the Dynamic Response to Insulin Signaling. Cell Rep. 2016, 16, 3062–3074. [Google Scholar] [CrossRef] [PubMed]
- Parrish, J.Z.; Xu, P.; Kim, C.C.; Jan, L.Y.; Jan, Y.N. The microRNA bantam functions in epithelial cells to regulate scaling growth of dendrite arbors in Drosophila sensory neurons. Neuron 2009, 63, 788–802. [Google Scholar] [CrossRef]
- Hać, A.; Pierzynowska, K.; Herman-Antosiewicz, A. S6k1 is indispensible for stress-induced microtubule acetylation and autophagic flux. Cells 2021, 10, 929. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.; Lee, H.K. Mammalian/mechanistic target of rapamycin (mTOR) complexes in neurodegeneration. Mol. Neurodegener. 2021, 16, 44. [Google Scholar] [CrossRef] [PubMed]
- Gerez, J.A.; Prymaczok, N.C.; Rockenstein, E.; Herrmann, U.S.; Schwarz, P.; Adame, A.; Enchev, R.I.; Courtheoux, T.; Boersema, P.J.; Riek, R.; et al. A cullin-RING ubiquitin ligase targets exogenous α-synuclein and inhibits Lewy body–like pathology. Sci. Transl. Med. 2019, 11, eaau6722. [Google Scholar] [CrossRef]
- Bhutani, S.; Das, A.; Maheshwari, M.; Lakhotia, S.C.; Jana, N.R. Dysregulation of core components of SCF complex in poly-glutamine disorders. Cell Death Dis. 2012, 3, e428. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.S.; Wong, A.K.Y.; Cheng, T.C.; Koon, A.C.; Chan, H.Y.E. FipoQ/FBXO33, a Cullin-1-based ubiquitin ligase complex component modulates ubiquitination and solubility of polyglutamine disease protein. J. Neurochem. 2019, 149, 781–798. [Google Scholar] [CrossRef]
- Switon, K.; Kotulska, K.; Janusz-Kaminska, A.; Zmorzynska, J.; Jaworski, J. Molecular neurobiology of mTOR. Neuroscience 2017, 341, 112–153. [Google Scholar] [CrossRef] [PubMed]
- Roscic, A.; Baldo, B.; Crochemore, C.; Marcellin, D.; Paganetti, P. Induction of autophagy with catalytic mTOR inhibitors reduces huntingtin aggregates in a neuronal cell model. J. Neurochem. 2011, 119, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.A.; Fishman-Jacob, T.; Youdim, M.B.H. Genetic reduction of the E3 ubiquitin ligase element, SKP1A and environmental manipulation to emulate cardinal features of Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18, S177–S179. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.A.; Fishman-Jacob, T.; Youdim, M.B.H. Targeting SKP1, an ubiquitin E3 ligase component found decreased in sporadic Parkinson’s disease. Neurodegener. Dis. 2012, 10, 220–223. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lottes, E.N.; Ciger, F.; Bhattacharjee, S.; Timmins, E.A.; Tete, B.; Tran, T.; Matta, J.; Patel, A.A.; Cox, D.N. CCT and Cullin1 Regulate the TORC1 Pathway to Promote Dendritic Arborization in Health and Disease. Cells 2024, 13, 1029. https://doi.org/10.3390/cells13121029
Lottes EN, Ciger F, Bhattacharjee S, Timmins EA, Tete B, Tran T, Matta J, Patel AA, Cox DN. CCT and Cullin1 Regulate the TORC1 Pathway to Promote Dendritic Arborization in Health and Disease. Cells. 2024; 13(12):1029. https://doi.org/10.3390/cells13121029
Chicago/Turabian StyleLottes, Erin N., Feyza Ciger, Shatabdi Bhattacharjee, Emily A. Timmins, Benoit Tete, Tommy Tran, Jais Matta, Atit A. Patel, and Daniel N. Cox. 2024. "CCT and Cullin1 Regulate the TORC1 Pathway to Promote Dendritic Arborization in Health and Disease" Cells 13, no. 12: 1029. https://doi.org/10.3390/cells13121029