
Advancements in Research on Genetic Kidney Diseases Using Human-Induced Pluripotent Stem Cell-Derived Kidney Organoids

 , , , , and
, , , , and
Abstract
:1. Introduction
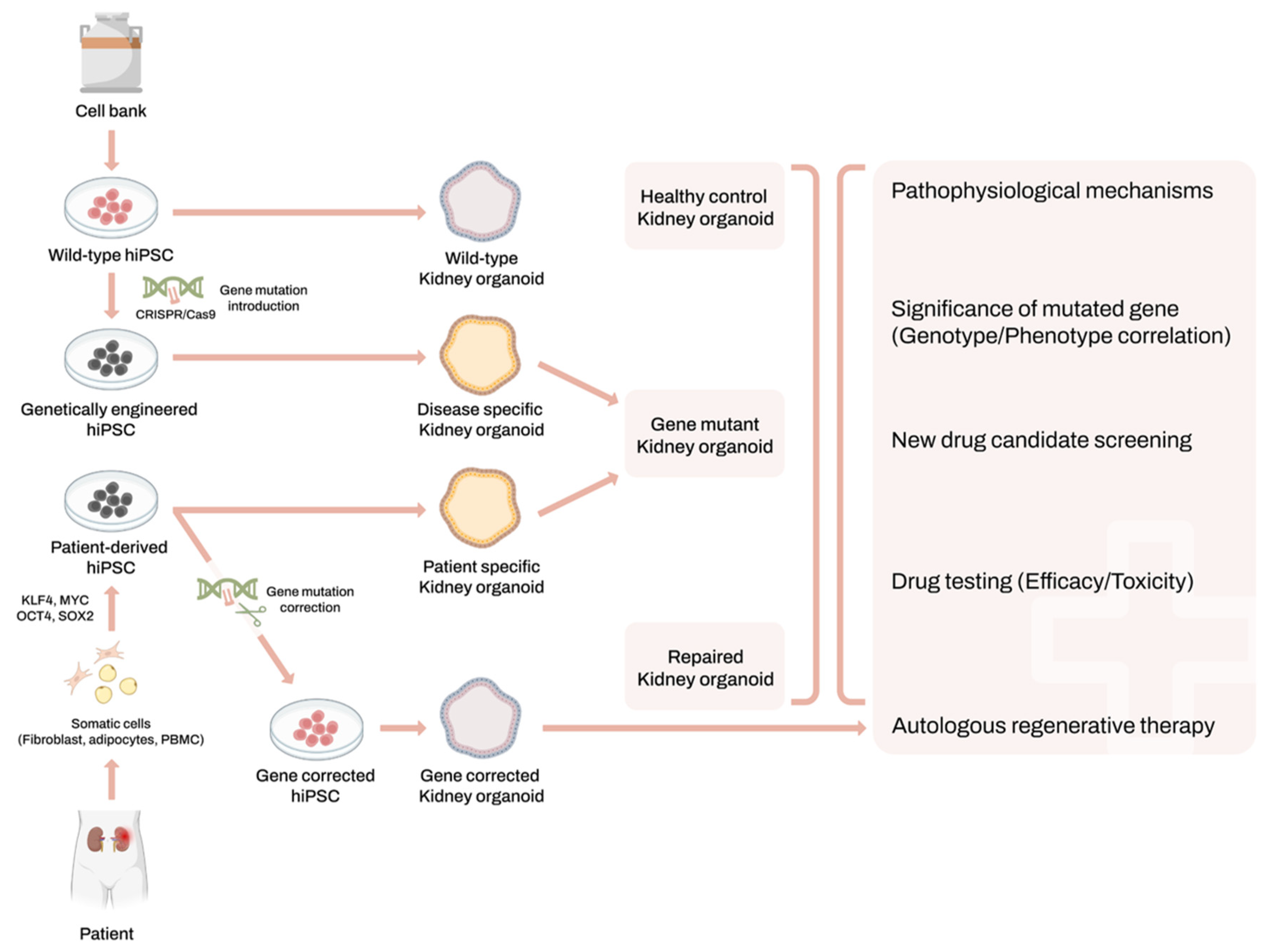
2. Human-Induced Pluripotent Stem Cells and Kidney Organoids System
3. Applications of Kidney Organoids for Genetic Kidney Disease Modeling
3.1. Autosomal Dominant Polycystic Kidney Disease
3.2. Autosomal Recessive Polycystic Kidney Disease
3.3. Fabry Disease Nephropathy
3.4. Gitelman Syndrome
3.5. Karyomegalic Interstitial Nephritis
3.6. Alport Syndrome
3.7. APOL1 Nephropathy
3.8. Autosomal Dominant Tubulointerstitial Kidney Disease (ADTKD)
3.9. Nephronophthisis (NPHP)
3.10. Autosomal Recessive Renal Tubular Dysgenesis (AR-RTD)
3.11. Nephrotic Syndrome
4. The Limitation of Kidney Organoids and Future Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.-M.; Yang, C.-W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Kovesdy, C.P. Epidemiology of chronic kidney disease: An update 2022. Kidney Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Devuyst, O.; Knoers, N.V.; Remuzzi, G.; Schaefer, F. Rare inherited kidney diseases: Challenges, opportunities, and perspectives. Lancet 2014, 383, 1844–1859. [Google Scholar] [CrossRef] [PubMed]
- Participants, K.C. Genetics in chronic kidney disease: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2022, 101, 1126–1141. [Google Scholar] [CrossRef]
- Hong, Y.A.; Ban, T.H.; Kang, C.-Y.; Hwang, S.D.; Choi, S.R.; Lee, H.; Jung, H.-Y.; Kim, K.; Kwon, Y.E.; Kim, S.H.; et al. Trends in epidemiologic characteristics of end-stage renal disease from 2019 Korean Renal Data System (KORDS). Kidney Res. Clin. Pract. 2021, 40, 52–61. [Google Scholar] [CrossRef] [PubMed]
- KORDS (Korea Renal Data System) Annual Data Report, Status and Trends in Epidemiologic Characteristics of End-Stage Renal Disease in Korea. 2023. Available online: https://www.ksn.or.kr/bbs/?code=report_eng (accessed on 20 April 2024).
- Domingo-Gallego, A.; Pybus, M.; Bullich, G.; Furlano, M.; Ejarque Vila, L.; Lorente-Grandoso, L.; Ruiz, P.; Fraga, G.; González, M.; Piñero-Fernández, J.; et al. Clinical utility of genetic testing in early-onset kidney disease: Seven genes are the main players. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transplant. Assoc.-Eur. Ren. Assoc. 2021, 37, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Giovanella, S.; Ligabue, G.; Chester, J.; Magistroni, R. Genomic Approaches for Monogenic Kidney Diseases: A Comparative Review of Diagnostic Methods and Precision Medicine Implications. Appl. Sci. 2023, 13, 12733. [Google Scholar] [CrossRef]
- Yousef Yengej, F.A.; Jansen, J.; Rookmaaker, M.B.; Verhaar, M.C.; Clevers, H. Kidney Organoids and Tubuloids. Cells 2020, 9, 1326. [Google Scholar] [CrossRef] [PubMed]
- Dilmen, E.; Orhon, I.; Jansen, J.; Hoenderop, J.G.J. Advancements in kidney organoids and tubuloids to study (dys)function. Trends Cell Biol. 2024, 34, 299–311. [Google Scholar] [CrossRef]
- Freedman, B.S. Modeling Kidney Disease with iPS Cells. Biomark. Insights 2015, 10, 153–169. [Google Scholar] [CrossRef]
- Rowe, R.G.; Daley, G.Q. Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet. 2019, 20, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Long, H.Y.; Qian, Z.P.; Lan, Q.; Xu, Y.J.; Da, J.J.; Yu, F.X.; Zha, Y. Human pluripotent stem cell-derived kidney organoids: Current progress and challenges. World J. Stem Cells 2024, 16, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Chambers, B.E.; Weaver, N.E.; Wingert, R.A. The “3Ds” of Growing Kidney Organoids: Advances in Nephron Development, Disease Modeling, and Drug Screening. Cells 2023, 12, 549. [Google Scholar] [CrossRef] [PubMed]
- Nishinakamura, R. Advances and challenges toward developing kidney organoids for clinical applications. Cell Stem Cell 2023, 30, 1017–1027. [Google Scholar] [CrossRef]
- Romero-Guevara, R.; Ioannides, A.; Xinaris, C. Kidney Organoids as Disease Models: Strengths, Weaknesses and Perspectives. Front. Physiol. 2020, 11, 563981. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef]
- Volarevic, V.; Markovic, B.S.; Gazdic, M.; Volarevic, A.; Jovicic, N.; Arsenijevic, N.; Armstrong, L.; Djonov, V.; Lako, M.; Stojkovic, M. Ethical and Safety Issues of Stem Cell-Based Therapy. Int. J. Med. Sci. 2018, 15, 36–45. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Park, I.H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008, 451, 141–146. [Google Scholar] [CrossRef]
- Takasato, M.; Er, P.X.; Chiu, H.S.; Little, M.H. Generation of kidney organoids from human pluripotent stem cells. Nat. Protoc. 2016, 11, 1681–1692. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, A.; Kaku, Y.; Ohmori, T.; Sharmin, S.; Ogawa, M.; Sasaki, H.; Nishinakamura, R. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell 2014, 14, 53–67. [Google Scholar] [CrossRef]
- Takasato, M.; Er, P.X.; Chiu, H.S.; Maier, B.; Baillie, G.J.; Ferguson, C.; Parton, R.G.; Wolvetang, E.J.; Roost, M.S.; Chuva de Sousa Lopes, S.M.; et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 2015, 526, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, A.; Nishinakamura, R. Higher-Order Kidney Organogenesis from Pluripotent Stem Cells. Cell Stem Cell 2017, 21, 730–746.e736. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.S.; Brooks, C.R.; Lam, A.Q.; Fu, H.; Morizane, R.; Agrawal, V.; Saad, A.F.; Li, M.K.; Hughes, M.R.; Werff, R.V.; et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat. Commun. 2015, 6, 8715. [Google Scholar] [CrossRef] [PubMed]
- Morizane, R.; Lam, A.Q.; Freedman, B.S.; Kishi, S.; Valerius, M.T.; Bonventre, J.V. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat. Biotechnol. 2015, 33, 1193–1200. [Google Scholar] [CrossRef] [PubMed]
- Little, M.H.; Combes, A.N. Kidney organoids: Accurate models or fortunate accidents. Genes Dev. 2019, 33, 1319–1345. [Google Scholar] [CrossRef]
- Tsujimoto, H.; Kasahara, T.; Sueta, S.I.; Araoka, T.; Sakamoto, S.; Okada, C.; Mae, S.I.; Nakajima, T.; Okamoto, N.; Taura, D.; et al. A Modular Differentiation System Maps Multiple Human Kidney Lineages from Pluripotent Stem Cells. Cell Rep. 2020, 31, 107476. [Google Scholar] [CrossRef] [PubMed]
- Uchimura, K.; Wu, H.; Yoshimura, Y.; Humphreys, B.D. Human Pluripotent Stem Cell-Derived Kidney Organoids with Improved Collecting Duct Maturation and Injury Modeling. Cell Rep. 2020, 33, 108514. [Google Scholar] [CrossRef]
- Tang, X.Y.; Wu, S.; Wang, D.; Chu, C.; Hong, Y.; Tao, M.; Hu, H.; Xu, M.; Guo, X.; Liu, Y. Human organoids in basic research and clinical applications. Signal Transduct. Target. Ther. 2022, 7, 168. [Google Scholar] [CrossRef]
- Lee, S.-G.; Kim, Y.-J.; Son, M.-Y.; Oh, M.-S.; Kim, J.; Ryu, B.; Kang, K.-R.; Baek, J.; Chung, G.; Woo, D.H.; et al. Generation of human iPSCs derived heart organoids structurally and functionally similar to heart. Biomaterials 2022, 290, 121860. [Google Scholar] [CrossRef]
- Novelli, G.; Spitalieri, P.; Murdocca, M.; Centanini, E.; Sangiuolo, F. Organoid factory: The recent role of the human induced pluripotent stem cells (hiPSCs) in precision medicine. Front. Cell Dev. Biol. 2023, 10, 1059579. [Google Scholar] [CrossRef] [PubMed]
- Mae, S.I.; Ryosaka, M.; Toyoda, T.; Matsuse, K.; Oshima, Y.; Tsujimoto, H.; Okumura, S.; Shibasaki, A.; Osafune, K. Generation of branching ureteric bud tissues from human pluripotent stem cells. Biochem. Biophys. Res. Commun. 2018, 495, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Czerniecki, S.M.; Cruz, N.M.; Harder, J.L.; Menon, R.; Annis, J.; Otto, E.A.; Gulieva, R.E.; Islas, L.V.; Kim, Y.K.; Tran, L.M.; et al. High-Throughput Screening Enhances Kidney Organoid Differentiation from Human Pluripotent Stem Cells and Enables Automated Multidimensional Phenotyping. Cell Stem Cell 2018, 22, 929–940.e924. [Google Scholar] [CrossRef] [PubMed]
- Rooney, K.M.; Woolf, A.S.; Kimber, S.J. Towards Modelling Genetic Kidney Diseases with Human Pluripotent Stem Cells. Nephron 2021, 145, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Little, M.H.; Quinlan, C. Advances in our understanding of genetic kidney disease using kidney organoids. Pediatr. Nephrol. 2020, 35, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Cruz, N.M.; Freedman, B.S. CRISPR Gene Editing in the Kidney. Am. J. Kidney Dis. 2018, 71, 874–883. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, C.W.; Ritsma, L.; Avramut, M.C.; Wiersma, L.E.; van den Berg, B.M.; Leuning, D.G.; Lievers, E.; Koning, M.; Vanslambrouck, J.M.; Koster, A.J.; et al. Renal Subcapsular Transplantation of PSC-Derived Kidney Organoids Induces Neo-vasculogenesis and Significant Glomerular and Tubular Maturation In Vivo. Stem Cell Rep. 2018, 10, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Usui, J.; Kobayashi, T.; Yamaguchi, T.; Knisely, A.S.; Nishinakamura, R.; Nakauchi, H. Generation of kidney from pluripotent stem cells via blastocyst complementation. Am. J. Pathol. 2012, 180, 2417–2426. [Google Scholar] [CrossRef]
- Yamanaka, S.; Tajiri, S.; Fujimoto, T.; Matsumoto, K.; Fukunaga, S.; Kim, B.S.; Okano, H.J.; Yokoo, T. Generation of interspecies limited chimeric nephrons using a conditional nephron progenitor cell replacement system. Nat. Commun. 2017, 8, 1719. [Google Scholar] [CrossRef]
- Matsui, K.; Yamanaka, S.; Chen, S.; Matsumoto, N.; Morimoto, K.; Kinoshita, Y.; Inage, Y.; Saito, Y.; Takamura, T.; Fujimoto, T.; et al. Long-term viable chimeric nephrons generated from progenitor cells are a reliable model in cisplatin-induced toxicity. Commun. Biol. 2023, 6, 1097. [Google Scholar] [CrossRef] [PubMed]
- Cruz, N.M.; Song, X.; Czerniecki, S.M.; Gulieva, R.E.; Churchill, A.J.; Kim, Y.K.; Winston, K.; Tran, L.M.; Diaz, M.A.; Fu, H.; et al. Organoid cystogenesis reveals a critical role of microenvironment in human polycystic kidney disease. Nat. Mater. 2017, 16, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Shamshirgaran, Y.; Jonebring, A.; Svensson, A.; Leefa, I.; Bohlooly, Y.M.; Firth, M.; Woollard, K.J.; Hofherr, A.; Rogers, I.M.; Hicks, R. Rapid target validation in a Cas9-inducible hiPSC derived kidney model. Sci. Rep. 2021, 11, 16532. [Google Scholar] [CrossRef] [PubMed]
- Kuraoka, S.; Tanigawa, S.; Taguchi, A.; Hotta, A.; Nakazato, H.; Osafune, K.; Kobayashi, A.; Nishinakamura, R. PKD1-Dependent Renal Cystogenesis in Human Induced Pluripotent Stem Cell-Derived Ureteric Bud/Collecting Duct Organoids. J. Am. Soc. Nephrol. 2020, 31, 2355–2371. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, C.; Gong, X.; Zhang, T.; Lian, M.M.; Chew, E.G.Y.; Cardilla, A.; Suzuki, K.; Wang, H.; Yuan, Y.; et al. Kidney organoid models reveal cilium-autophagy metabolic axis as a therapeutic target for PKD both in vitro and in vivo. Cell Stem Cell 2024, 31, 52–70.e8. [Google Scholar] [CrossRef] [PubMed]
- Low, J.H.; Li, P.; Chew, E.G.Y.; Zhou, B.; Suzuki, K.; Zhang, T.; Lian, M.M.; Liu, M.; Aizawa, E.; Rodriguez Esteban, C.; et al. Generation of Human PSC-Derived Kidney Organoids with Patterned Nephron Segments and a De Novo Vascular Network. Cell Stem Cell 2019, 25, 373–387.e9. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kim, H.W.; Nam, S.A.; Lee, J.Y.; Cho, H.J.; Kim, T.M.; Kim, Y.K. Human kidney organoids reveal the role of glutathione in Fabry disease. Exp. Mol. Med. 2021, 53, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Fang, X.; Lee, H.; Shin, Y.J.; Koh, E.S.; Chung, S.; Park, H.S.; Lim, S.W.; Lee, K.I.; Lee, J.Y.; et al. Modeling of Fabry disease nephropathy using patient derived human induced pluripotent stem cells and kidney organoid system. J. Transl. Med. 2023, 21, 138. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.W.; Fang, X.; Cui, S.; Lee, H.; Shin, Y.J.; Ko, E.J.; Lee, K.I.; Lee, J.Y.; Chung, B.H.; Yang, C.W. CRISPR-Cas9-Mediated Correction of SLC12A3 Gene Mutation Rescues the Gitelman’s Disease Phenotype in a Patient-Derived Kidney Organoid System. Int. J. Mol. Sci. 2023, 24, 3019. [Google Scholar] [CrossRef]
- Lim, S.W.; Shin, Y.J.; Cui, S.; Ko, E.J.; Lee, K.I.; Lee, J.Y.; Chung, B.H.; Yang, C.W. Generation of a human induced pluripotent stem cell line (CMCi002-A) from a patient with Gitelman’s syndrome. Stem Cell Res. 2020, 49, 102110. [Google Scholar] [CrossRef]
- Lim, S.W.; Na, D.; Lee, H.; Fang, X.; Cui, S.; Shin, Y.J.; Lee, K.I.; Lee, J.Y.; Yang, C.W.; Chung, B.H. Modeling of FAN1-Deficient Kidney Disease Using a Human Induced Pluripotent Stem Cell-Derived Kidney Organoid System. Cells 2023, 12, 2319. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, R.; Toyohara, K.; Watanabe, K.; Otsuki, T.; Araoka, T.; Mae, S.I.; Horinouchi, T.; Yamamura, T.; Okita, K.; Hotta, A.; et al. iPSC-derived type IV collagen alpha5-expressing kidney organoids model Alport syndrome. Commun. Biol. 2023, 6, 854. [Google Scholar] [CrossRef] [PubMed]
- Morais, M.; Tian, P.; Lawless, C.; Murtuza-Baker, S.; Hopkinson, L.; Woods, S.; Mironov, A.; Long, D.A.; Gale, D.P.; Zorn, T.M.T.; et al. Kidney organoids recapitulate human basement membrane assembly in health and disease. Elife 2022, 11, e73486. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.; Radmanesh, B.; Chung, B.H.; Donnan, M.D.; Yi, D.; Dadi, A.; Smith, K.D.; Himmelfarb, J.; Li, M.; Freedman, B.S.; et al. Profiling APOL1 Nephropathy Risk Variants in Genome-Edited Kidney Organoids with Single-Cell Transcriptomics. Kidney360 2020, 1, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Riella, C.V.; Chung, H.; Shah, S.S.; Wang, M.; Magraner, J.M.; Ribas, G.T.; Ribas, H.T.; Zhang, J.-Y.; Alper, S.L.; et al. DGAT2 Inhibition Potentiates Lipid Droplet Formation to Reduce Cytotoxicity in APOL1 Kidney Risk Variants. J. Am. Soc. Nephrol. 2022, 33, 889–907. [Google Scholar] [CrossRef]
- Song, H.; Dumas, S.J.; Ma, L.; Wang, G.; Witjas, F.; Berg, C.W.V.d.; Rocco, M.V.; Freedman, B.I.; Rabelink, T.J.; Spijker, S. APOL1 Risk Variants Induce Mitochondrial Dysfunction in Patient-Derived Kidney Organoids: SA-PO789. J. Am. Soc. Nephrol. 2023, 34, 947–948. [Google Scholar] [CrossRef]
- Forbes, T.A.; Howden, S.E.; Lawlor, K.; Phipson, B.; Maksimovic, J.; Hale, L.; Wilson, S.; Quinlan, C.; Ho, G.; Holman, K.; et al. Patient-iPSC-Derived Kidney Organoids Show Functional Validation of a Ciliopathic Renal Phenotype and Reveal Underlying Pathogenetic Mechanisms. Am. J. Hum. Genet. 2018, 102, 816–831. [Google Scholar] [CrossRef] [PubMed]
- Cornec-Le Gall, E.; Torres, V.E.; Harris, P.C. Genetic Complexity of Autosomal Dominant Polycystic Kidney and Liver Diseases. J. Am. Soc. Nephrol. 2018, 29, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Kuppe, C.; Perales-Paton, J.; Hayat, S.; Kranz, J.; Abdallah, A.T.; Nagai, J.; Li, Z.; Peisker, F.; Saritas, T.; et al. Adult human kidney organoids originate from CD24+ cells and represent an advanced model for adult polycystic kidney disease. Nat. Genet. 2022, 54, 1690–1701. [Google Scholar] [CrossRef]
- Vishy, C.E.; Thomas, C.; Vincent, T.; Crawford, D.K.; Goddeeris, M.M.; Freedman, B.S. Genetics of cystogenesis in base-edited human organoids reveal therapeutic strategies for polycystic kidney disease. Cell Stem Cell 2024, 31, 537–553.e5. [Google Scholar] [CrossRef]
- Porath, B.; Gainullin, V.G.; Cornec-Le Gall, E.; Dillinger, E.K.; Heyer, C.M.; Hopp, K.; Edwards, M.E.; Madsen, C.D.; Mauritz, S.R.; Banks, C.J.; et al. Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am. J. Hum. Genet. 2016, 98, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Galeano, M.C.R.; Ott, E.; Kaeslin, G.; Kausalya, P.J.; Kramer, C.; Ortiz-Brüchle, N.; Hilger, N.; Metzis, V.; Hiersche, M.; et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat. Genet. 2017, 49, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Primers 2018, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Guay-Woodford, L.M.; Muecher, G.; Hopkins, S.D.; Avner, E.D.; Germino, G.G.; Guillot, A.P.; Herrin, J.; Holleman, R.; Irons, D.A.; Primack, W.; et al. The severe perinatal form of autosomal recessive polycystic kidney disease maps to chromosome 6p21.1-p12: Implications for genetic counseling. Am. J. Hum. Genet. 1995, 56, 1101–1107. [Google Scholar] [PubMed]
- Goggolidou, P.; Richards, T. The genetics of Autosomal Recessive Polycystic Kidney Disease (ARPKD). Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166348. [Google Scholar] [CrossRef] [PubMed]
- Cordido, A.; Vizoso-Gonzalez, M.; Garcia-Gonzalez, M.A. Molecular Pathophysiology of Autosomal Recessive Polycystic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 6523. [Google Scholar] [CrossRef] [PubMed]
- Zarate, Y.A.; Hopkin, R.J. Fabry’s disease. Lancet 2008, 372, 1427–1435. [Google Scholar] [CrossRef]
- Cui, S.; Shin, Y.J.; Fang, X.; Lee, H.; Eum, S.H.; Ko, E.J.; Lim, S.W.; Shin, E.; Lee, K.I.; Lee, J.Y.; et al. CRISPR/Cas9-mediated A4GALT suppression rescues Fabry disease phenotypes in a kidney organoid model. Transl. Res. 2023, 258, 35–46. [Google Scholar] [CrossRef]
- Kok, K.; Zwiers, K.C.; Boot, R.G.; Overkleeft, H.S.; Aerts, J.; Artola, M. Fabry Disease: Molecular Basis, Pathophysiology, Diagnostics and Potential Therapeutic Directions. Biomolecules 2021, 11, 271. [Google Scholar] [CrossRef]
- Blanchard, A.; Bockenhauer, D.; Bolignano, D.; Calo, L.A.; Cosyns, E.; Devuyst, O.; Ellison, D.H.; Karet Frankl, F.E.; Knoers, N.V.; Konrad, M.; et al. Gitelman syndrome: Consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2017, 91, 24–33. [Google Scholar] [CrossRef]
- Gitelman, H.J.; Graham, J.B.; Welt, L.G. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans. Assoc. Am. Physicians 1966, 79, 221–235. [Google Scholar] [PubMed]
- Burry, A.F. Extreme dysplasia in renal epithelium of a young woman dying from hepatocarcinoma. J. Pathol. 1974, 113, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Na, D.H.; Lim, S.W.; Kim, B.M.; Kim, K.W.; Shin, Y.J.; Chae, H.; Ko, E.J.; Yang, C.W.; Kim, M.; Chung, B.H. Generation of a human induced pluripotent stem cell line (CMCi001-A) from a patient with karyomegalic interstitial nephritis with homozygous frameshift deletion mutation c.1985_1994del10 of the FANCD2/FANCI-Associated Nuclease 1 gene. Stem Cell Res 2020, 46, 101876. [Google Scholar] [CrossRef]
- Bekheirnia, M.R.; Reed, B.; Gregory, M.C.; McFann, K.; Shamshirsaz, A.A.; Masoumi, A.; Schrier, R.W. Genotype-phenotype correlation in X-linked Alport syndrome. J. Am. Soc. Nephrol. 2010, 21, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, J.A.; Lemmink, H.H.; Hughes, A.E.; Hill, C.M.; Smeets, H.J.; Doherty, C.C.; Maxwell, A.P. Autosomal dominant Alport syndrome linked to the type IV collage alpha 3 and alpha 4 genes (COL4A3 and COL4A4). Nephrol. Dial. Transplant. 1997, 12, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Lennon, R.; Byron, A.; Humphries, J.D.; Randles, M.J.; Carisey, A.; Murphy, S.; Knight, D.; Brenchley, P.E.; Zent, R.; Humphries, M.J. Global analysis reveals the complexity of the human glomerular extracellular matrix. J. Am. Soc. Nephrol. 2014, 25, 939–951. [Google Scholar] [CrossRef]
- Friedman, D.J.; Pollak, M.R. Genetics of kidney failure and the evolving story of APOL1. J. Clin. Investig. 2011, 121, 3367–3374. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.J.; Pollak, M.R. APOL1 Nephropathy: From Genetics to Clinical Applications. Clin. J. Am. Soc. Nephrol. 2021, 16, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Devuyst, O.; Olinger, E.; Weber, S.; Eckardt, K.-U.; Kmoch, S.; Rampoldi, L.; Bleyer, A.J. Autosomal dominant tubulointerstitial kidney disease. Nat. Rev. Dis. Primers 2019, 5, 60. [Google Scholar] [CrossRef]
- Eckardt, K.U.; Alper, S.L.; Antignac, C.; Bleyer, A.J.; Chauveau, D.; Dahan, K.; Deltas, C.; Hosking, A.; Kmoch, S.; Rampoldi, L.; et al. Autosomal dominant tubulointerstitial kidney disease: Diagnosis, classification, and management—A KDIGO consensus report. Kidney Int. 2015, 88, 676–683. [Google Scholar] [CrossRef]
- Przepiorski, A.; Sander, V.; Tran, T.; Hollywood, J.A.; Sorrenson, B.; Shih, J.H.; Wolvetang, E.J.; McMahon, A.P.; Holm, T.M.; Davidson, A.J. A Simple Bioreactor-Based Method to Generate Kidney Organoids from Pluripotent Stem Cells. Stem Cell Rep. 2018, 11, 470–484. [Google Scholar] [CrossRef] [PubMed]
- Mae, S.I.; Ryosaka, M.; Sakamoto, S.; Matsuse, K.; Nozaki, A.; Igami, M.; Kabai, R.; Watanabe, A.; Osafune, K. Expansion of Human iPSC-Derived Ureteric Bud Organoids with Repeated Branching Potential. Cell Rep. 2020, 32, 107963. [Google Scholar] [CrossRef] [PubMed]
- Dvela-Levitt, M.; Kost-Alimova, M.; Emani, M.; Kohnert, E.; Thompson, R.; Sidhom, E.H.; Rivadeneira, A.; Sahakian, N.; Roignot, J.; Papagregoriou, G.; et al. Small Molecule Targets TMED9 and Promotes Lysosomal Degradation to Reverse Proteinopathy. Cell 2019, 178, 521–535.e23. [Google Scholar] [CrossRef]
- Wolf, M.T.F.; Bonsib, S.M.; Larsen, C.P.; Hildebrandt, F. Nephronophthisis: A pathological and genetic perspective. Pediatr. Nephrol. 2023, 39, 1977–2000. [Google Scholar] [CrossRef] [PubMed]
- Gubler, M.C.; Antignac, C. Renin-angiotensin system in kidney development: Renal tubular dysgenesis. Kidney Int. 2010, 77, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Pode-Shakked, N.; Slack, M.; Sundaram, N.; Schreiber, R.; McCracken, K.W.; Dekel, B.; Helmrath, M.; Kopan, R. RAAS-deficient organoids indicate delayed angiogenesis as a possible cause for autosomal recessive renal tubular dysgenesis. Nat. Commun. 2023, 14, 8159. [Google Scholar] [CrossRef] [PubMed]
- Veissi, S.; Smeets, B.; van den Heuvel, L.P.; Schreuder, M.F.; Jansen, J. Nephrotic syndrome in a dish: Recent developments in modeling in vitro. Pediatr. Nephrol. 2020, 35, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Shabaka, A.; Tato Ribera, A.; Fernandez-Juarez, G. Focal Segmental Glomerulosclerosis: State-of-the-Art and Clinical Perspective. Nephron 2020, 144, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Tanigawa, S.; Islam, M.; Sharmin, S.; Naganuma, H.; Yoshimura, Y.; Haque, F.; Era, T.; Nakazato, H.; Nakanishi, K.; Sakuma, T.; et al. Organoids from Nephrotic Disease-Derived iPSCs Identify Impaired NEPHRIN Localization and Slit Diaphragm Formation in Kidney Podocytes. Stem Cell Rep. 2018, 11, 727–740. [Google Scholar] [CrossRef]
- Ohmori, T.; De, S.; Tanigawa, S.; Miike, K.; Islam, M.; Soga, M.; Era, T.; Shiona, S.; Nakanishi, K.; Nakazato, H.; et al. Impaired NEPHRIN localization in kidney organoids derived from nephrotic patient iPS cells. Sci. Rep. 2021, 11, 3982. [Google Scholar] [CrossRef]
- Jansen, J.; van den Berge, B.T.; van den Broek, M.; Maas, R.J.; Daviran, D.; Willemsen, B.; Roverts, R.; van der Kruit, M.; Kuppe, C.; Reimer, K.C.; et al. Human pluripotent stem cell-derived kidney organoids for personalized congenital and idiopathic nephrotic syndrome modeling. Development 2022, 149, dev200198. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Buerger, F.; Forbes, T.A.; Klambt, V.; Schneider, R.; Deutsch, K.; Kitzler, T.M.; Howden, S.E.; Scurr, M.; Tan, K.S.; et al. Recessive NOS1AP variants impair actin remodeling and cause glomerulopathy in humans and mice. Sci. Adv. 2021, 7, eabe1386. [Google Scholar] [CrossRef] [PubMed]
- Xinaris, C. Organoids for replacement therapy: Expectations, limitations and reality. Curr. Opin. Organ. Transplant. 2019, 24, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Uchimura, K.; Donnelly, E.L.; Kirita, Y.; Morris, S.A.; Humphreys, B.D. Comparative Analysis and Refinement of Human PSC-Derived Kidney Organoid Differentiation with Single-Cell Transcriptomics. Cell Stem Cell 2018, 23, 869–881.e8. [Google Scholar] [CrossRef]
- Gabbin, B.; Meraviglia, V.; Angenent, M.L.; Ward-van Oostwaard, D.; Sol, W.; Mummery, C.L.; Rabelink, T.J.; van Meer, B.J.; van den Berg, C.W.; Bellin, M. Heart and kidney organoids maintain organ-specific function in a microfluidic system. Mater. Today Bio 2023, 23, 100818. [Google Scholar] [CrossRef] [PubMed]
- Song, S.-S.; Park, H.-J.; Kim, Y.K.; Kang, S.-W. Revolutionizing biomedical research: The imperative need for heart–kidney-connected organoids. APL Bioeng. 2024, 8, 010902. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, K.T.; Vanslambrouck, J.M.; Higgins, J.W.; Chambon, A.; Bishard, K.; Arndt, D.; Er, P.X.; Wilson, S.B.; Howden, S.E.; Tan, K.S.; et al. Cellular extrusion bioprinting improves kidney organoid reproducibility and conformation. Nat. Mater. 2021, 20, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.; Song, C.J.; Nguyen, T.; Cheng, S.Y.; McMahon, J.A.; Yang, R.; Guo, Q.; Der, B.; Lindstrom, N.O.; Lin, D.C.; et al. A scalable organoid model of human autosomal dominant polycystic kidney disease for disease mechanism and drug discovery. Cell Stem Cell 2022, 29, 1083–1101.e7. [Google Scholar] [CrossRef]
- Oishi, H.; Tabibzadeh, N.; Morizane, R. Advancing preclinical drug evaluation through automated 3D imaging for high-throughput screening with kidney organoids. Biofabrication 2024, 16, 035003. [Google Scholar] [CrossRef]
- Brassard, J.A.; Nikolaev, M.; Hübscher, T.; Hofer, M.; Lutolf, M.P. Recapitulating macro-scale tissue self-organization through organoid bioprinting. Nat. Mater. 2021, 20, 22–29. [Google Scholar] [CrossRef]
- Chen, E.P.; Toksoy, Z.; Davis, B.A.; Geibel, J.P. 3D Bioprinting of Vascularized Tissues for in vitro and in vivo Applications. Front. Bioeng. Biotechnol. 2021, 9, 664188. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Disease | Gene | Phenotype and Key Finding | Refs. |
|---|---|---|---|
| Autosomal Dominant Polycystic Kidney Disease | PKD1 or PKD2 | Cyst formation from tubules Cyst formation from tubules | [26,35,43] [44] |
| Cyst formation from Ureteric bud | [45] | ||
| GANAB, DZIP1L | Cyst formation from tubules | [46] | |
| Autosomal Recessive Polycystic Kidney Disease | PKHD1 | Cyst formation from tubules | [47] |
| Fabry Disease Nephropathy | GLA | Deformation of podocytes and tubular cells with accumulation of Gb3, increased oxidative stress and apoptosis | [48] |
| Decreased α-Gal A activity and increased Gb3 deposition according to disease severity | [49] | ||
| Gitelman Syndrome | SLC12A3 | Decreased expression of NCCT protein and RNA | [50,51] |
| Karyomegalic Interstitial Nephritis | FAN1 | DNA repair impairment | [45,52] |
| Alport Syndrome | COL4A5 | Altered expression of Type 4 Collagen α5 | [53,54] |
| APOL1 Nephropathy | APOL1 | Transcriptomic profiling of APOL1 mutant organoid | [55,56,57] |
| Nephronophthisis | IFT140 | Shortened, club-shaped primary cilia and Polarization defect | [58] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Na, D.H.; Cui, S.; Fang, X.; Lee, H.; Eum, S.H.; Shin, Y.J.; Lim, S.W.; Yang, C.W.; Chung, B.H. Advancements in Research on Genetic Kidney Diseases Using Human-Induced Pluripotent Stem Cell-Derived Kidney Organoids. Cells 2024, 13, 1190. https://doi.org/10.3390/cells13141190
Na DH, Cui S, Fang X, Lee H, Eum SH, Shin YJ, Lim SW, Yang CW, Chung BH. Advancements in Research on Genetic Kidney Diseases Using Human-Induced Pluripotent Stem Cell-Derived Kidney Organoids. Cells. 2024; 13(14):1190. https://doi.org/10.3390/cells13141190
Chicago/Turabian StyleNa, Do Hyun, Sheng Cui, Xianying Fang, Hanbi Lee, Sang Hun Eum, Yoo Jin Shin, Sun Woo Lim, Chul Woo Yang, and Byung Ha Chung. 2024. "Advancements in Research on Genetic Kidney Diseases Using Human-Induced Pluripotent Stem Cell-Derived Kidney Organoids" Cells 13, no. 14: 1190. https://doi.org/10.3390/cells13141190