
Post-GWAS Validation of Target Genes Associated with HbF and HbA2 Levels

 , , , ,
, , , , 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
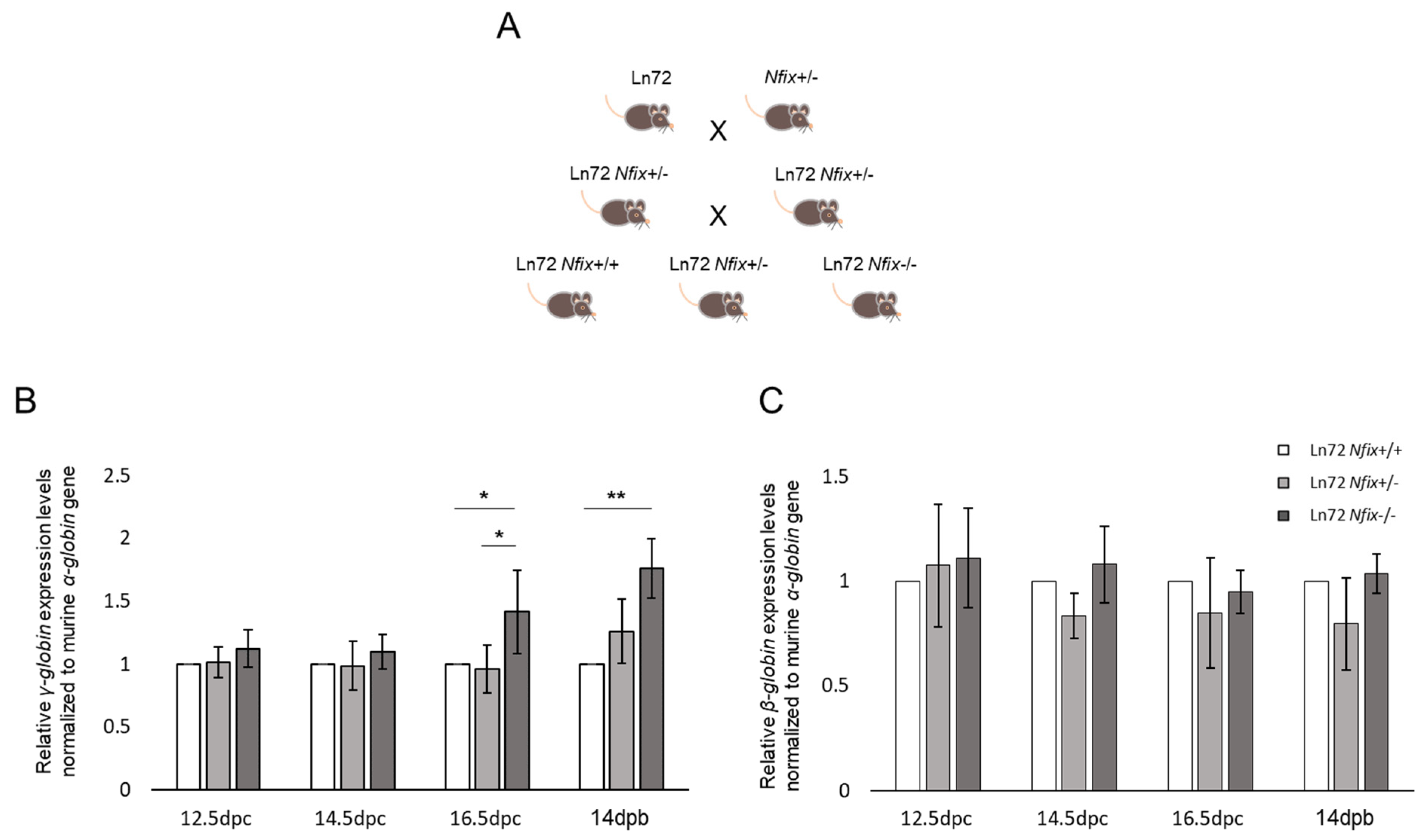
2.1. Mice
2.2. Real-Time
2.3. Hematology
2.4. Flow Cytometry
2.5. Constructs
2.6. Cell Culture and Transient Transfections
2.7. Luciferase Assay
2.8. Statistical Analysis
3. Results
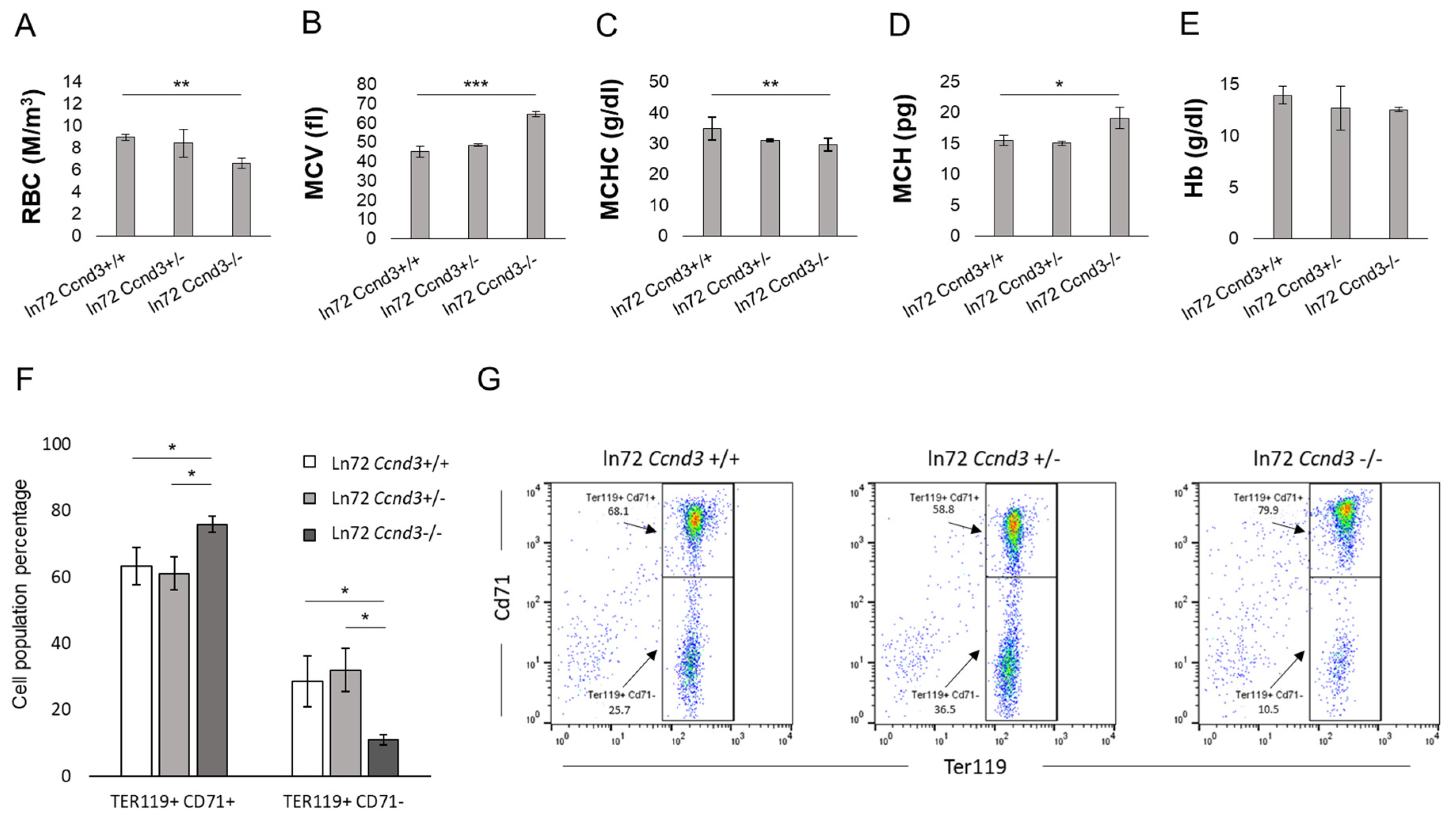
3.1. Ccnd3 Deprivation Increases the Expression of γ- and δ-Globin Genes
3.2. Ccnd3 Deprivation Affects Bone Marrow Erythropoiesis
3.3. Nfix Deprivation Increases the Expression of γ-Globin Gene
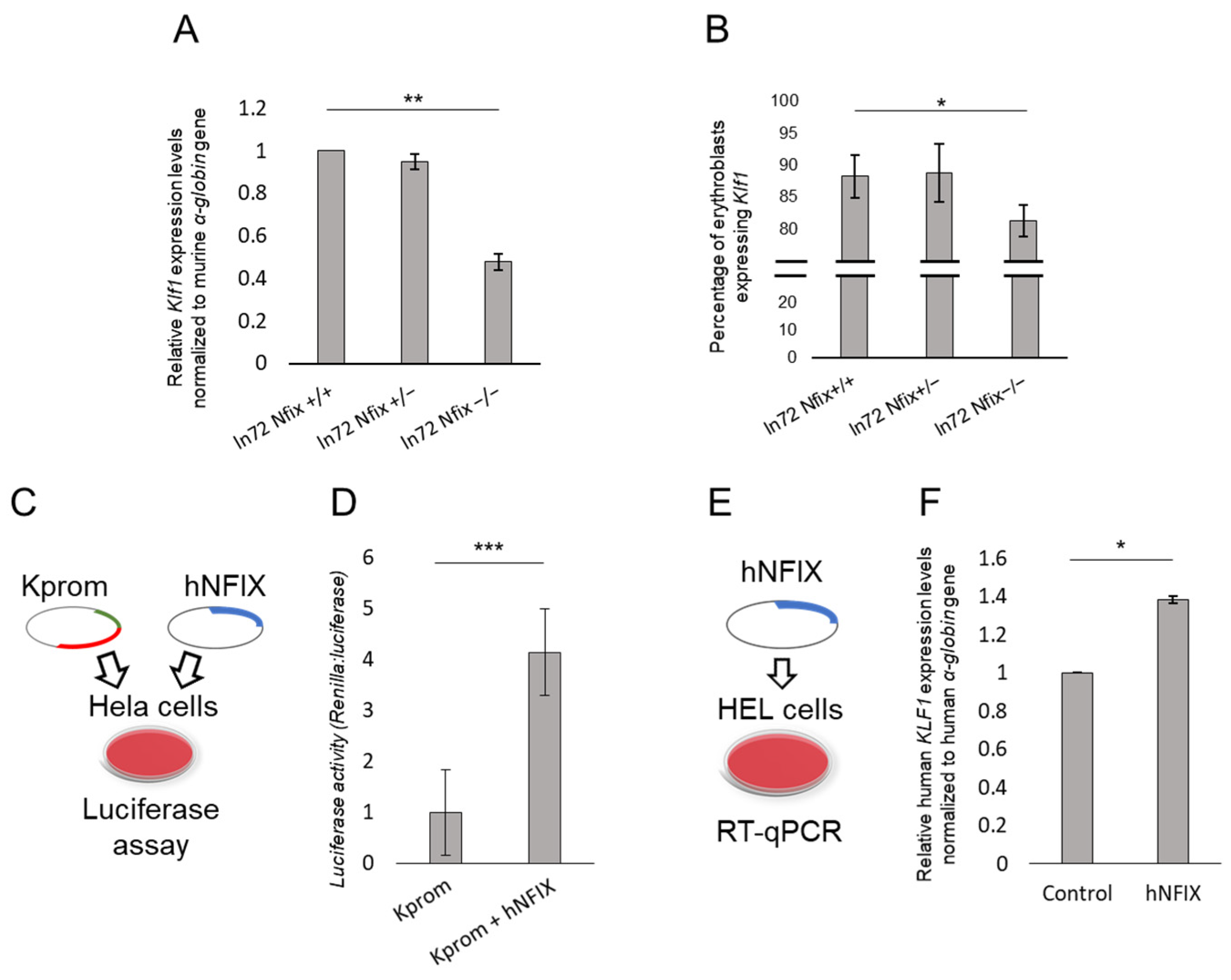
3.4. Nfix Affects Klf1 Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burton, P.R.; Clayton, D.G.; Cardon, L.R.; Craddock, N.; Deloukas, P.; Duncanson, A.; Kwiatkowski, D.P.; McCarthy, M.I.; Ouwehand, W.H.; Samani, N.J.; et al. Genome-Wide Association Study of 14,000 Cases of Seven Common Diseases and 3000 Shared Controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef]
- Visscher, P.M.; Brown, M.A.; McCarthy, M.I.; Yang, J. Five Years of GWAS Discovery. Am. J. Hum. Genet. 2012, 90, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Abdellaoui, A.; Yengo, L.; Verweij, K.J.H.; Visscher, P.M. 15 Years of GWAS Discovery: Realizing the Promise. Am. J. Hum. Genet. 2023, 110, 179–194. [Google Scholar] [CrossRef]
- Gallagher, M.D.; Chen-Plotkin, A.S. The Post-GWAS Era: From Association to Function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and Limitations of Genome-Wide Association Studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Hassell, K.L. Population Estimates of Sickle Cell Disease in the U.S. Am. J. Prev. Med. 2010, 38, S512–S521. [Google Scholar] [CrossRef] [PubMed]
- Orkin, S.H.; Higgs, D.R. Medicine. Sickle Cell Disease at 100 Years. Science 2010, 329, 291–292. [Google Scholar] [CrossRef] [PubMed]
- Piel, F.B.; Hay, S.I.; Gupta, S.; Weatherall, D.J.; Williams, T.N. Global Burden of Sickle Cell Anaemia in Children under Five, 2010–2050: Modelling Based on Demographics, Excess Mortality, and Interventions. PLoS Med. 2013, 10, e1001484. [Google Scholar] [CrossRef]
- Weatherall, D.J. The Inherited Diseases of Hemoglobin Are an Emerging Global Health Burden. Blood 2010, 115, 4331–4336. [Google Scholar] [CrossRef]
- Orkin, S.H. Molecular Medicine: Found in Translation. Med 2021, 2, 122–136. [Google Scholar] [CrossRef]
- Check Hayden, E. Promising Gene Therapies Pose Million-Dollar Conundrum. Nature 2016, 534, 305–306. [Google Scholar] [CrossRef]
- McGann, P.T.; Ware, R.E. Hydroxyurea for Sickle Cell Anemia: What Have We Learned and What Questions Still Remain? Curr. Opin. Hematol. 2011, 18, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Lettre, G.; Sankaran, V.G.; Bezerra, M.A.C.; Araújo, A.S.; Uda, M.; Sanna, S.; Cao, A.; Schlessinger, D.; Costa, F.F.; Hirschhorn, J.N.; et al. DNA Polymorphisms at the BCL11A, HBS1L-MYB, and Beta-Globin Loci Associate with Fetal Hemoglobin Levels and Pain Crises in Sickle Cell Disease. Proc. Natl. Acad. Sci. USA 2008, 105, 11869–11874. [Google Scholar] [CrossRef]
- Sedgewick, A.E.; Timofeev, N.; Sebastiani, P.; So, J.C.C.; Ma, E.S.K.; Chan, L.C.; Fucharoen, G.; Fucharoen, S.; Barbosa, C.G.; Vardarajan, B.N.; et al. BCL11A Is a Major HbF Quantitative Trait Locus in Three Different Populations with Beta-Hemoglobinopathies. Blood Cells Mol. Dis. 2008, 41, 255–258. [Google Scholar] [CrossRef]
- Uda, M.; Galanello, R.; Sanna, S.; Lettre, G.; Sankaran, V.G.; Chen, W.; Usala, G.; Busonero, F.; Maschio, A.; Albai, G.; et al. Genome-Wide Association Study Shows BCL11A Associated with Persistent Fetal Hemoglobin and Amelioration of the Phenotype of Beta-Thalassemia. Proc. Natl. Acad. Sci. USA 2008, 105, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Wang, X.; Maeda, M.; Canver, M.C.; Sher, F.; Funnell, A.P.W.; Fisher, C.; Suciu, M.; Martyn, G.E.; Norton, L.J.; et al. Transcription Factors LRF and BCL11A Independently Repress Expression of Fetal Hemoglobin. Science 2016, 351, 285–289. [Google Scholar] [CrossRef]
- Manchinu, M.F.; Marongiu, M.F.; Poddie, D.; Casu, C.; Latini, V.; Simbula, M.; Galanello, R.; Moi, P.; Cao, A.; Porcu, S.; et al. In Vivo Activation of the Human δ-Globin Gene: The Therapeutic Potential in β-Thalassemic Mice. Haematologica 2014, 99, 76–84. [Google Scholar] [CrossRef]
- Porcu, S.; Simbula, M.; Marongiu, M.F.; Perra, A.; Poddie, D.; Perseu, L.; Kowalik, M.A.; Littera, R.; Barella, S.; Caria, C.A.; et al. Delta-Globin Gene Expression Improves Sickle Cell Disease in a Humanised Mouse Model. Br. J. Haematol. 2021, 193, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Li, H.; Aerbajinai, W.; Kumkhaek, C.; Pirooznia, M.; Saxena, A.; Dagur, P.; Chin, K.; Rodgers, G.P. Kruppel-like Factor 1–GATA1 Fusion Protein Improves the Sickle Cell Disease Phenotype in Mice Both in Vitro and in Vivo. Blood 2022, 140, 2276–2289. [Google Scholar] [CrossRef]
- Menzel, S.; Jiang, J.; Silver, N.; Gallagher, J.; Cunningham, J.; Surdulescu, G.; Lathrop, M.; Farrall, M.; Spector, T.D.; Thein, S.L. The HBS1L-MYB Intergenic Region on Chromosome 6q23.3 Influences Erythrocyte, Platelet, and Monocyte Counts in Humans. Blood 2007, 110, 3624–3626. [Google Scholar] [CrossRef] [PubMed]
- Humphries, R.K.; Ley, T.; Turner, P.; Moulton, A.D.; Nienhuis, A.W. Differences in Human Alpha-, Beta- and Delta-Globin Gene Expression in Monkey Kidney Cells. Cell 1982, 30, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.H.; Adams, J.G. Hemoglobin A2: Origin, Evolution, and Aftermath. Blood 1991, 78, 2165–2177. [Google Scholar]
- Steinberg, M.H.; Rodgers, G.P. HbA2: Biology, Clinical Relevance and a Possible Target for Ameliorating Sickle Cell Disease. Br. J. Haematol. 2015, 170, 781–787. [Google Scholar] [CrossRef]
- Donze, D.; Jeancake, P.H.; Townes, T.M. Activation of Delta-Globin Gene Expression by Erythroid Krupple-like Factor: A Potential Approach for Gene Therapy of Sickle Cell Disease. Blood 1996, 88, 4051–4057. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.C.; Ebb, D.; Hardison, R.C.; Rodgers, G.P. Restoration of the CCAAT Box or Insertion of the CACCC Motif Activates [Corrected] Delta-Globin Gene Expression. Blood 1997, 90, 421–427. [Google Scholar] [CrossRef]
- Zhu, J.; Chin, K.; Aerbajinai, W.; Trainor, C.; Gao, P.; Rodgers, G.P. Recombinant Erythroid Kruppel-like Factor Fused to GATA1 up-Regulates Delta- and Gamma-Globin Expression in Erythroid Cells. Blood 2011, 117, 3045–3052. [Google Scholar] [CrossRef]
- Danjou, F.; Zoledziewska, M.; Sidore, C.; Steri, M.; Busonero, F.; Maschio, A.; Mulas, A.; Perseu, L.; Barella, S.; Porcu, E.; et al. Genome-Wide Association Analyses Based on Whole-Genome Sequencing in Sardinia Provide Insights into Regulation of Hemoglobin Levels. Nat. Genet. 2015, 47, 1264–1271. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. Living with or without Cyclins and Cyclin-Dependent Kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef]
- Sicinska, E.; Aifantis, I.; Le Cam, L.; Swat, W.; Borowski, C.; Yu, Q.; Ferrando, A.A.; Levin, S.D.; Geng, Y.; von Boehmer, H.; et al. Requirement for Cyclin D3 in Lymphocyte Development and T Cell Leukemias. Cancer Cell 2003, 4, 451–461. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell Cycle, CDKs and Cancer: A Changing Paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Ludwig, L.S.; Sicinska, E.; Xu, J.; Bauer, D.E.; Eng, J.C.; Patterson, H.C.; Metcalf, R.A.; Natkunam, Y.; Orkin, S.H.; et al. Cyclin D3 Coordinates the Cell Cycle during Differentiation to Regulate Erythrocyte Size and Number. Genes Dev. 2012, 26, 2075–2087. [Google Scholar] [CrossRef] [PubMed]
- Seiki, T.; Naito, M.; Hishida, A.; Takagi, S.; Matsunaga, T.; Sasakabe, T.; Hattori, Y.; Kawai, S.; Okada, R.; Yin, G.; et al. Association of Genetic Polymorphisms with Erythrocyte Traits: Verification of SNPs Reported in a Previous GWAS in a Japanese Population. Gene 2018, 642, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, R.A.; Zhao, S.; Anderson, M.W.; Lu, Z.S.; Galperin, I.; Marinelli, R.J.; Cherry, A.M.; Lossos, I.S.; Natkunam, Y. Characterization of D-Cyclin Proteins in Hematolymphoid Neoplasms: Lack of Specificity of Cyclin-D2 and D3 Expression in Lymphoma Subtypes. Mod. Pathol. 2010, 23, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Manzano, M.; Patil, A.; Waldrop, A.; Dave, S.S.; Behdad, A.; Gottwein, E. Gene Essentiality Landscape and Druggable Oncogenic Dependencies in Herpesviral Primary Effusion Lymphoma. Nat. Commun. 2018, 9, 3263. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, H.; Chen, J. Mechanisms of the CDK4/6 Inhibitor Palbociclib (PD 0332991) and Its Future Application in Cancer Treatment (Review). Oncol. Rep. 2018, 39, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Gronostajski, R.M. Roles of the NFI/CTF Gene Family in Transcription and Development. Gene 2000, 249, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Driller, K.; Pagenstecher, A.; Uhl, M.; Omran, H.; Berlis, A.; Gründer, A.; Sippel, A.E. Nuclear Factor I X Deficiency Causes Brain Malformation and Severe Skeletal Defects. Mol. Cell Biol. 2007, 27, 3855–3867. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.E.; Piper, M.; Plachez, C.; Yeh, Y.-T.; Baizer, J.S.; Osinski, J.M.; Litwack, E.D.; Richards, L.J.; Gronostajski, R.M. The Transcription Factor Nfix Is Essential for Normal Brain Development. BMC Dev. Biol. 2008, 8, 52. [Google Scholar] [CrossRef]
- Piper, M.; Gronostajski, R.; Messina, G. Nuclear Factor One X in Development and Disease. Trends Cell Biol. 2019, 29, 20–30. [Google Scholar] [CrossRef]
- Zenker, M.; Bunt, J.; Schanze, I.; Schanze, D.; Piper, M.; Priolo, M.; Gerkes, E.H.; Gronostajski, R.M.; Richards, L.J.; Vogt, J.; et al. Variants in Nuclear Factor I Genes Influence Growth and Development. Am. J. Med. Genet. 2019, 181, 611–626. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.E.; Graham, C.B.; Scott, C.R.; Smith, D.W. Syndrome of Accelerated Skeletal Maturation and Relative Failure to Thrive: A Newly Recognized Clinical Growth Disorder. J. Pediatr. 1971, 78, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Malan, V.; Rajan, D.; Thomas, S.; Shaw, A.C.; Louis dit Picard, H.; Layet, V.; Till, M.; van Haeringen, A.; Mortier, G.; Nampoothiri, S.; et al. Distinct Effects of Allelic NFIX Mutations on Nonsense-Mediated mRNA Decay Engender Either a Sotos-like or a Marshall-Smith Syndrome. Am. J. Hum. Genet. 2010, 87, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Strouboulis, J.; Dillon, N.; Grosveld, F. Developmental Regulation of a Complete 70-Kb Human Beta-Globin Locus in Transgenic Mice. Genes Dev. 1992, 6, 1857–1864. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Socolovsky, M.; Murrell, M.; Liu, Y.; Pop, R.; Porpiglia, E.; Levchenko, A. Negative Autoregulation by FAS Mediates Robust Fetal Erythropoiesis. PLoS Biol. 2007, 5, e252. [Google Scholar] [CrossRef]
- Qin, K.; Huang, P.; Feng, R.; Keller, C.A.; Peslak, S.A.; Khandros, E.; Saari, M.S.; Lan, X.; Mayuranathan, T.; Doerfler, P.A.; et al. Dual Function NFI Factors Control Fetal Hemoglobin Silencing in Adult Erythroid Cells. Nat. Genet. 2022, 54, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Caria, C.A.; Faà, V.; Ristaldi, M.S. Krüppel-Like Factor 1: A Pivotal Gene Regulator in Erythropoiesis. Cells 2022, 11, 3069. [Google Scholar] [CrossRef]
- Manchinu, M.F.; Simbula, M.; Caria, C.A.; Musu, E.; Perseu, L.; Porcu, S.; Steri, M.; Poddie, D.; Frau, J.; Cocco, E.; et al. Delta-Globin Gene Expression Is Enhanced in Vivo by Interferon Type I. Front. Med. 2020, 7, 163. [Google Scholar] [CrossRef]
- Hofrichter, J.; Ross, P.D.; Eaton, W.A. Kinetics and Mechanism of Deoxyhemoglobin S Gelation: A New Approach to Understanding Sickle Cell Disease. Proc. Natl. Acad. Sci. USA 1974, 71, 4864–4868. [Google Scholar] [CrossRef]
- Bunn, H.F.; Forget, B.G. Hemoglobin: Molecular, Genetic and Clinical Aspects; Saunders: Philadelphia, PA, USA, 1986; ISBN 978-0-7216-2181-4. [Google Scholar]
- Mozzarelli, A.; Hofrichter, J.; Eaton, W.A. Delay Time of Hemoglobin S Polymerization Prevents Most Cells from Sickling in Vivo. Science 1987, 237, 500–506. [Google Scholar] [CrossRef]
- Eaton, W.A.; Hofrichter, J. Sickle Cell Hemoglobin Polymerization. In Advances in Protein Chemistry; Elsevier: Amsterdam, The Netherlands, 1990; Volume 40, pp. 63–279. ISBN 978-0-12-034240-2. [Google Scholar]
- Poillon, W.N.; Kim, B.C.; Rodgers, G.P.; Noguchi, C.T.; Schechter, A.N. Sparing Effect of Hemoglobin F and Hemoglobin A2 on the Polymerization of Hemoglobin S at Physiologic Ligand Saturations. Proc. Natl. Acad. Sci. USA 1993, 90, 5039–5043. [Google Scholar] [CrossRef]
- Steinberg, M.H.; Chui, D.H.K.; Dover, G.J.; Sebastiani, P.; Alsultan, A. Fetal Hemoglobin in Sickle Cell Anemia: A Glass Half Full? Blood 2014, 123, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Ferrone, F.A. The Delay Time in Sickle Cell Disease after 40 Years: A Paradigm Assessed: The Delay Time in Sickle Cell Disease. Am. J. Hematol. 2015, 90, 438–445. [Google Scholar] [CrossRef]
- Eaton, W.A.; Bunn, H.F. Treating Sickle Cell Disease by Targeting HbS Polymerization. Blood 2017, 129, 2719–2726. [Google Scholar] [CrossRef]
- Estepp, J.H.; Smeltzer, M.P.; Kang, G.; Li, C.; Wang, W.C.; Abrams, C.; Aygun, B.; Ware, R.E.; Nottage, K.; Hankins, J.S. A Clinically Meaningful Fetal Hemoglobin Threshold for Children with Sickle Cell Anemia during Hydroxyurea Therapy. Am. J. Hematol. 2017, 92, 1333–1339. [Google Scholar] [CrossRef] [PubMed]
- Powars, D.R.; Weiss, J.N.; Chan, L.S.; Schroeder, W.A. Is There a Threshold Level of Fetal Hemoglobin That Ameliorates Morbidity in Sickle Cell Anemia? Blood 1984, 63, 921–926. [Google Scholar] [CrossRef]
- Steinberg, M.H. Fetal Hemoglobin in Sickle Hemoglobinopathies: High HbF Genotypes and Phenotypes. J. Clin. Med. 2020, 9, 3782. [Google Scholar] [CrossRef] [PubMed]
- Nagel, R.L.; Bookchin, R.M.; Johnson, J.; Labie, D.; Wajcman, H.; Isaac-Sodeye, W.A.; Honig, G.R.; Schilirò, G.; Crookston, J.H.; Matsutomo, K. Structural Bases of the Inhibitory Effects of Hemoglobin F and Hemoglobin A2 on the Polymerization of Hemoglobin S. Proc. Natl. Acad. Sci. USA 1979, 76, 670–672. [Google Scholar] [CrossRef]
- Adachi, K.; Pang, J.; Reddy, L.R.; Bradley, L.E.; Chen, Q.; Trifillis, P.; Schwartz, E.; Surrey, S. Polymerization of Three Hemoglobin A2 Variants Containing Val6 and Inhibition of Hemoglobin S Polymerization by Hemoglobin A2. J. Biol. Chem. 1996, 271, 24557–24563. [Google Scholar] [CrossRef]
- Thein, S.L.; Menzel, S.; Peng, X.; Best, S.; Jiang, J.; Close, J.; Silver, N.; Gerovasilli, A.; Ping, C.; Yamaguchi, M.; et al. Intergenic Variants of HBS1L-MYB Are Responsible for a Major Quantitative Trait Locus on Chromosome 6q23 Influencing Fetal Hemoglobin Levels in Adults. Proc. Natl. Acad. Sci. USA 2007, 104, 11346–11351. [Google Scholar] [CrossRef] [PubMed]
- Soranzo, N.; Spector, T.D.; Mangino, M.; Kühnel, B.; Rendon, A.; Teumer, A.; Willenborg, C.; Wright, B.; Chen, L.; Li, M.; et al. A Genome-Wide Meta-Analysis Identifies 22 Loci Associated with Eight Hematological Parameters in the HaemGen Consortium. Nat. Genet. 2009, 41, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Menzel, S.; Garner, C.; Rooks, H.; Spector, T.D.; Thein, S.L. HbA2 Levels in Normal Adults Are Influenced by Two Distinct Genetic Mechanisms. Br. J. Haematol. 2013, 160, 101–105. [Google Scholar] [CrossRef] [PubMed]
- McGann, P.T.; Ware, R.E. Hydroxyurea Therapy for Sickle Cell Anemia. Expert Opin. Drug Saf. 2015, 14, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Baliga, B.S.; Pace, B.S.; Chen, H.H.; Shah, A.K.; Yang, Y.M. Mechanism for Fetal Hemoglobin Induction by Hydroxyurea in Sickle Cell Erythroid Progenitors. Am. J. Hematol. 2000, 65, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Wojda, U.; Noel, P.; Miller, J.L. Fetal and Adult Hemoglobin Production during Adult Erythropoiesis: Coordinate Expression Correlates with Cell Proliferation. Blood 2002, 99, 3005–3013. [Google Scholar] [CrossRef] [PubMed]
- Chaand, M.; Fiore, C.; Johnston, B.; D’Ippolito, A.; Moon, D.H.; Carulli, J.P.; Shearstone, J.R. Erythroid Lineage Chromatin Accessibility Maps Facilitate Identification and Validation of NFIX as a Fetal Hemoglobin Repressor. Commun. Biol. 2023, 6, 640. [Google Scholar] [CrossRef]
- Toogood, P.L.; Harvey, P.J.; Repine, J.T.; Sheehan, D.J.; VanderWel, S.N.; Zhou, H.; Keller, P.R.; McNamara, D.J.; Sherry, D.; Zhu, T.; et al. Discovery of a Potent and Selective Inhibitor of Cyclin-Dependent Kinase 4/6. J. Med. Chem. 2005, 48, 2388–2406. [Google Scholar] [CrossRef]
- Padda, I.S.; Mahtani, A.U.; Patel, P.; Parmar, M. Small Interfering RNA (siRNA) Therapy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caria, C.A.; Faà, V.; Porcu, S.; Marongiu, M.F.; Poddie, D.; Perseu, L.; Meloni, A.; Vaccargiu, S.; Ristaldi, M.S. Post-GWAS Validation of Target Genes Associated with HbF and HbA2 Levels. Cells 2024, 13, 1185. https://doi.org/10.3390/cells13141185
Caria CA, Faà V, Porcu S, Marongiu MF, Poddie D, Perseu L, Meloni A, Vaccargiu S, Ristaldi MS. Post-GWAS Validation of Target Genes Associated with HbF and HbA2 Levels. Cells. 2024; 13(14):1185. https://doi.org/10.3390/cells13141185
Chicago/Turabian StyleCaria, Cristian Antonio, Valeria Faà, Susanna Porcu, Maria Franca Marongiu, Daniela Poddie, Lucia Perseu, Alessandra Meloni, Simona Vaccargiu, and Maria Serafina Ristaldi. 2024. "Post-GWAS Validation of Target Genes Associated with HbF and HbA2 Levels" Cells 13, no. 14: 1185. https://doi.org/10.3390/cells13141185
APA StyleCaria, C. A., Faà, V., Porcu, S., Marongiu, M. F., Poddie, D., Perseu, L., Meloni, A., Vaccargiu, S., & Ristaldi, M. S. (2024). Post-GWAS Validation of Target Genes Associated with HbF and HbA2 Levels. Cells, 13(14), 1185. https://doi.org/10.3390/cells13141185