

ADAR1 Is Essential for Smooth Muscle Homeostasis and Vascular Integrity


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Histopathology Staining
2.3. Western Blotting
2.4. Co-Immunoprecipitation (Co-IP)
2.5. Proximity Ligation Assay (PLA)
2.6. Flowcytometry and Tissue Digestion
2.7. Transmission Electron Microscopy (TEM)
2.8. Aortic Elastin Architecture Preservation and Scanning Electron Microscopy (SEM)
2.9. High Throughput RNA Sequencing and Gene Expression Analysis
2.10. Human Aorta Tissue Specimens
2.11. TUNEL Analysis
2.12. Statistical Analysis
3. Results
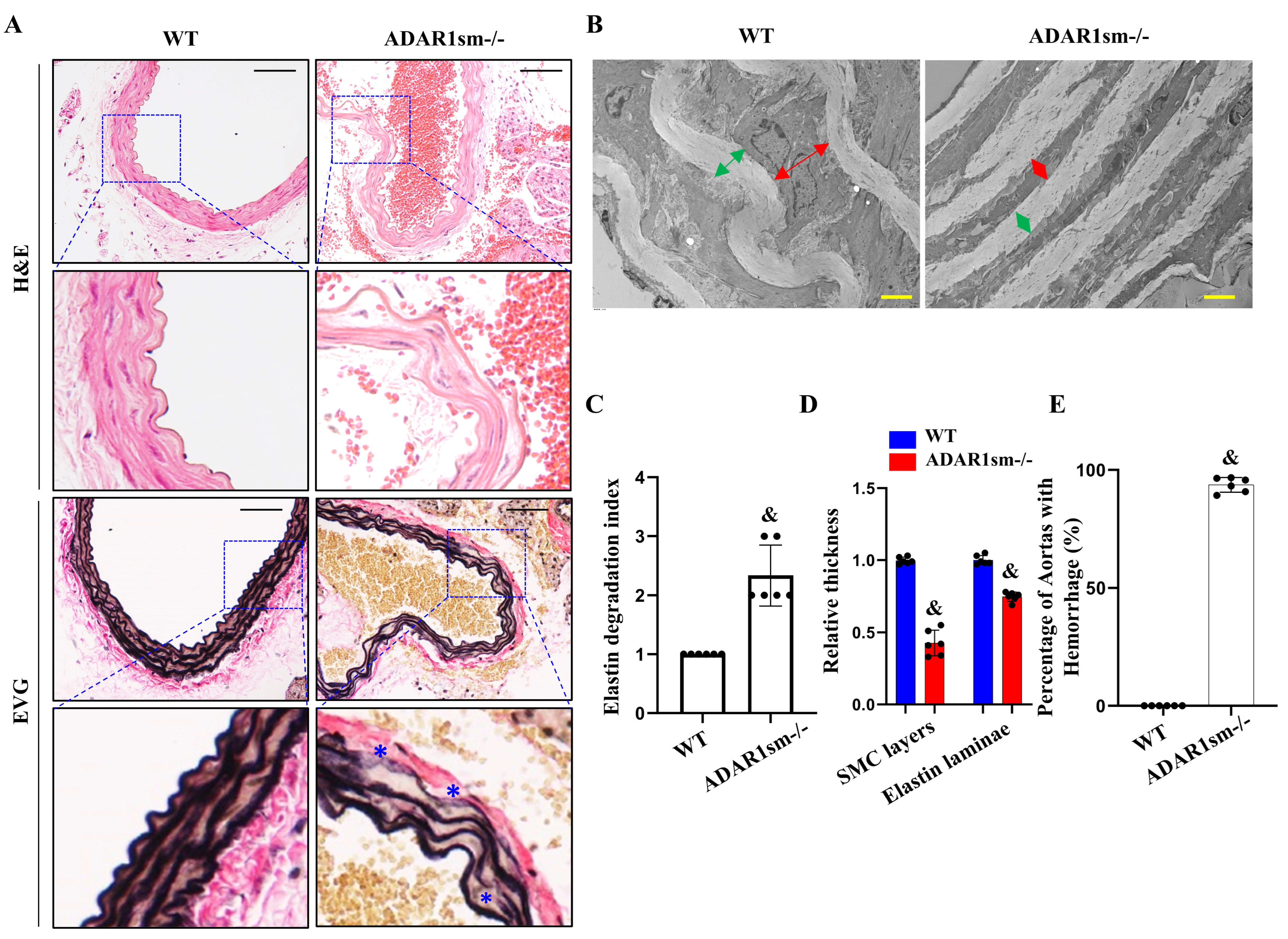
3.1. ADAR1sm-/- Causes Lethality in Mice Due to Extensive Hemorrhages
3.2. ADAR1sm-/- Impairs Elastin Fiber Integrity
3.3. ADAR1sm-/- Impairs the SMC and Elastic Laminae Interaction
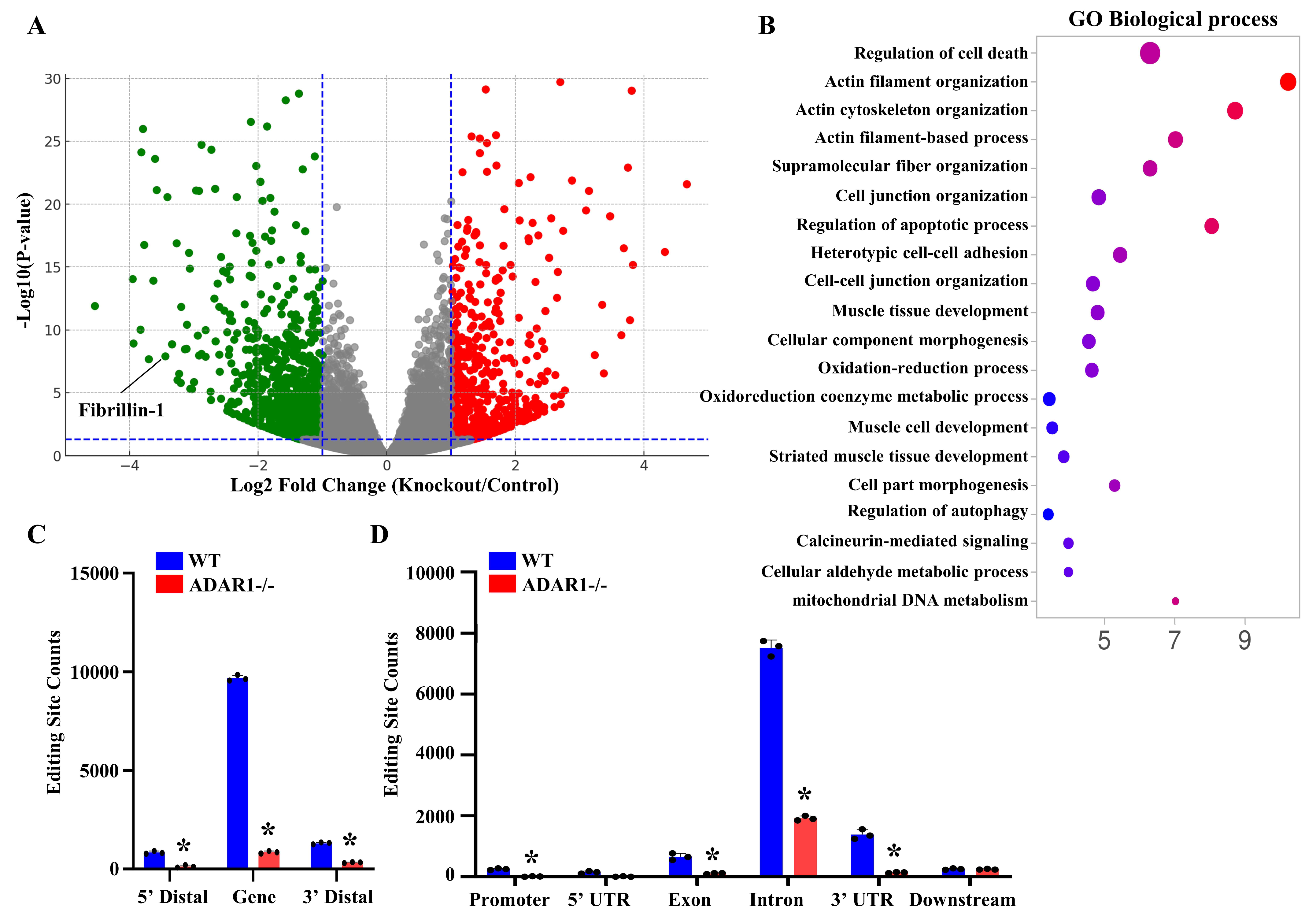
3.4. ADAR1sm-/- Alters the Expression and RNA Editing Profiles of Genes Related to Vascular Homeostasis
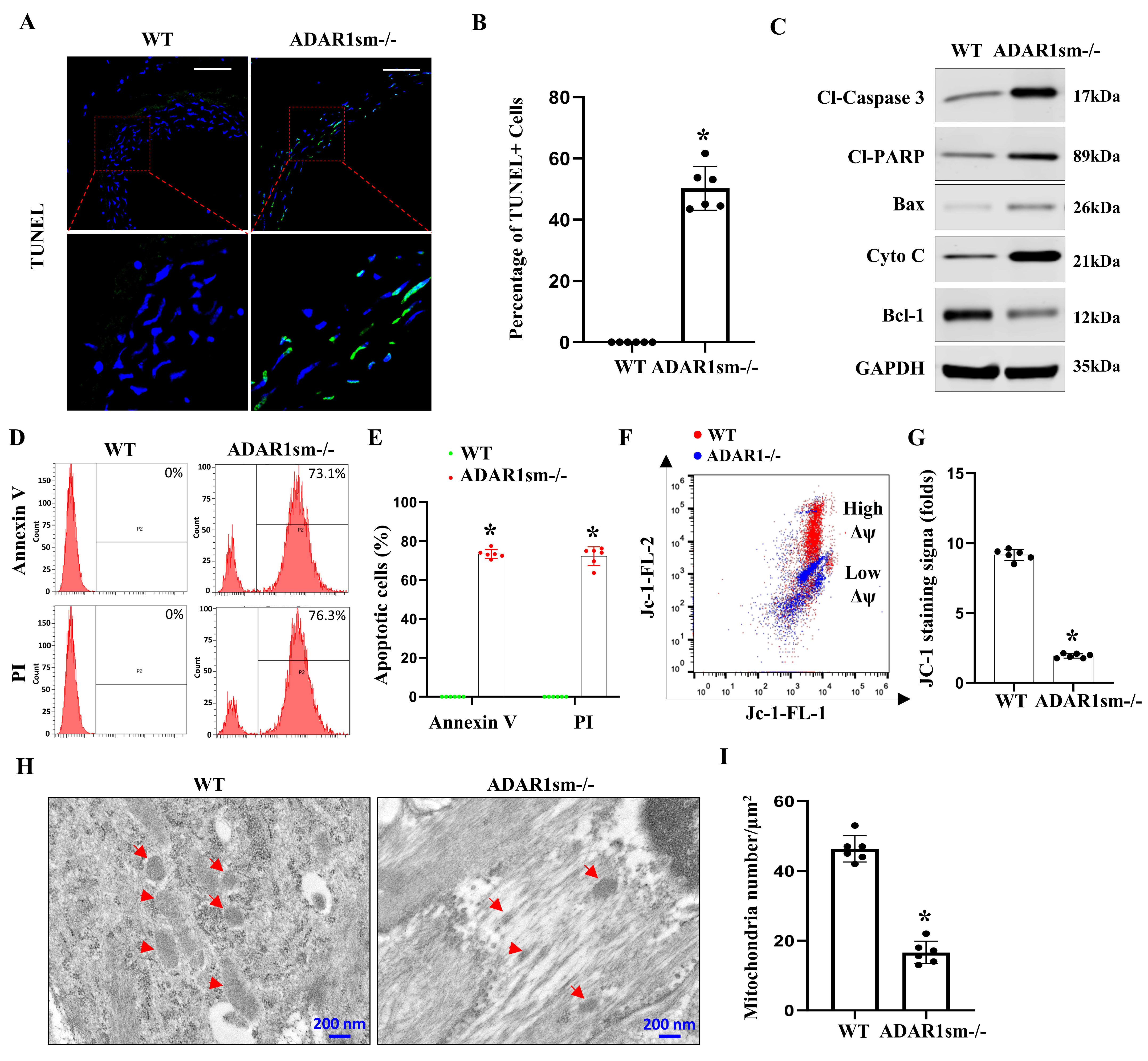
3.5. ADAR1sm-/- Leads to SMC Apoptosis through the Mitochondrial Pathway
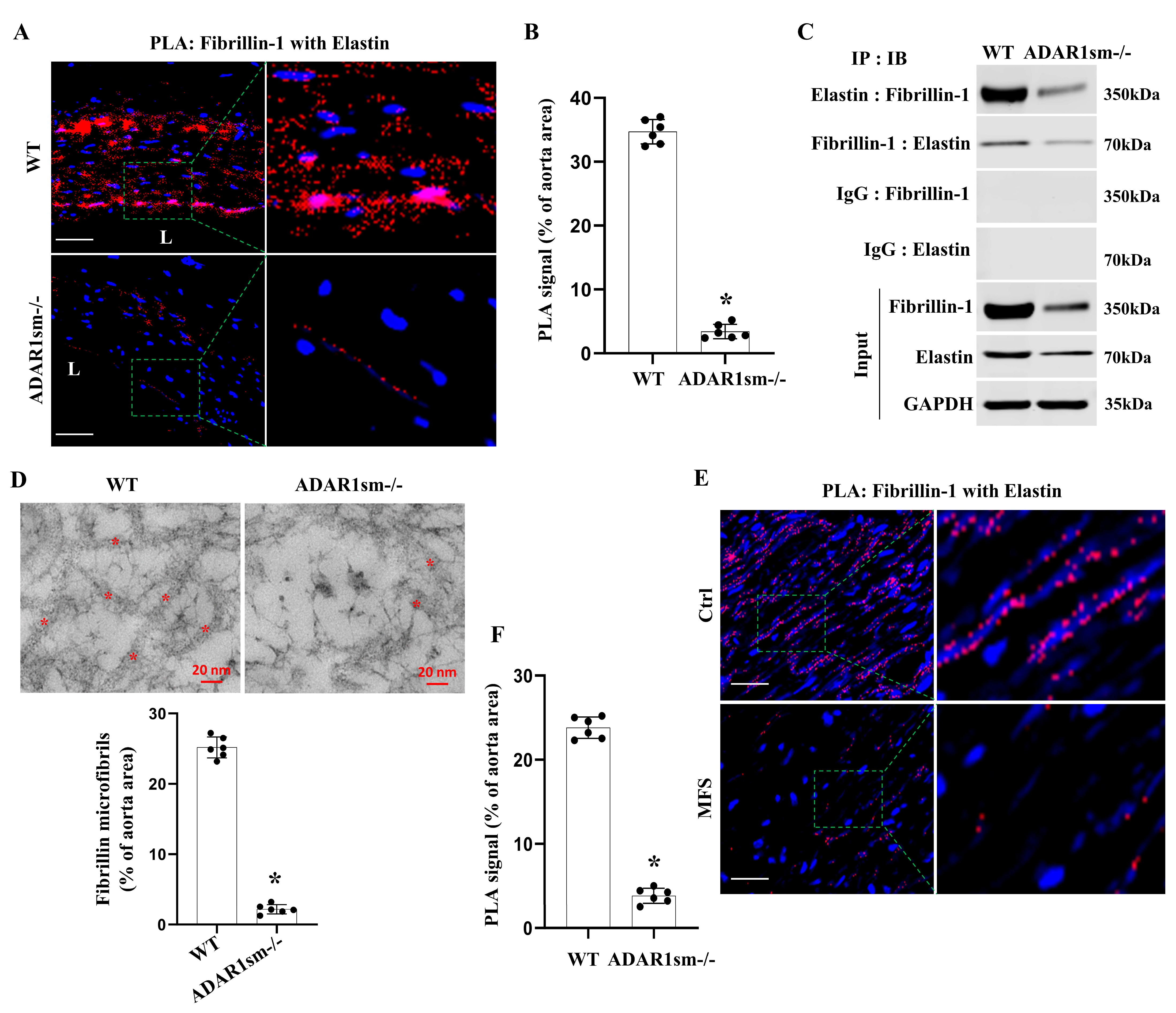
3.6. ADAR1sm-/- Disrupts the Fibrillin-1 and Elastin Interaction in the Aorta ECM
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Daugherty, A.; Powell, J.T. Recent highlights of ATVB: Aneurysms. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Blaise, R.; Mateo, V.; Rouxel, C.; Zaccarini, F.; Glorian, M.; Bereziat, G.; Golubkov, V.S.; Limon, I. Wild-type amyloid beta 1–40 peptide induces vascular smooth muscle cell death independently from matrix metalloprotease activity. Aging Cell 2012, 11, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Borges, B.E.; Appel, M.H.; Cofre, A.R.; Prado, M.L.; Steclan, C.A.; Esnard, F.; Zanata, S.M.; Laurindo, F.R.; Nakao, L.S. The flavo-oxidase QSOX1 supports vascular smooth muscle cell migration and proliferation: Evidence for a role in neointima growth. Biochim. Biophys. Acta 2015, 1852, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Hernando, C.; Jozsef, L.; Jenkins, D.; Di Lorenzo, A.; Sessa, W.C. Absence of Akt1 reduces vascular smooth muscle cell migration and survival and induces features of plaque vulnerability and cardiac dysfunction during atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2033–2040. [Google Scholar] [CrossRef]
- Han, D.G.; Ahn, C.B.; Lee, J.H.; Hwang, Y.; Kim, J.H.; Park, K.Y.; Lee, J.W.; Son, K.H. Optimization of Electrospun Poly(caprolactone) Fiber Diameter for Vascular Scaffolds to Maximize Smooth Muscle Cell Infiltration and Phenotype Modulation. Polymers 2019, 11, 643. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Dong, W.; Lin, Z.; Lu, J.; Wan, H.; Zhou, Z.; Liu, Z. CCN4 regulates vascular smooth muscle cell migration and proliferation. Mol. Cells 2013, 36, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Murgai, M.; Ju, W.; Eason, M.; Kline, J.; Beury, D.W.; Kaczanowska, S.; Miettinen, M.M.; Kruhlak, M.; Lei, H.; Shern, J.F.; et al. KLF4-dependent perivascular cell plasticity mediates pre-metastatic niche formation and metastasis. Nat. Med. 2017, 23, 1176–1190. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.D.; Narine, A.; Shaver, P.R.; Fox, J.C.; Vuncannon, J.R.; Tulis, D.A. AMP-activated protein kinase inhibits vascular smooth muscle cell proliferation and migration and vascular remodeling following injury. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H369–H381. [Google Scholar] [CrossRef] [PubMed]
- Iizasa, H. Pathophysiological role of A-to-I RNA editing enzyme ADAR1 in human diseases. Seikagaku 2016, 88, 593–599. [Google Scholar]
- Jiao, Y.; Xu, Y.; Liu, C.; Miao, R.; Liu, C.; Wang, Y.; Liu, J. The role of ADAR1 through and beyond its editing activity in cancer. Cell Commun. Signal. 2024, 22, 42. [Google Scholar] [CrossRef]
- Kleinova, R.; Rajendra, V.; Leuchtenberger, A.F.; Lo Giudice, C.; Vesely, C.; Kapoor, U.; Tanzer, A.; Derdak, S.; Picardi, E.; Jantsch, M.F. The ADAR1 editome reveals drivers of editing-specificity for ADAR1-isoforms. Nucleic Acids Res. 2023, 51, 4191–4207. [Google Scholar] [CrossRef] [PubMed]
- Liddicoat, B.J.; Hartner, J.C.; Piskol, R.; Ramaswami, G.; Chalk, A.M.; Kingsley, P.D.; Sankaran, V.G.; Wall, M.; Purton, L.E.; Seeburg, P.H.; et al. Adenosine-to-inosine RNA editing by ADAR1 is essential for normal murine erythropoiesis. Exp. Hematol. 2016, 44, 947–963. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Sakurai, M.; Shiromoto, Y.; Nishikura, K. Functions of the RNA Editing Enzyme ADAR1 and Their Relevance to Human Diseases. Genes 2016, 7, 129. [Google Scholar] [CrossRef]
- Ota, H.; Sakurai, M.; Gupta, R.; Valente, L.; Wulff, B.E.; Ariyoshi, K.; Iizasa, H.; Davuluri, R.V.; Nishikura, K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 2013, 153, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Pfaller, C.K.; Li, Z.; George, C.X.; Samuel, C.E. Protein kinase PKR and RNA adenosine deaminase ADAR1: New roles for old players as modulators of the interferon response. Curr. Opin. Immunol. 2011, 23, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Radetskyy, R.; Daher, A.; Gatignol, A. ADAR1 and PKR, interferon stimulated genes with clashing effects on HIV-1 replication. Cytokine Growth Factor Rev. 2018, 40, 48–58. [Google Scholar] [CrossRef]
- Shiromoto, Y.; Sakurai, M.; Qu, H.; Kossenkov, A.V.; Nishikura, K. Processing of Alu small RNAs by DICER/ADAR1 complexes and their RNAi targets. RNA 2020, 26, 1801–1814. [Google Scholar] [CrossRef]
- Yu, Z.; Luo, R.; Li, Y.; Li, X.; Yang, Z.; Peng, J.; Huang, K. ADAR1 inhibits adipogenesis and obesity by interacting with Dicer to promote the maturation of miR-155-5P. J. Cell Sci. 2022, 135, jcs259333. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jin, J.; Jiang, J.; Wang, Y. Adenosine deaminase acting on RNA 1 (ADAR1) as crucial regulators in cardiovascular diseases: Structures, pathogenesis, and potential therapeutic approach. Front. Pharmacol. 2023, 14, 1194884. [Google Scholar] [CrossRef]
- Cai, D.; Sun, C.; Murashita, T.; Que, X.; Chen, S.Y. ADAR1 Non-Editing Function in Macrophage Activation and Abdominal Aortic Aneurysm. Circ. Res. 2023, 132, e78–e93. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, H.; Lin, W.; Shuai, P. ADAR1 overexpression is associated with cervical cancer progression and angiogenesis. Diagn. Pathol. 2017, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Fei, J.; Cui, X.B.; Wang, J.N.; Dong, K.; Chen, S.Y. ADAR1-Mediated RNA Editing, a Novel Mechanism Controlling Phenotypic Modulation of Vascular Smooth Muscle Cells. Circ. Res. 2016, 119, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Shigeyasu, K.; Okamoto, K.; Matsuoka, H.; Masuyama, H. ADAR1 and AZIN1 RNA editing function as an oncogene and contributes to immortalization in endometrial cancer. Gynecol. Oncol. 2022, 166, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Shiromoto, Y.; Sakurai, M.; Towers, M.; Zhang, Q.; Wu, S.; Havas, A.; Wang, L.; Berger, S.; Adams, P.D.; et al. ADAR1 downregulation by autophagy drives senescence independently of RNA editing by enhancing p16(INK4a) levels. Nat. Cell Biol. 2022, 24, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Y.; Zhang, J.; Liu, L.; Chen, Y.; Yang, X.; Liao, X.; He, M.; Jia, Z.; Fan, J.; et al. ADAR1 regulates vascular remodeling in hypoxic pulmonary hypertension through N1-methyladenosine modification of circCDK17. Acta Pharm. Sin. B 2023, 13, 4840–4855. [Google Scholar] [CrossRef] [PubMed]
- Hartner, J.C.; Schmittwolf, C.; Kispert, A.; Müller, A.M.; Higuchi, M.; Seeburg, P.H. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J. Biol. Chem. 2004, 279, 4894–4902. [Google Scholar] [CrossRef] [PubMed]
- Hartner, J.C.; Walkley, C.R.; Lu, J.; Orkin, S.H. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat. Immunol. 2009, 10, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Miyakoda, M.; Yang, W.; Khillan, J.; Stachura, D.L.; Weiss, M.J.; Nishikura, K. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J. Biol. Chem. 2004, 279, 4952–4961. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Sun, C.; Zhang, G.; Que, X.; Fujise, K.; Weintraub, N.L.; Chen, S.Y. A Novel Mechanism Underlying Inflammatory Smooth Muscle Phenotype in Abdominal Aortic Aneurysm. Circ. Res. 2021, 129, e202–e214. [Google Scholar] [CrossRef]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef]
- Aneja, R.; Liu, M.; Yates, C.; Gao, J.; Dong, X.; Zhou, B.; Vangapandu, S.N.; Zhou, J.; Joshi, H.C. Multidrug resistance-associated protein-overexpressing teniposide-resistant human lymphomas undergo apoptosis by a tubulin-binding agent. Cancer Res. 2008, 68, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.; Barrientos, A. Sucrose Gradient Sedimentation Analysis of Mitochondrial Ribosomes. Methods Mol. Biol. 2021, 2192, 211–226. [Google Scholar] [PubMed]
- Hu, D.; Yin, C.; Mohanta, S.K.; Weber, C.; Habenicht, A.J. Preparation of Single Cell Suspensions from Mouse Aorta. Bio-Protocol 2016, 6, e1832. [Google Scholar] [CrossRef] [PubMed]
- Fujino, M. Ultrastructural studies of the mouse aorta and its endothelial pinocytosis in diet-induced arteriosclerosis. Acta Pathol. Jpn. 1983, 33, 1115–1130. [Google Scholar]
- Ushiki, T. Preserving the original architecture of elastin components in the formic acid-digested aorta by an alternative procedure for scanning electron microscopy. J. Electron Microsc. 1992, 41, 60–63. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, D.; Chen, S.-Y. ADAR1 Is Essential for Smooth Muscle Homeostasis and Vascular Integrity. Cells 2024, 13, 1257. https://doi.org/10.3390/cells13151257
Cai D, Chen S-Y. ADAR1 Is Essential for Smooth Muscle Homeostasis and Vascular Integrity. Cells. 2024; 13(15):1257. https://doi.org/10.3390/cells13151257
Chicago/Turabian StyleCai, Dunpeng, and Shi-You Chen. 2024. "ADAR1 Is Essential for Smooth Muscle Homeostasis and Vascular Integrity" Cells 13, no. 15: 1257. https://doi.org/10.3390/cells13151257
APA StyleCai, D., & Chen, S.-Y. (2024). ADAR1 Is Essential for Smooth Muscle Homeostasis and Vascular Integrity. Cells, 13(15), 1257. https://doi.org/10.3390/cells13151257