
The Role of Fatty Acid Synthase in the Vascular Smooth Muscle Cell to Foam Cell Transition


 , and
, and 
Abstract
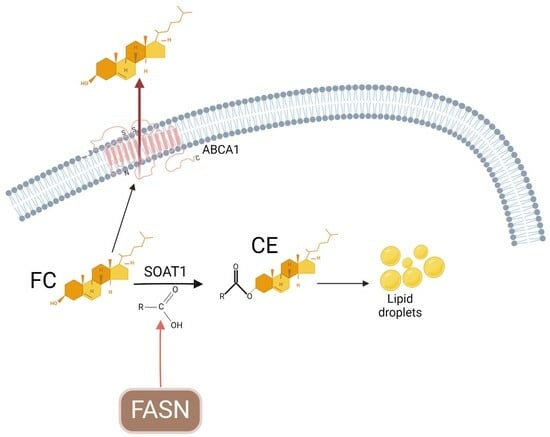
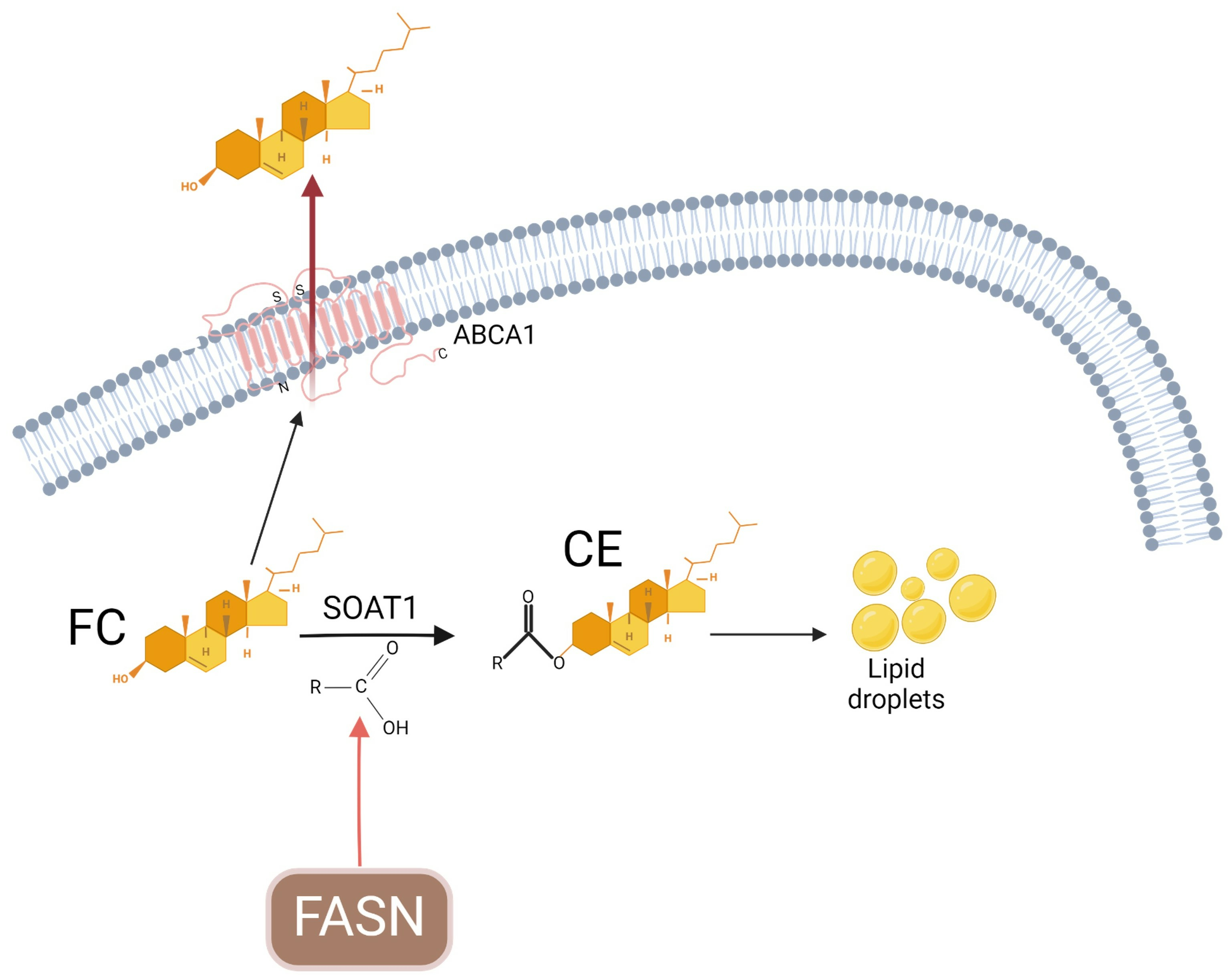{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Human Coronary Arteries
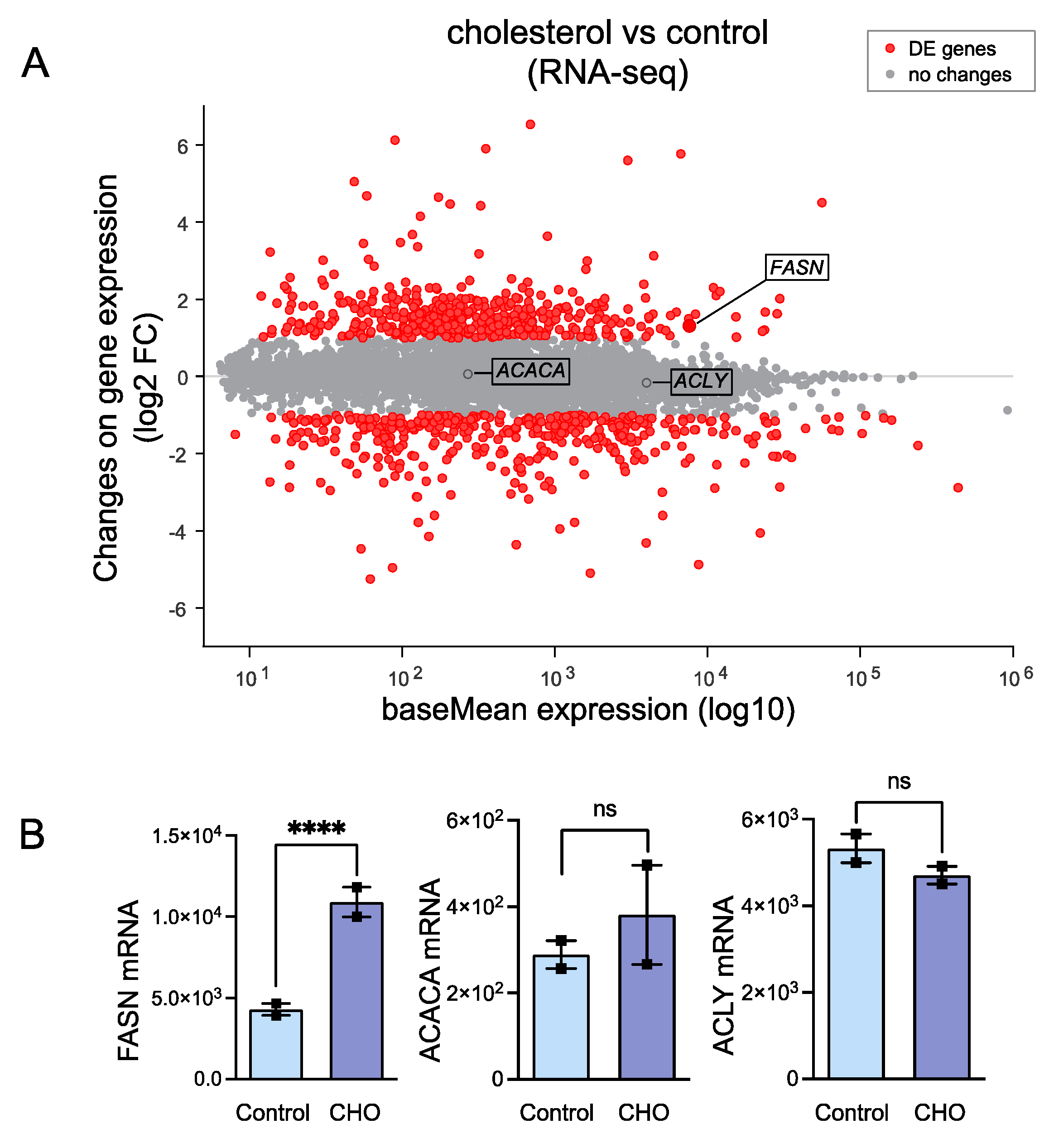
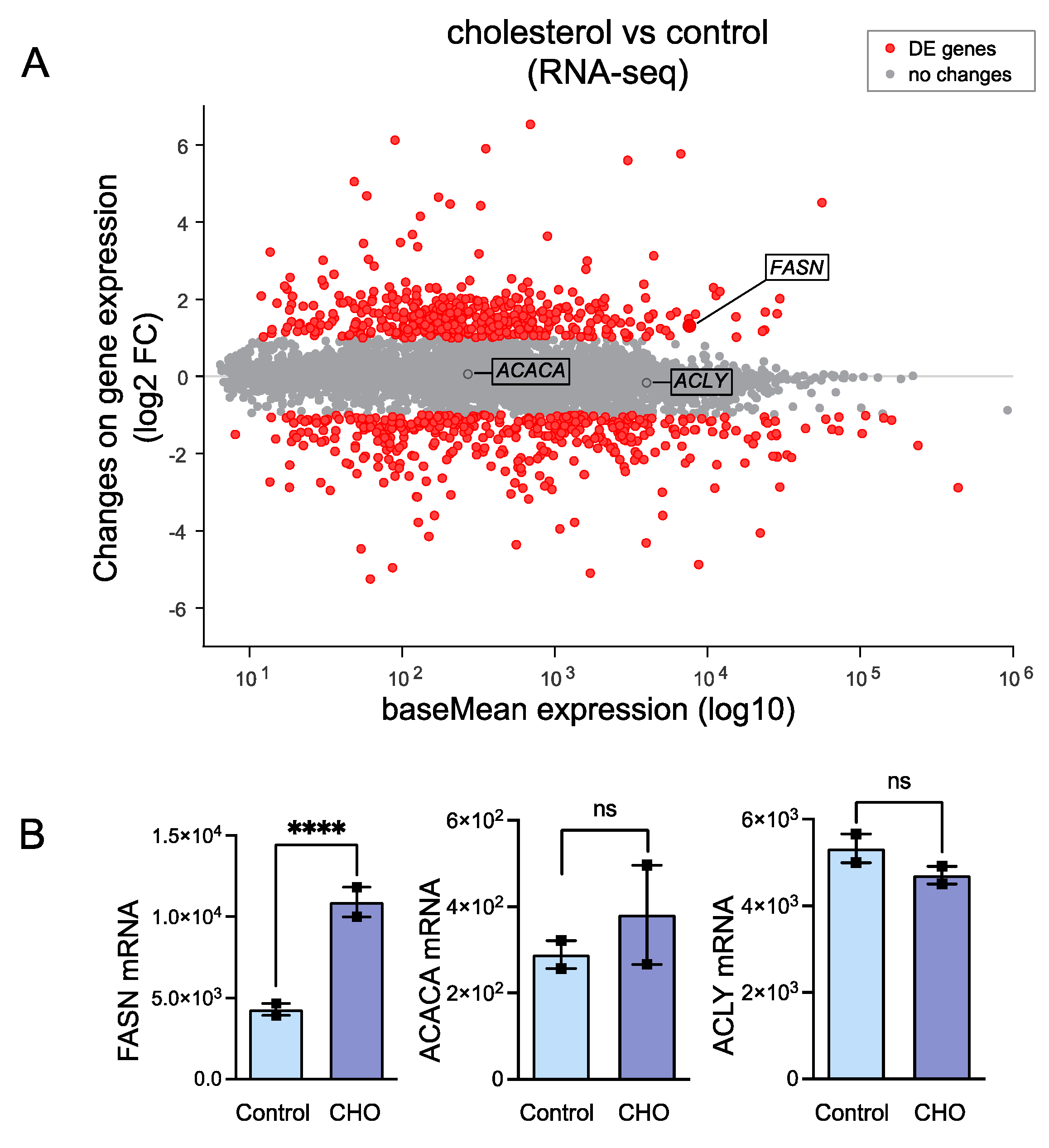
2.2. RNA-Seq in Cholesterol-Treated HASMCs
2.3. Cell Culture and Cholesterol Treatment
2.4. Cell Transfection
2.5. Preparation of Bovine Serum Albumin (BSA)–Palmitate Conjugate and Cell Treatment
2.6. Intracellular Neutral Lipid Staining
2.7. Western Blots
2.8. Statistical Analysis
3. Results
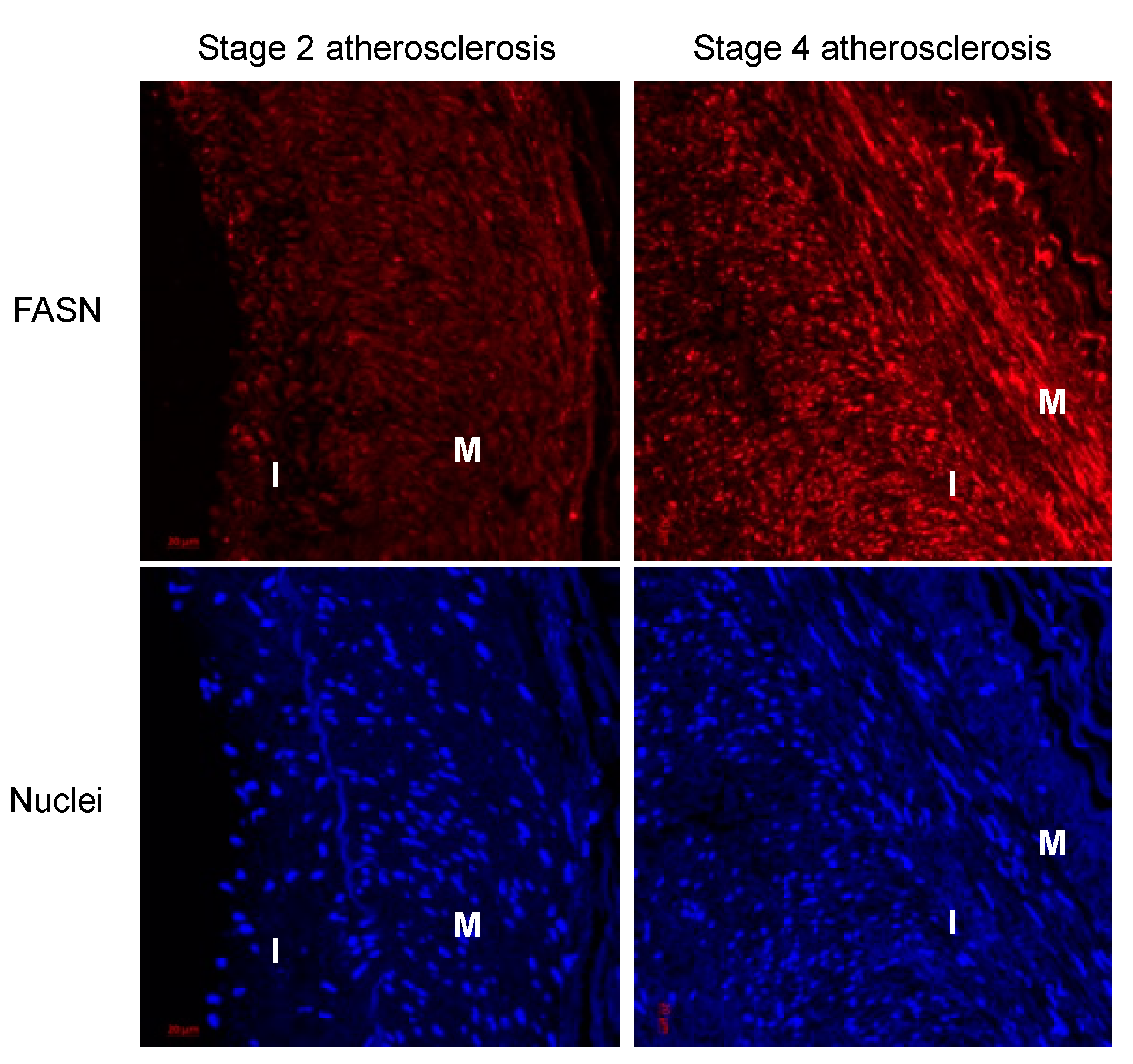
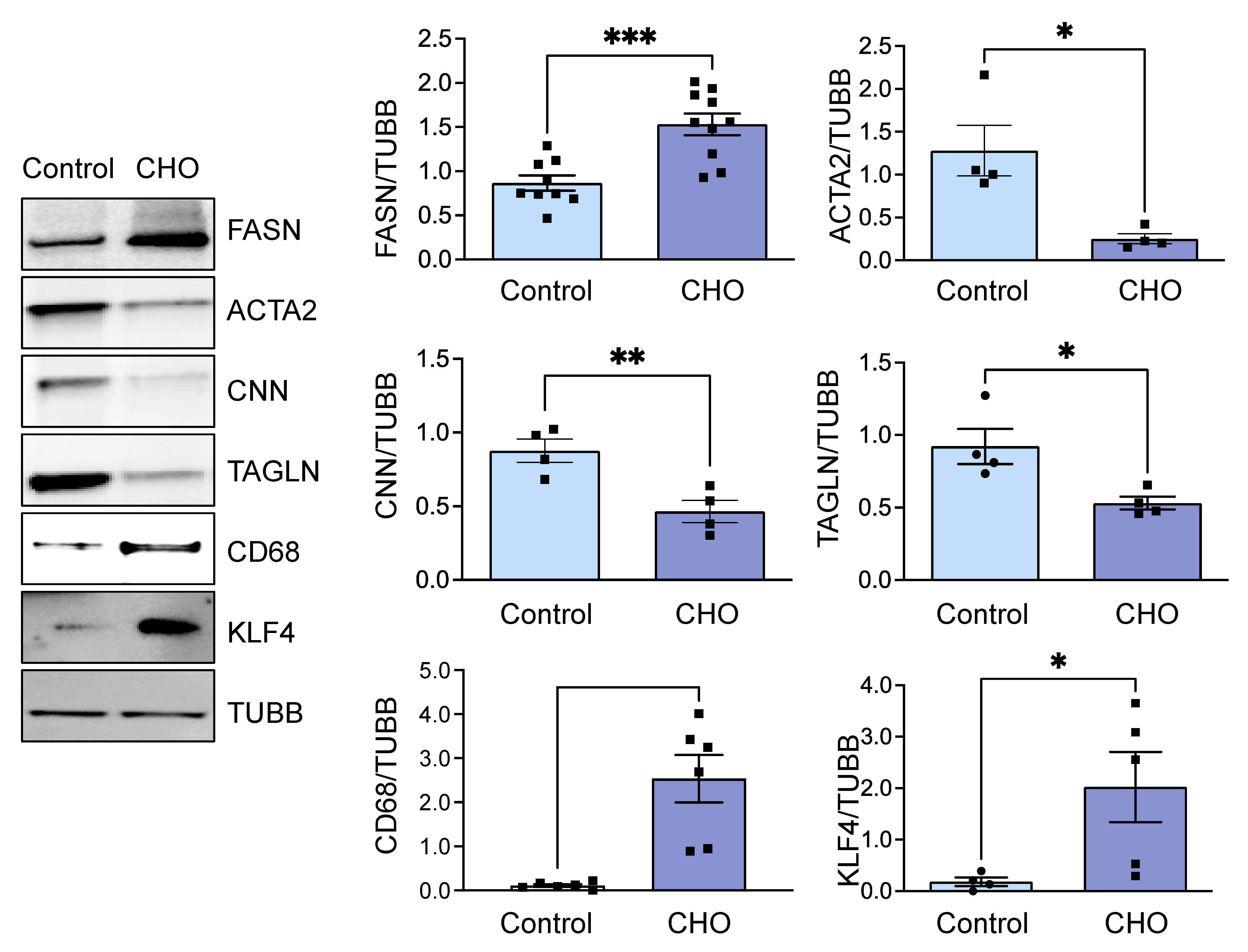
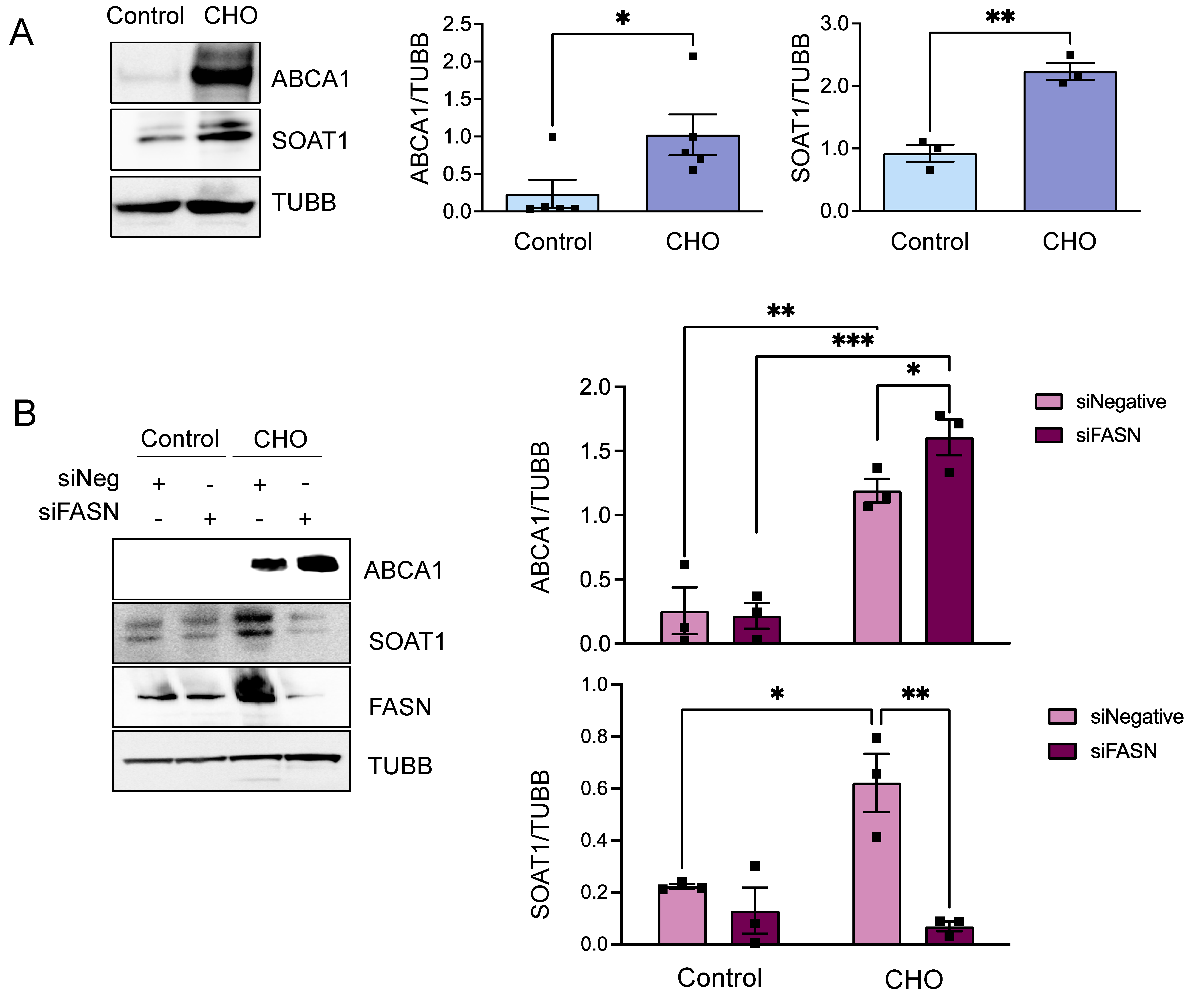
3.1. Cholesterol-Induced Phenotypic Switching Is Accompanied by the Upregulation of the Fatty Acid Synthase (FASN) in VSMC In Vivo and In Vitro
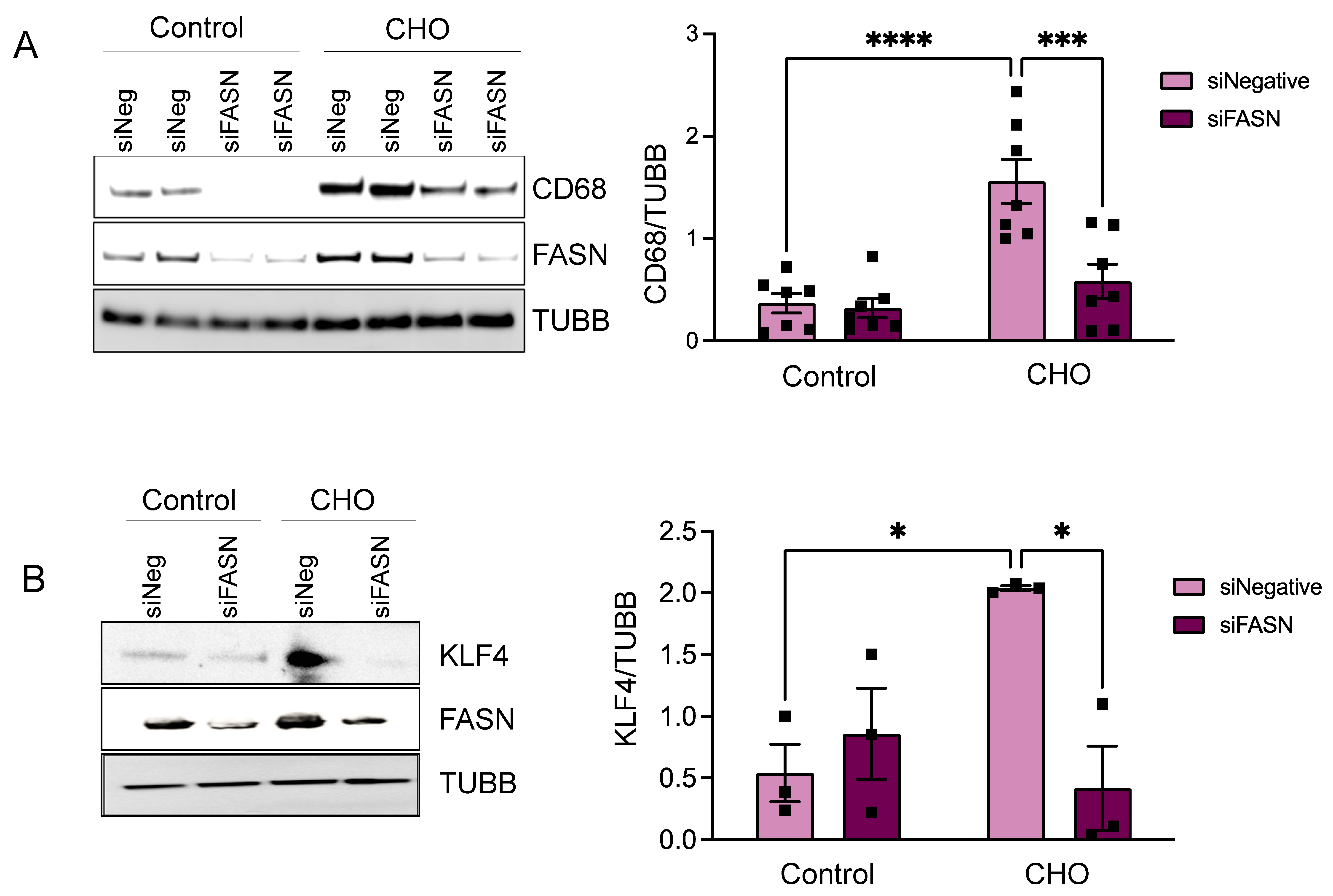
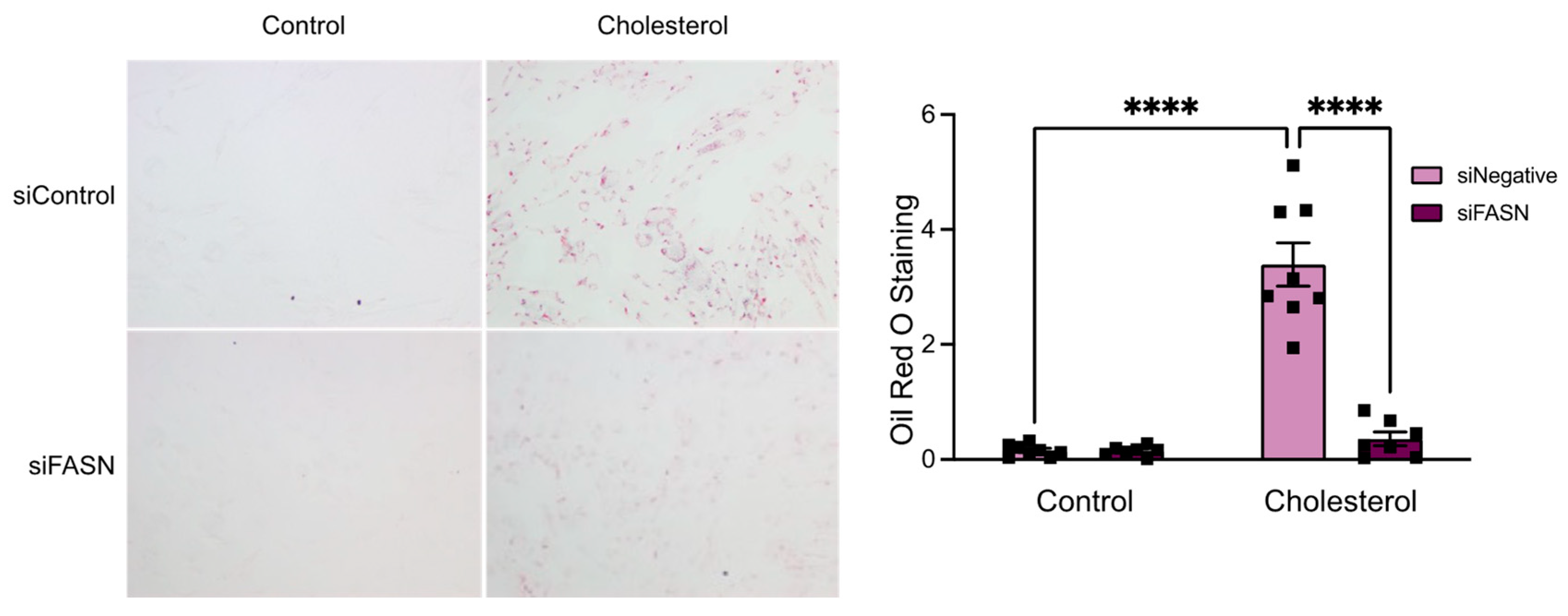
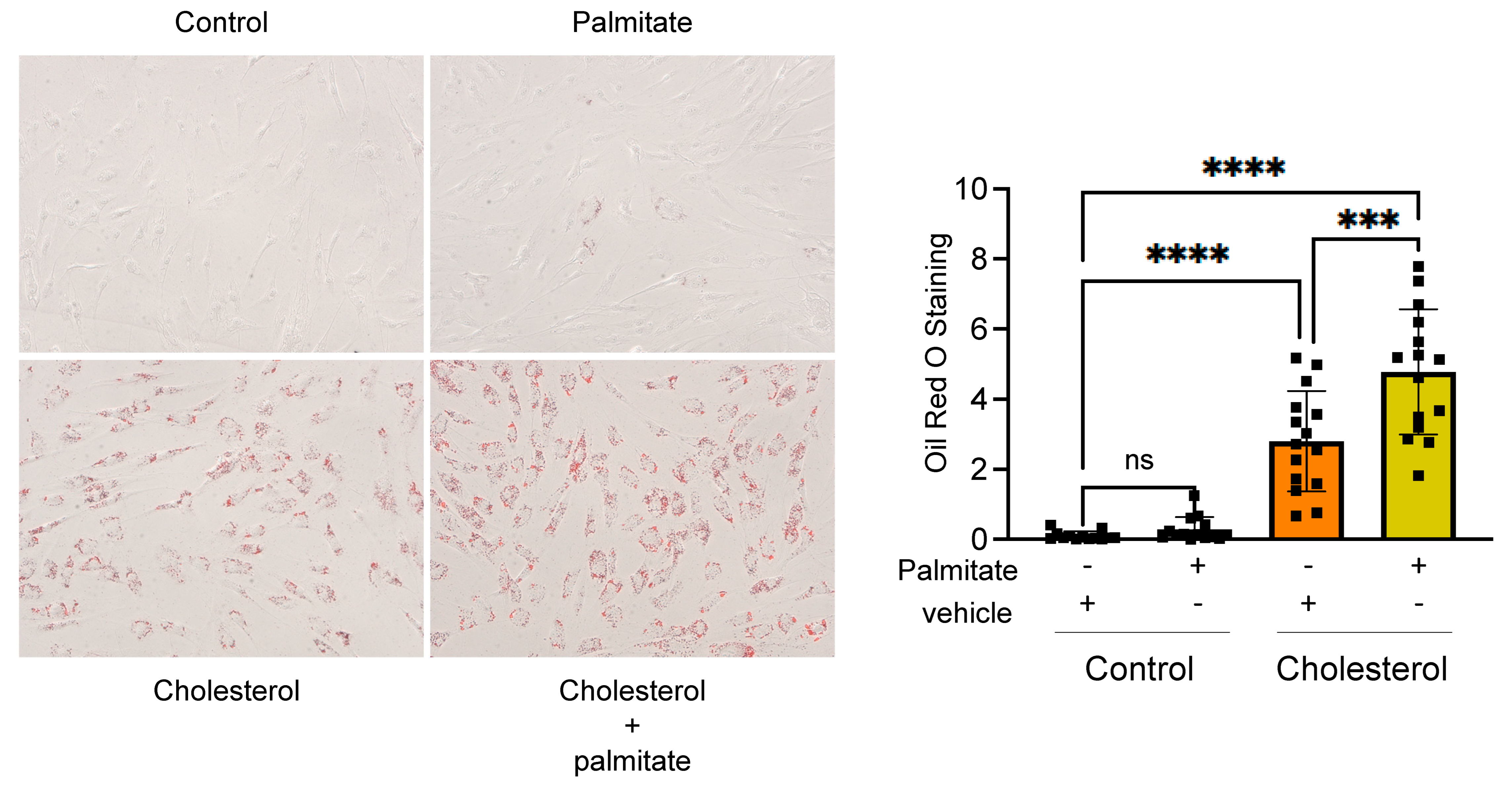
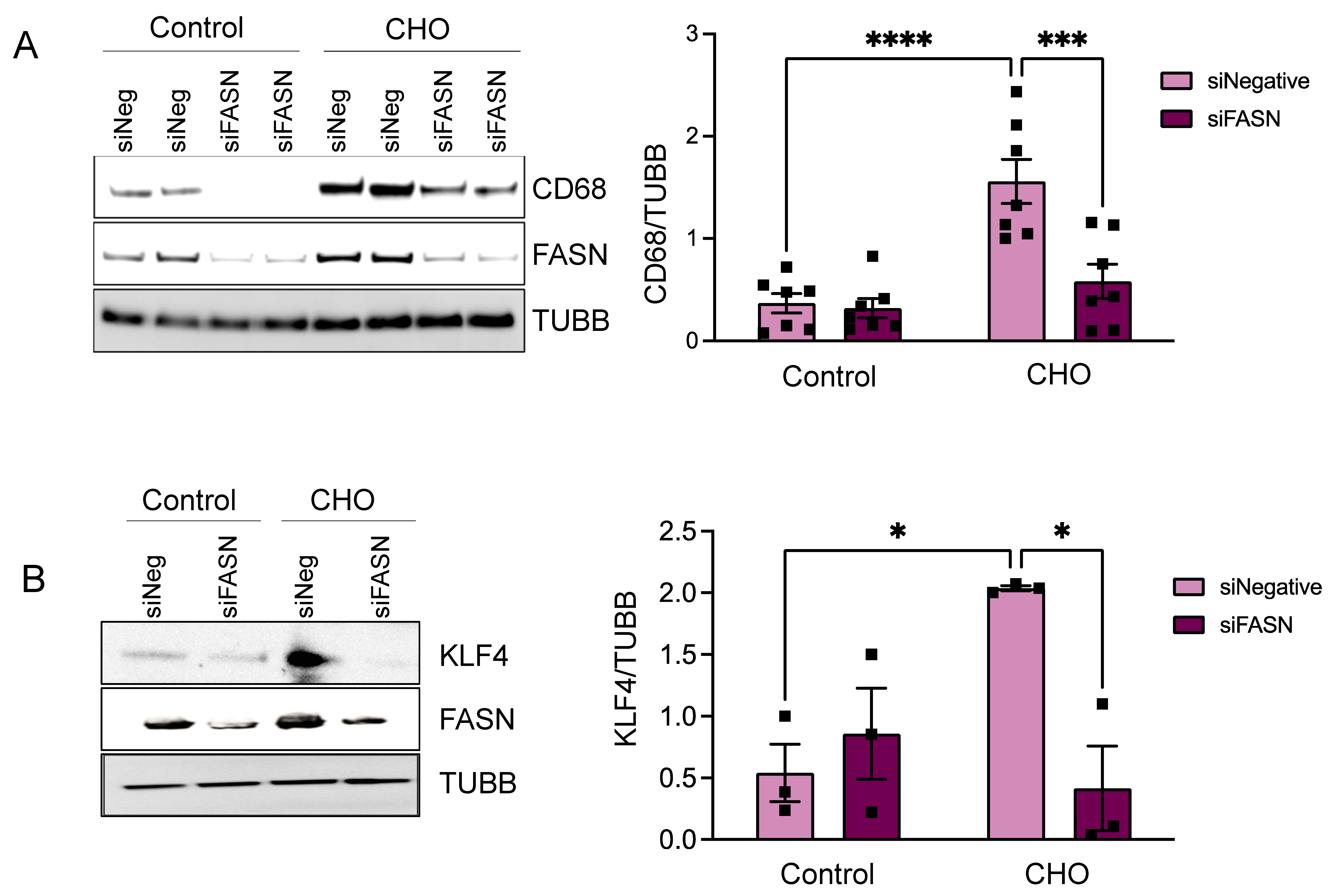
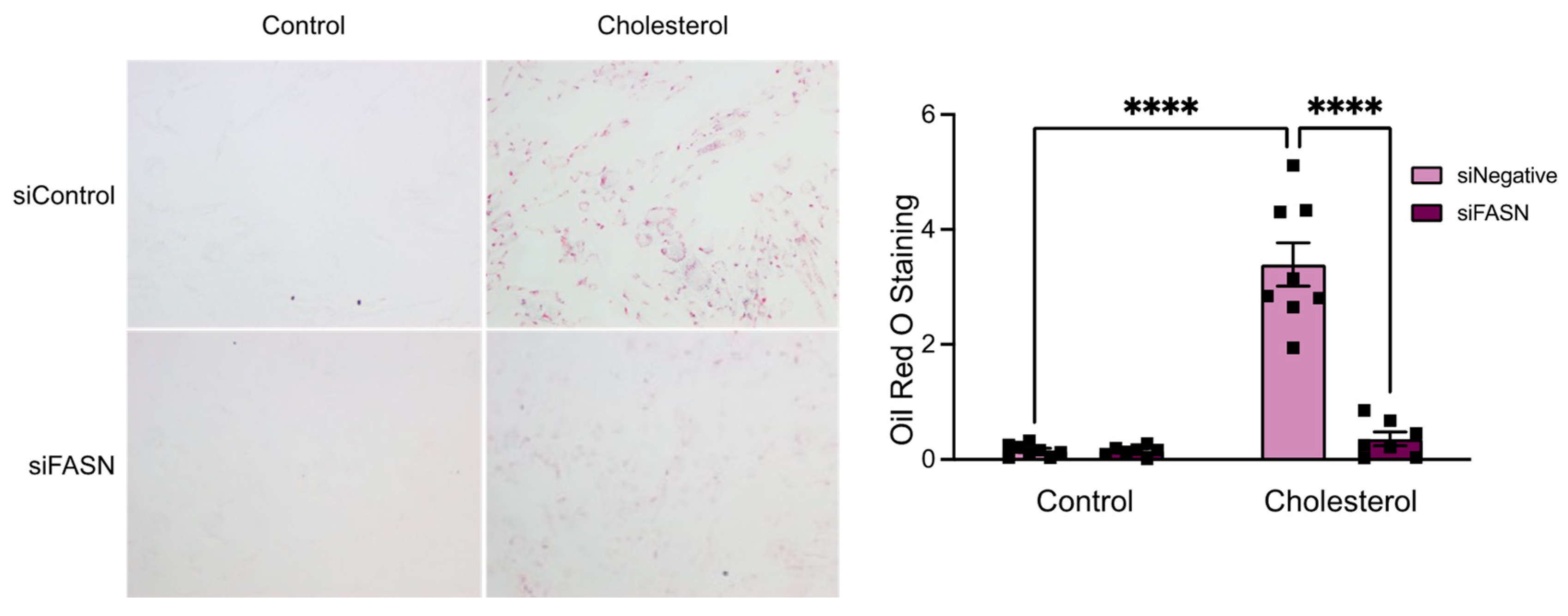
3.2. FASN Deficiency Inhibits Cholesterol-Induced VSMC-Derived Foam Cell Formation
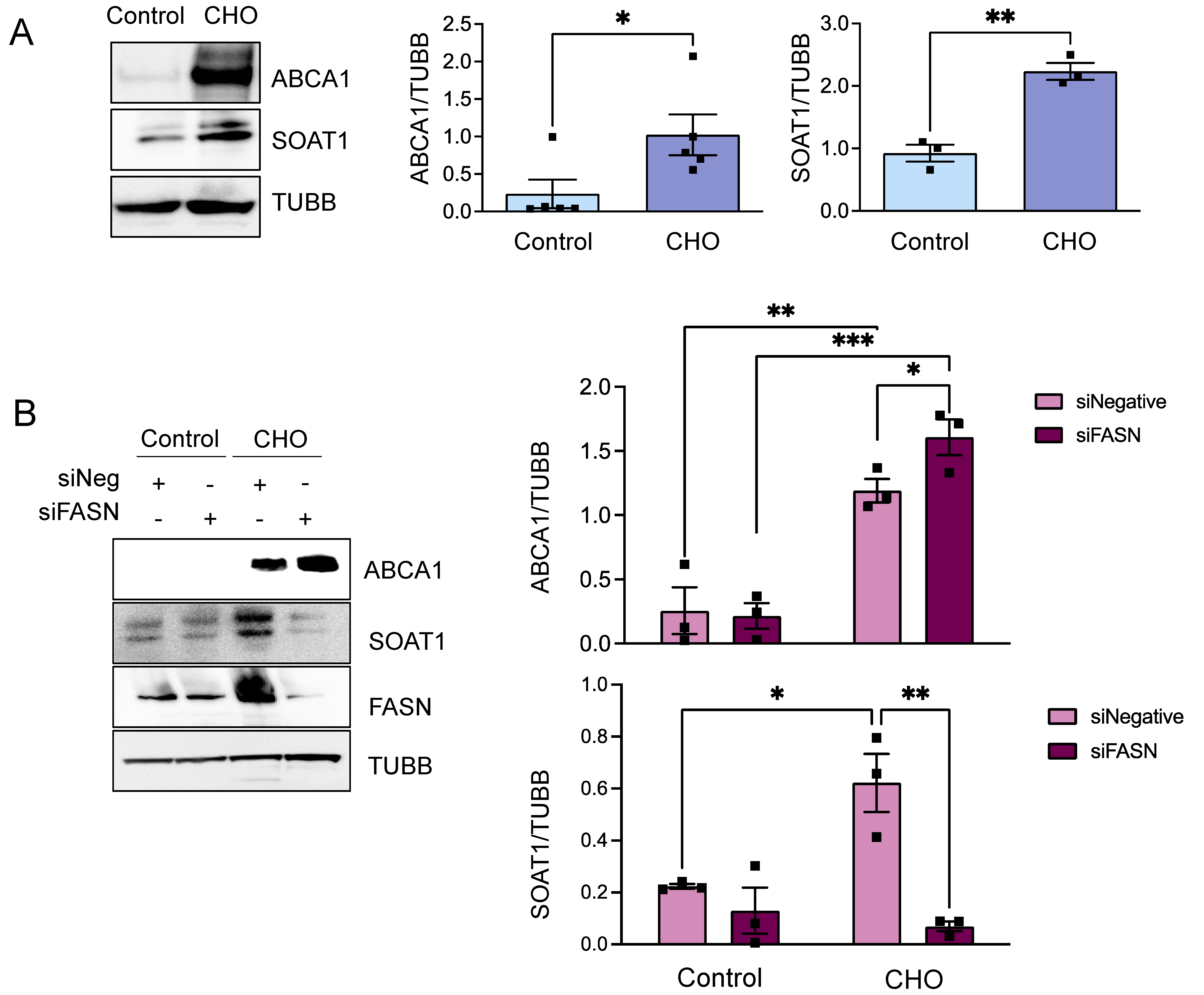
3.3. FASN Regulates Proteins from the Cholesterol Efflux Pathway
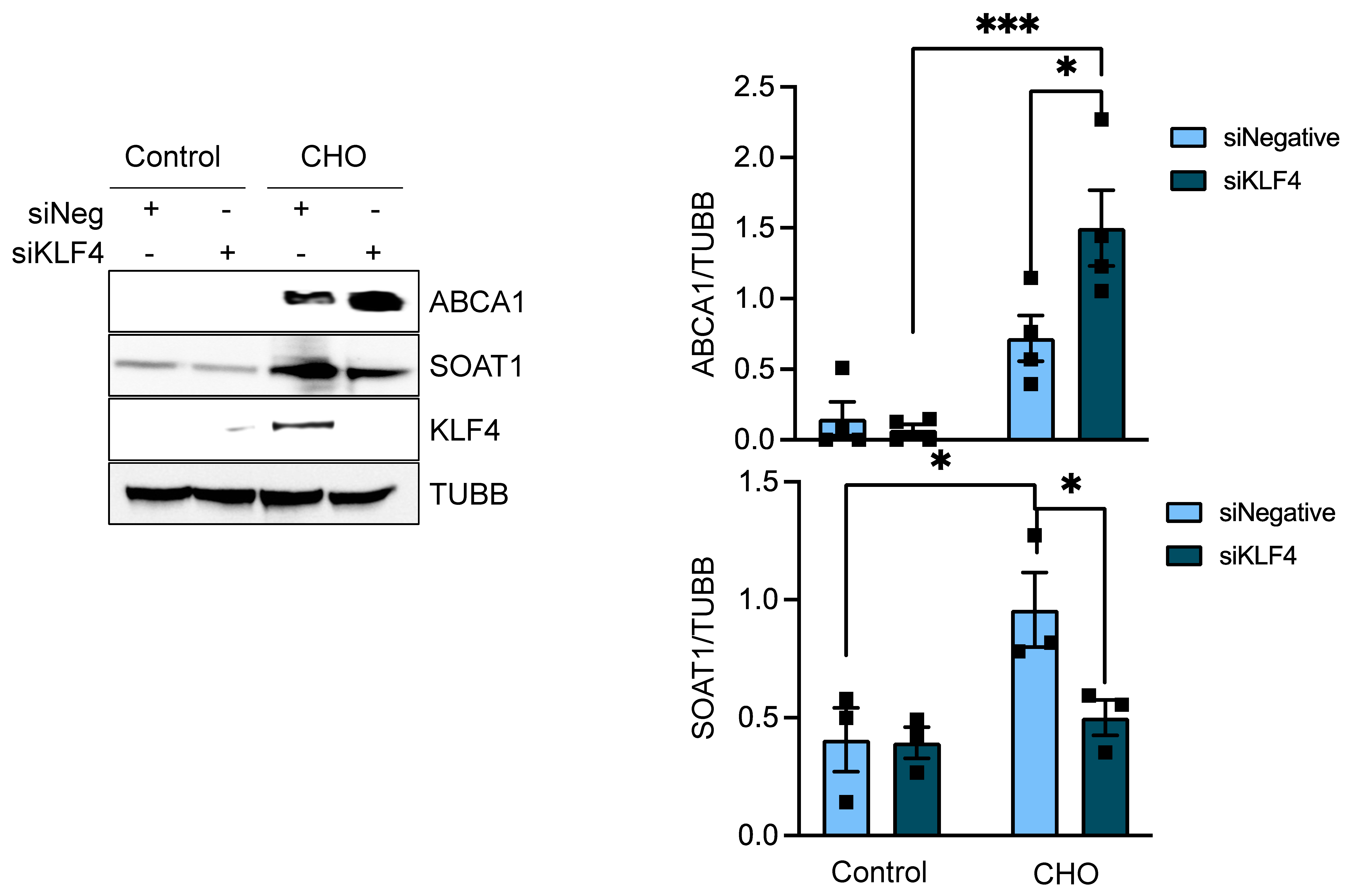
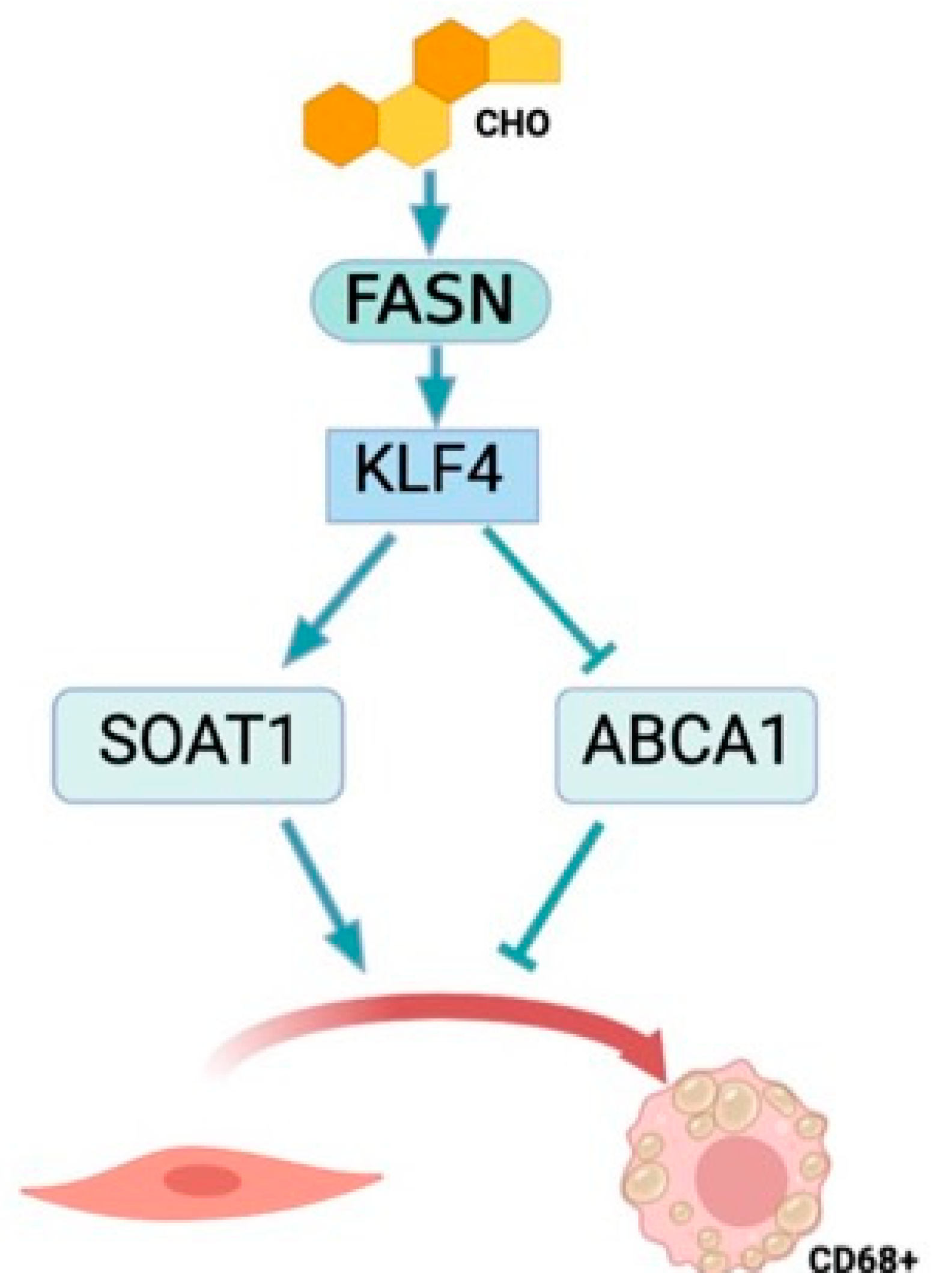
3.4. KLF4 Is the Downstream Effector of FASN
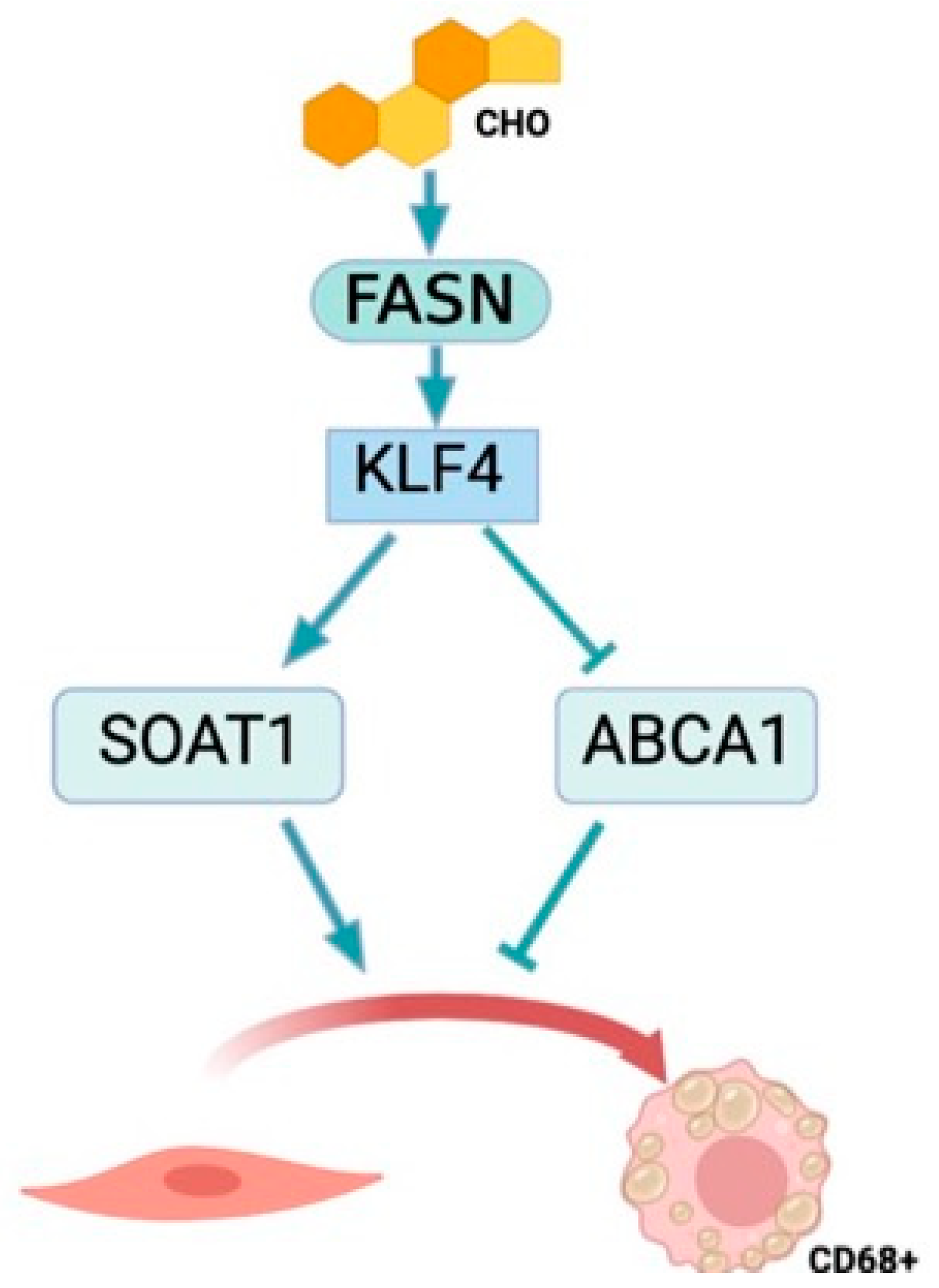
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alencar, G.F.; Owsiany, K.M.; Karnewar, S.; Sukhavasi, K.; Mocci, G.; Nguyen, A.T.; Williams, C.M.; Shamsuzzaman, S.; Mokry, M.; Henderson, C.A.; et al. Stem Cell Pluripotency Genes Klf4 and Oct4 Regulate Complex SMC Phenotypic Changes Critical in Late-Stage Atherosclerotic Lesion Pathogenesis. Circulation 2020, 142, 2045–2059. [Google Scholar] [CrossRef]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Javadifar, A.; Rastgoo, S.; Banach, M.; Jamialahmadi, T.; Johnston, T.P.; Sahebkar, A. Foam Cells as Therapeutic Targets in Atherosclerosis with a Focus on the Regulatory Roles of Non-Coding RNAs. Int. J. Mol. Sci. 2021, 22, 2529. [Google Scholar] [CrossRef]
- Murray, C.J.; Lopez, A.D. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet 1997, 349, 1436–1442. [Google Scholar] [CrossRef]
- Drechsler, M.; Duchene, J.; Soehnlein, O. Chemokines control mobilization, recruitment, and fate of monocytes in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1050–1055. [Google Scholar] [CrossRef]
- Wang, Y.; Dubland, J.A.; Allahverdian, S.; Asonye, E.; Sahin, B.; Jaw, J.E.; Sin, D.D.; Seidman, M.A.; Leeper, N.J.; Francis, G.A. Smooth Muscle Cells Contribute the Majority of Foam Cells in ApoE (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 876–887. [Google Scholar] [CrossRef]
- Jawien, A.; Bowen-Pope, D.F.; Lindner, V.; Schwartz, S.M.; Clowes, A.W. Platelet-derived growth factor promotes smooth muscle migration and intimal thickening in a rat model of balloon angioplasty. J. Clin. Investig. 1992, 89, 507–511. [Google Scholar] [CrossRef]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of Intimal Smooth Muscle Cells to Cholesterol Accumulation and Macrophage-Like Cells in Human Atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef]
- Yang, C.; Xiao, X.; Huang, L.; Zhou, F.; Chen, L.H.; Zhao, Y.Y.; Qu, S.L.; Zhang, C. Role of Kruppel-like factor 4 in atherosclerosis. Clin. Chim. Acta 2021, 512, 135–141. [Google Scholar] [CrossRef]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ. Res. 2014, 115, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Ghaleb, A.M.; Yang, V.W. Kruppel-like factor 4 (KLF4): What we currently know. Gene 2017, 611, 27–37. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Gelissen, I.C.; Ammit, A.J. Regulation of ATP binding cassette transporter A1 (ABCA1) expression: Cholesterol-dependent and - independent signaling pathways with relevance to inflammatory lung disease. Respir. Res. 2020, 21, 250. [Google Scholar] [CrossRef] [PubMed]
- Gonen, A.; Miller, Y.I. From Inert Storage to Biological Activity-In Search of Identity for Oxidized Cholesteryl Esters. Front. Endocrinol. 2020, 11, 602252. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.G.; Yang, Z.; Chakravarthy, M.V.; Lodhi, I.J.; Wei, X.; Turk, J.; Semenkovich, C.F. Macrophage fatty-acid synthase deficiency decreases diet-induced atherosclerosis. J. Biol. Chem. 2010, 285, 23398–23409. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, D.; Weiss, D.; Lassegue, B.; Clempus, R.E.; Szocs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef]
- Stary, H.C.; Chandler, A.B.; Dinsmore, R.E.; Fuster, V.; Glagov, S.; Insull, W., Jr.; Rosenfeld, M.E.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1512–1531. [Google Scholar] [CrossRef]
- Zhang, Z.; Huang, J.; Wang, Y.; Shen, W. Transcriptome analysis revealed a two-step transformation of vascular smooth muscle cells to macrophage-like cells. Atherosclerosis 2022, 346, 26–35. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Shrestha, C.; Ito, T.; Kawahara, K.; Shrestha, B.; Yamakuchi, M.; Hashiguchi, T.; Maruyama, I. Saturated fatty acid palmitate induces extracellular release of histone H3: A possible mechanistic basis for high-fat diet-induced inflammation and thrombosis. Biochem. Biophys. Res. Commun. 2013, 437, 573–578. [Google Scholar] [CrossRef]
- Cousin, S.P.; Hugl, S.R.; Wrede, C.E.; Kajio, H.; Myers, M.G., Jr.; Rhodes, C.J. Free fatty acid-induced inhibition of glucose and insulin-like growth factor I-induced deoxyribonucleic acid synthesis in the pancreatic beta-cell line INS-1. Endocrinology 2001, 142, 229–240. [Google Scholar] [CrossRef]
- Rong, J.X.; Shapiro, M.; Trogan, E.; Fisher, E.A. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc. Natl. Acad. Sci. USA 2003, 100, 13531–13536. [Google Scholar] [CrossRef]
- Liu, X.; Ducasa, G.M.; Mallela, S.K.; Kim, J.J.; Molina, J.; Mitrofanova, A.; Wilbon, S.S.; Ge, M.; Fontanella, A.; Pedigo, C.; et al. Sterol-O-acyltransferase-1 has a role in kidney disease associated with diabetes and Alport syndrome. Kidney Int. 2020, 98, 1275–1285. [Google Scholar] [CrossRef]
- Cao, K.; Zhang, T.; Li, Z.; Song, M.; Li, A.; Yan, J.; Guo, S.; Wang, L.; Huang, S.; Li, Z.; et al. Glycolysis and de novo fatty acid synthesis cooperatively regulate pathological vascular smooth muscle cell phenotypic switching and neointimal hyperplasia. J. Pathol. 2023, 259, 388–401. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogan, B.J.; Williams, H.C.; Holden, C.M.; Patel, V.; Joseph, G.; Fierro, C.; Sepulveda, H.; Taylor, W.R.; Rezvan, A.; San Martin, A. The Role of Fatty Acid Synthase in the Vascular Smooth Muscle Cell to Foam Cell Transition. Cells 2024, 13, 658. https://doi.org/10.3390/cells13080658
Bogan BJ, Williams HC, Holden CM, Patel V, Joseph G, Fierro C, Sepulveda H, Taylor WR, Rezvan A, San Martin A. The Role of Fatty Acid Synthase in the Vascular Smooth Muscle Cell to Foam Cell Transition. Cells. 2024; 13(8):658. https://doi.org/10.3390/cells13080658
Chicago/Turabian StyleBogan, Bethany J., Holly C. Williams, Claire M. Holden, Vraj Patel, Giji Joseph, Christopher Fierro, Hugo Sepulveda, W. Robert Taylor, Amir Rezvan, and Alejandra San Martin. 2024. "The Role of Fatty Acid Synthase in the Vascular Smooth Muscle Cell to Foam Cell Transition" Cells 13, no. 8: 658. https://doi.org/10.3390/cells13080658
APA StyleBogan, B. J., Williams, H. C., Holden, C. M., Patel, V., Joseph, G., Fierro, C., Sepulveda, H., Taylor, W. R., Rezvan, A., & San Martin, A. (2024). The Role of Fatty Acid Synthase in the Vascular Smooth Muscle Cell to Foam Cell Transition. Cells, 13(8), 658. https://doi.org/10.3390/cells13080658